

SUMMARY
Genomes of the major human helminth parasites, and indeed many others of agricultural significance, are now the research focus of intensive genome sequencing and annotation. A draft genome sequence of the filarial parasite Brugia malayi was reported in 2007 and draft genomes of two of the human schistosomes, Schistosoma japonicum and S. mansoni reported in 2009. These genome data provide the basis for a comprehensive understanding of the molecular mechanisms involved in schistosome nutrition and metabolism, host-dependent development and maturation, immune evasion and invertebrate evolution. In addition, new potential vaccine candidates and drug targets will likely be predicted. However, testing these predictions is often not straightforward with schistosomes because of the difficulty and expense in maintenance of the developmental cycle. To facilitate this goal, several developmental stages can be maintained in vitro for shorter or longer intervals of time, and these are amenable to manipulation. Our research interests focus on experimental studies of schistosome gene functions, and more recently have focused on development of transgenesis and RNA interference with the longer term aim of heritable gene manipulation. Here we review methods to isolate and culture developmental stages of Schistosoma mansoni, including eggs, sporocysts, schistosomules and adults, in particular as these procedures relate to approaches for gene manipulation. We also discuss recent advances in genetic manipulation of schistosomes including the deployment of square wave electroporation to introduce reporter genes into cultured schistosomes.
Keywords: Schistosome, sporocyst, schistosomule, egg, in vitro culture, electroporation
SCHISTOSOME GENOMICS
The helminth parasites of humans belong to the phyla Platyhelminthes (the flatworms) and Nematoda (the roundworms). The flatworms include flukes (trematodes) and tapeworms (cestodes). Schistosomes are blood flukes – the adult forms reside in the venules of the intestines (Schistosoma japonicum, S. mansoni) or urogenital system (S. haematobium). Unlike other platyhelminths, schistosomes are dioecious, and exhibit marked sexual dimorphism as adults. Advances in molecular genetics and immunology hold the promise to control the spread of schistosomiasis and to combat the morbidity and mortality associated with this great neglected tropical disease (Hotez et al. 2008). Control of schistosomiasis largely relies on chemotherapy with praziquantel, and the wide-scale use of this drug has led to concerns that drug resistance might develop (see Utzinger and Keiser, 2004). As a consequence, the WHO Schistosoma Genome Project was initiated in the early 1990s with the declared goal to identify target genes in order to be able to address fundamental questions on the biology and physiology of the parasites, and to guide the rational development of new interventions (Hu et al. 2004; Han et al. 2009). Schistosomes have comparatively large genomes, estimated at 398 megabase pairs (MB) for the haploid genome of S. japonicum (Schistosoma japonicum Genome Sequencing and Functional Analysis Consortium et al. 2009) and 363 MB for S. mansoni (Berriman et al. 2009) arrayed on seven pairs of autosomes and one pair of sex chromosomes. S. haematobium, the other major schistosome species parasitizing humans probably has a genome of similar size, based on similarity of the karyotypes (Hirai et al. 2000). The schistosome genomes represent the first of any of the Lophotrochozoa to be published. Analysis of the genomes revealed wide-spread domain structure reduction, complex signal transduction and sensory pathways, proliferation of mini-exons, unusual intron size distribution, large numbers of protease encoding genes, and other singular features (Schistosoma japonicum Genome Sequencing and Functional Analysis Consortium et al. 2009; Berriman et al. 2009). The reports of the draft genomes of S. japonicum and S. mansoni followed the reasonably comprehensive descriptions of the transcriptomes and proteomes (Hu et al. 2003; Verjovski-Almeida et al. 2003; Liu et al. 2006, 2008; Jolly et al. 2007; Hokke et al. 2007; Gobert et al. 2009). Less is known about S. haematobium although since more people are infected with this pathogen than both S. japonicum and S. mansoni combined (see Hotez et al. 2008), and because it also causes bladder cancer (Bouvard et al. 2009), genomics and transcriptomics study of S. haematobium clearly deserves more attention.
CULTURE METHODS, MOLECULAR TOOLS TO INVESTIGATE GENE FUNCTION
Despite this abundance of sequence data, functional analysis of potential target genes will not be possible until reliable methods for reverse genetics in schistosomes become available. Whereas reverse genetics tools including gene silencing by RNAi and transgenesis are finding utility in schistosomes (Morales et al. 2008; Rinaldi et al. 2009) in vitro culture techniques remain necessary to undertake these gene manipulations and other approaches. Transformation and gene manipulation in schistosomes have been reviewed (e.g. Beckmann et al. 2007; Brindley and Pearce, 2007) and informative reviews are available also on the general maintenance of the schistosome life cycle (e.g. Hackett, 1993; Lewis, 1998). To date, tools and protocols for gene manipulation of schistosomes remain rudimentary, especially when compared to established transgenesis systems and techniques in other pathogens such as some parasitic protozoa and for vector mosquitoes (Crabb and Gilson, 2007; Terenius et al. 2008), as well as in mammalian systems (e.g. Sasaki et al. 2009). The fundamental nature of the blood flukes – the complex developmental cycle, large size, multicellular tissues and complex organization, and the absence of immortalized cell lines and inability to rear the entire life cycle in vitro, have hindered development of tractable transgenesis models. Although the entire developmental cycle of the human schistosomes cannot be maintained in vitro, laboratory maintenance of the developmental cycles of all three human schistosomes can be accomplished using rodents as the mammalian hosts and natural host snail species as the intermediate hosts (Lewis, 1998). In addition, a number of the developmental stages, both mammalian and molluscan parasitic stages can be maintained in the laboratory.
Schistosomes are large, multicellular eukaryotes, and though aceolomate, they possess complex organ systems including a gut, female and male reproductive tissues, muscles, nervous tissues with eyespots and so forth. The blood-stage forms are covered by a syncytial tegument that is bounded at the parasite-host interface with a double lipid bilayer. Furthermore, the developmental stages differ dramatically in appearance and structure, cell numbers, ratio of germ to soma, and morphology. All these features pose challenges for genetic manipulation and certainly for germ line transgenesis. However, genetic manipulation and germ line transgenesis are worthwhile goals because they would facilitate a deep understanding of the molecular biology of schistosomes, roles of molecules in host-parasite interaction and, ultimately, to identify molecules that could be targeted/disrupted with drugs or vaccines. Here we review recent approaches to maintenance of developmental stages of S. mansoni, specifically in relation to genetic manipulation of these parasites for subsequent investigation of genetic transformation and transgenesis (Kines et al. 2006, 2008; Morales et al. 2007) as well as transient gene silencing by RNAi (Morales et al. 2008; Rinaldi et al. 2009).
ADULTS
S. mansoni worms commence shedding eggs from 42 days after infection of mice; many eggs are discharged with the faeces although a large percentage of all eggs produced eventually become entrapped in the capillary beds of the liver and other organs. We recover adult S. mansoni from experimentally infected mice six to 12 weeks after infection by portal perfusion, as described (e.g. Smithers and Terry, 1965; Duvall and DeWitt, 1967; Lewis, 1998). In brief, we euthanize infected mice with an overdose of sodium pentobarbital administered along with heparin into the peritoneal cavity (Duvall and DeWitt, 1967). This causes the schistosomes to shift from the mesenteric veins to the liver and inhibits blood clots, both of which enhance efficiency of perfusion of the worms from the vasculature. After death, the mouse is thoroughly wetted with 70% ethanol using a pump-spray to minimize potential for microbial contamination (and contamination with mouse hair) in the subsequent procedures. We make an incision with scissors in the abdomen after which the skin is peeled back to expose the peritoneum. Thereafter we cut through the rib-cage and diaphragm to expose the heart, and position the mouse over a 9 cm diameter Petri dish in order to collect adults soon to be flushed from the mesenteric veins. The hepatic portal vein (enlarged in schistosome-infected mice because of portal hypertension) of the mouse is located and severed with scissors. Occasionally, a bolus of adults immediately comes out of the vein and it is convenient to rinse the mouse viscera close to the portal vein in order to collect them in order to recover as many adult worms as possible before the perfusion. We insert a 20-gauge needle fitted to (6.35 mm internal diameter) tubing from a peristaltic pump (e.g. Simon Varistaltic pump model no. 72-312-000), connected to a reservoir of at least one litre of perfusion buffer (150 mM sodium chloride, 15 mM sodium citrate, pH to 7.0 with sodium hydroxide) (=1×SSC) into the left ventricle. We inject perfusion buffer into the heart at a flow rate of~5 ml per min and perfuse the vasculature of the mouse with ~10 ml buffer. If the portal perfusion is effective, the liver changes from a deep maroon to greyish colour. The adult schistosomes are discharged from the severed hepatic portal vein into the Petri dish along with the blood and perfusate. A second perfusion can be undertaken in order to recover as many adult worms as possible. After perfusion, we rinse the mouse viscera with additional perfusion buffer (~2 ml discharged from the pump) with the aim of flushing out additional adult schistosomes that may have been perfused from the portal system but which lodged on the exterior of the viscera or within the peritoneal cavity. We remove the liver from the perfused mouse, taking care not to perforate the stomach or intestines, and transfer the liver to a sterile 50 ml conical tube.
After the infected mice have been necropsied and the schistosomes perfused, we transfer the pooled perfusion buffer, adult schistosomes and blood to a sterile beaker (e.g. 600 ml beaker) until the adults can be prepared for in vitro culture. The adults should be removed as soon as practical (preferably within an hour) from the bloody perfusion buffer. We wash the worms by gravity using phosphate buffered saline (1× PBS) with 1× antibiotic/antimycotic (Invitrogen, Carlsbad, CA, catalogue no. 15240-062). This is performed several times until the PBS is apparently free of blood and other visible contaminants such as mouse hair. Thereafter, we culture the worms in DMEM supplemented with 10% foetal calf serum (FCS) serum and 1× antibiotic/antimycotic (Invitrogen) under 5% CO2 in air at 37 °C (Table 1). For long-term culture (>two days), we supplement the culture of adult schistosomes with washed human erythrocytes, e.g. one μl erythrocytes (50% suspension)/ml culture medium (Kines et al. 2008). In general, we culture from several to 20 adult worms (mixed sexes) in 3–5 ml of culture medium in 6-well plates (Sarstedt Inc, Newton NC, catalogue no. 83.1839).
Table 1.
Culture conditions and media for developmental stages of Schistosoma mansoni
| Developmental stage | Medium | Atmosphere/Temperature | Key references |
|---|---|---|---|
| Egg | Dulbecco’s modified Eagle’s medium (DMEM) with 10% foetal bovine serum (FBS), 1× penicillin and streptomycin | 5% CO2 in air; 37 °C | Rinaldi et al. (2009) |
| Primary sporocyst | MEMSJ-E | 5% CO2, 5% O2; 90% N2; 27 °C | Kawanaka et al. (1983, 1985) |
| Schistosomule | Modified Basch’s medium supplemented with human erythrocytes | 5% CO2 in air; 37 °C | Basch (1981) |
| Adult | Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum, 1× antibiotic/antimycotic, and human erythrocytes | 5% CO2 in air; 37 °C |
Morales et al. (2008) Kines et al. (2008) |
EGGS
This method is based on the procedure of Dalton et al. (1997). We carry out the procedures in a biological safety cabinet in order to enhance aseptic cultivation of the eggs. We transfer livers from infected mice at necropsy to a 50 ml conical tube (above), including three to five infected livers per tube. We fill the tube to the 40 ml mark with perfusion buffer and retain the tube on ice until all the livers are processed. Afterwards, we remove the livers from the conical tube with sterile forceps, and if possible, remove the gall bladder and fibrous tissues. We transfer the liver to a 9 cm Petri dish that contains~5 ml 70% ethanol where the liver is rinsed thoroughly. We replace the dish and ethanol for each liver. After this, we rinse the liver in Dulbecco’s phosphate buffered saline (DPBS) supplemented to 1× antibiotic/antimycotic, and then transfer the livers to a sterile Petri dish. Here we chop/mince the liver to a fine consistency using a sterile razor blade or scalpel (we often process 10 livers at a time), and then transfer the chopped livers to a 50 ml conical tube, three to five chopped livers per tube. We also rinse the Petri dish with DPBS and transfer this wash to the chopped livers in the 50 ml tube. We fill the tube to the 40 ml mark with DPBS, then add 5 ml of 5% clostridial collagenase solution (25 mg) (Sigma-Aldrich, St. Louis, MO, catalogue no. C5138). The collagenase is included to release the eggs from granulomas by proteolysis of the interstitial matrix of mouse liver tissue. We prepare and sterile filter the collagenase freshly each time. We also include 500 μl of polymixin B (100K Units) (Sigma-Aldrich, P4932-1MU), a gram negative bactericidal antibiotic. If processing fewer livers, we adjust the concentrations of collagenase and polymixin B accordingly. We cap the 50 ml tube tightly and seal the exterior of cap with several layers of tightly bound Parafilm. The capped tube containing the liver/collagenase digest is incubated at 37 °C for 15–18 hours, with gentle shaking. Subsequently, we centrifuge the tube containing the digested livers at 400× g for 5 min at RT. After decanting the supernatant, we resuspend the pelleted material (containing the schistosome eggs) by refilling the tube to 50 ml with DPBS containing 1× antibiotic/antimycotic. (We have seen liver cells on top of the pellet of eggs; these can gently decanted without appreciable loss of schistosome eggs.) We resuspend the pellet material containing the eggs by refilling the tube to 50 ml with DPBS containing 1× antibiotic/antimycotic, and repeat the wash procedure three more times. Afterwards, the pellet is resuspended in 25 ml DPBS. Using a 10 ml serological pipette, we force the suspension through a sterile 250 μm sieve (Arthur H. Thomas Co., Philadelphia, PA) into a sterile beaker (e.g. 600 ml beaker). The resulting filtrate is forced through a 150 μM sieve (Arthur H. Thomas Co.) into a second sterile beaker, transferred to a 50 ml conical tube, and centrifuged at 400× g for 5 minutes at room temperature. In parallel, we prepare a Percoll column: 8 ml of sterile Percoll mixed with 30 ml of sterile filtered 0.25 M sucrose in a 50 ml conical tube. After the filtrate is centrifuged, we decant the supernatant and add three ml of DPBS to the pellet. The resultant slurry is mixed thoroughly after which it is applied gently, using a serological pipette, to the top of the Percoll gradient, minimizing disruption to the surface of the Percoll/sucrose solution. The Percoll gradient is centrifuged at 800× g for 10 minutes in a swinging bucket rotor (e.g. Eppendorf 5810 R, rotor A-4-62). By aspiration starting at the top, we remove the surface layer of the gradient that contains liver cells. We transfer the eggs (pelleted at the bottom of the tube) to a new tube and resuspend in DPBS. These are washed three times in DPBS, and then resuspended in 0.5 ml DPBS. We apply this to the surface of a second Percoll column, prepared by combining 2.5 ml Percoll with 7.5 ml of sterile filtered 0.25 M sucrose in a 15 ml conical tube. The eggs are centrifuged through this second Percoll column, and washed as above. We remove as much of the DPBS as practical, and suspend the eggs in Dulbecco’s modified Eagle’s medium (DMEM) with 10% foetal bovine serum (FBS) and 1× penicillin/streptomycin (Invitrogen, catalogue no. 15140-122). We split the eggs into 2 ml aliquots in a 6-well plate and culture at 37 °C, 5% CO2 (Fig. 1A–D) (Table 1).
Fig. 1.

Cultured eggs of Schistosoma mansoni. These eggs were isolated from livers of experimentally infected mice. Panel A and B: micrographs illustrating a mixed population, mature and immature eggs, three days after isolation from mouse liver. Scales bars=50 μm. Panel C: two immature eggs (black arrow-heads) and a mature egg containing the miracidium (black arrow); scale bar=20 μm. Panel D: a mature egg containing miracidium (black arrow) and an empty egg shell after hatching has taken place (black arrow-head); Scale bar=20 μm. The micrographs were taken using a digital camera (Zeiss AxioCam ICc3) fitted to an inverted microscope (Zeiss Axio Observer A1).
Dalton et al. (1997) demonstrated using Southern hybridization analysis that eggs purified with this Percoll-based procedure are free of detectable contaminating host tissues, and therefore can be confidently employed in molecular studies. The eggs can be maintained in culture for a maximum of 10 days in DMEM media containing 10% FBS and 1× penicillin/streptomycin. We recommend carrying out manipulations of cultured eggs before they reach seven days age of culture. After this, they begin to lose viability; after three weeks in culture, we have observed they will not hatch upon transfer to distilled water (below).
MIRACIDIA
To isolate miracidia, we resuspend the S. mansoni eggs in 5 ml sterile distilled H20 (dH20) at 23 °C, 1000 to 2000 eggs per ml. The eggs can be aliquoted in one ml volumes into the wells of a sterile 24-well tissue culture plate (Sarstedt), after which an additional one ml of dH20 can be added to the well. We then locate the 24-well plate directly under a fluorescent light (low heat) source to induce hatching of the eggs (Lewis, 1998) (Fig. 2A, B). We remove one ml of supernatant from the surface (to avoid non hatched eggs) every 30 min or so, using a pipette, and transfer water containing miracidia into a 50 ml conical tube, on ice. This process is continued at 30 minute intervals for two to three hours to harvest miracidia. After the final harvest, the miracidia can be counted; they will have settled to the bottom of the 50 ml tube because of the ice cold temperature. However, if they remain non-concentrated, the tube can be centrifuged at 800× g for 10 min at 4 °C after which the supernatant is removed quickly by suction pipetting. At that point, we resuspend the miracidia in a minimal volume of dH2O, where they can be counted by sampling several small aliquots, e.g. 5 μl miracidia added to 15 μl 95% ethanol, counted using a microscope.
Fig. 2.

Miracidia of Schistosoma mansoni hatching from eggs and transforming into mother sporocysts in vitro. Panel A: mature egg ready to hatch (arrow-head) and two immature eggs. Panel B: two miracidia after hatching. Panel C: miracidium losing its ciliated plates (arrow-head). Panel D: in vitro cultured mother sporocyst (black arrow). Scale bars: 20 μm. The micrographs were taken using a digital camera (Zeiss AxioCam ICc3) fitted to a Zeiss Axio Observer A1 microscope.
SPOROCYSTS
To establish cultures on in vitro transformed sporocysts, we transfer miracidia from water (above) into sporocyst media in 6-well plates (Sarstedt) at a concentration of 500 to 2000 miracidia per well. For this, we employ a chemically defined medium termed MEMSE-J (Kawanaka et al. 1983, 1985) (Table 1). The formula for this medium is presented in Table 2; we prepare MEMSE-J in one litre batches, and store in aliquots of 50 ml at −20 °C until needed. Regardless of the concentration of the miracidia, we include ≤500 μl of water containing the miracidia into the 6-well plate, and increase the volume per well to 5 ml with sporocyst medium. We maintain the 6-well plate containing the developing sporocysts in a hypoxia chamber (e.g. Billups-Rothenberg, Del Mar, CA catalogue no. MIC-101) under 5%O2, 5% CO2, 90%N2 at 27 °C (Bixler et al. 2001). It is important to maintain the atmospheric pressure in the chamber below 2 psi (<15 kPa) in order not to damage the chamber’s atmospheric seal. After the miracidia have shed the ciliated plates (Fig. 2B–D), at which point they are referred to as in vitro cultured sporocysts, they can be maintained for at least 14 days. In addition, other investigators have maintained in vitro cultured sporocysts in co-cultures with Bge cells (ATCC, Manassas, VA, catalogue no. CRL-1494), a cell line from the pulmonate snail Biomphalaria glabrata (see Coustau and Yoshino, 2000), or Bge conditioned culture medium. This can facilitate longer term, perhaps continuous, culture of the sporocysts and even production of cercariae (Ivanchenko et al. 1999), although this is challenging. In vitro cultured sporocysts can be transplanted into B. glabrata snails to establish lines of S. mansoni (see Kapp et al. 2003).
Table 2.
Composition of MEMSJ mediuma for culture of Schistosoma mansoni primary sporocysts
| Componentb | Concentration (mg/L) | |
|---|---|---|
| 1. | L-Arginine | 126 |
| 2. | L-Cysteine | 24 |
| 3. | L-Histidine.HCL.H2O | 42 |
| 4. | L-Isoleucine | 52 |
| 5. | L-Leucine | 52 |
| 6. | L-Lysine. HCl | 72·5 |
| 7. | L-Methionine | 15 |
| 8. | L-Phenylalanine | 32 |
| 9. | L-Threonine | 48 |
| 10. | L-Tryptophan | 10 |
| 11. | L-Tyrosine | 36 |
| 12. | L-Valine | 46 |
| 13. | L-Glutamine | 300 |
| 14. | Glycine | 10 |
| 15 | L-Serine | 46 |
| 16. | Choline chloride | 3 |
| 17. | Nicotinamide | 1 |
| 18. | NaCl | 6800 |
| 19. | KCl | 400 |
| 20. | CaCl2 | 200 |
| 21. | MgCl2.6H2O | 200 |
| 22. | NaH2PO4.2H2O | 150 |
| 23. | NaHCO3 | 2000 |
| 24. | Phenol red | 10 |
| 25. | Penicillin/streptomycin NaOH to pH 7·2 | 1× |
Adapted from Kawanaka et al. (1983).
Components 1–12 are the same as Invitrogen’s 1× MEM amino acid solution (catalogue no. 11130-051).
SCHISTOSOMULA
We obtain Biomphalaria glabrata snails infected with the NMRI (Puerto Rican) strain of Schistosoma mansoni from Dr. Fred Lewis, Biomedical Research Institute (Rockville, MD) and culture the snails in aquaria at 25 °C. Cercariae begin to exit infected snails into the aquarium water from about 35 days after infection with miracidia (Lewis, 1998). We wash the shells of the infected snails with deionized water (or aged tap water) delivered in a jet stream from a domestic garden-type weed sprayer to remove rotifers. For this task, we transfer the snails from aquaria into a kitchen-style mesh colander, and pressure spray them over a sink in order to remove rotifers and other commensals that tend to establish on the shells of the snails. Rotifers release metabolites injurious to schistosome cercariae (Stirewalt and Lewis, 1981). Subsequently, the snails are transferred to 100–200 ml of de-chlorinated water, and the snails in the beaker illuminated from about 20 cm above with a fluorescent light (60 watt equivalent) source. After one to two hours, the water containing the cercariae is transferred into a second sterile beaker avoiding any debris in the bottom of the beaker. To remove residual debris, we pour the cercarial water through a stainless steel screen (e.g. Millipore catalogue no. XX1004730, 47 μm) into a 50 ml conical tube(s). The snails are returned to aquaria. We concentrate the cercariae by centrifugation (400×g/10 min) and wash them ×3 with somule wash, DPBS supplemented with 1× antibiotic/antimycotic or similar wash buffer, e.g. Correnti and Pearce (2004). The cercariae are resuspended in ~4 ml of somule wash in a sterile Petri dish. A 22- gauge double-hub, emulsifying needle with a stabilizing bar (Popper & Sons, New Hyde, New York, catalogue no. 7975) is fitted to a sterile 10 ml Luerlock syringe. A second 10 ml syringe is used to draw up the cercariae. An additional three ml of somule wash is added to the Petri dish and this is also drawn up into the syringe. This syringe is fitted to the open end of the emulsifying needle. The tails of cercariae are sheared o. by ~20 passes back and forth through the emulsifying needle. (Because of obvious biohazard with this procedure, which involves manipulation of thousands of cercariae, we recommend that protective clothing and gloves, including protection for the face and the eyes be worn.) Thereafter, the schistosomule bodies are isolated from the sheared tails by Percoll gradient centrifugation (Lazdins et al. 1982). After the cercariae have been sheared, the contents of the syringes are dispensed into a 15 ml conical tube and concentrated by centrifugation 450× g for 5 min. The Percoll gradient is prepared in 15 ml conical tubes; 10 ml of the Percoll gradient solution is added to each tube (12 ml Percoll, 2 ml 10× PBS, 0.5 ml 100 mM Hepes, 0.5 ml antibiotic antimycotic, 5 ml sterile water). The cercarial bodies [schistosomula] are resuspended in one ml somule wash. 0.5 ml of the schistosomula suspension is applied to each column carefully in order not, or only minimally, to disturb the surface of the Percoll gradient. We centrifuge the gradient at 450× g for 10 minutes, with the brake at its least forceful setting (e.g. setting number 1). The tails will be at the top of the column, the schistosomule bodies at the bottom. Thereafter, we remove the top layers of the gradient first; the schistosomula should be pelleted at the bottom of the tube. (Cercarial tails can be retained and stored at −80 °C for later use, for example as a source of genomic DNA.) We wash the schistosomules three times in wash medium and transfer them to modified Basch’s medium at 37 °C under 5% CO2 in air (Tables 1, 3) (Basch, 1981). For procedures where we have investigated the activity of transgenes and/or schistosome gene promoters driving transgenes, we and others have cultured schistosomules for >15 days (Correnti et al. 2005, 2007; Morales et al. 2007) (Fig. 3A–D).
Table 3.
Composition of modified Basch’s medium Basch’s medium
| Basch’s medium (Basch, 1981) | Working concentration |
|---|---|
| Basal Medium Eagle (Invitrogen catalogue no. 21010) | to final volume |
| Lactalbumin hydrolysate | 1 g/L |
| Glucose | 1 g/L |
| Hypoxanthine | 0·5 μM |
| Serotonin | 1 μM |
| Insulin | 8 μg/ml |
| Hydrocortisone | 1 μM |
| Triiodothyronine | 0·2 μM |
| MEM vitamins (Invitrogen cat. 11120-052) | 0·5× |
| Schneider’s medium | 5% |
| Foetal bovine serum | 10% |
| Hepes | 10 mM |
| Antibiotic/antimycotic (Invitrogen cat. 15240-062) | 1× |
Fig. 3.
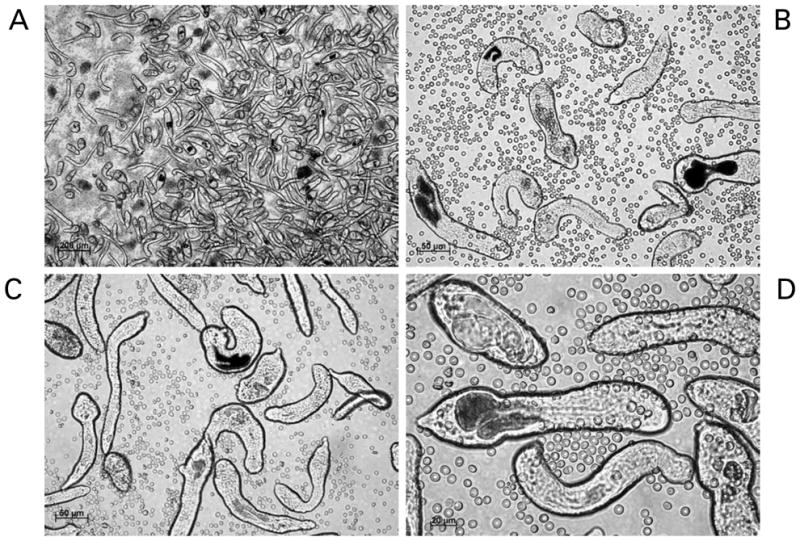
Schistosomules of Schistosoma mansoni. The schistosomules were mechanically transformed from cercariae and cultured for 14 days. Black pigmented guts are evident in many of the larvae. Erythrocytes have been added to the cultures. Panels A to D: schistosomules viewed at 5, 20, 20 and 40× magnification, respectively. Scale bars are shown in each panel. The micrographs were taken using a digital camera (Zeiss AxioCam ICc3) fitted to a Zeiss Axio Observer A1 inverted microscope.
We change media on cultured schistosomules every second day, if feasible. For this, schistosomules are transferred to 15 ml conical tubes. The well is washed with somule wash and added to the tube until the tube contains ~10 ml. The tube is centrifuged for 5 min at 100×g at RT or 4 °C. The supernatant is removed and the somules are resuspended in 4 ml of prewarmed somule media and returned to the 6-well plate. At day two or three after transformation from cercariae, washed human erythrocytes are included in the culture; 1–2 μl of red cells (50% suspension inDMEM) added to each well (Fig. 3). Once a week the old erythrocytes are removed and replaced with fresh blood cells. The somules are transferred from the plate to a 15 ml conical tube as above. The somules will settle by gravity for 30 minutes at 37 °C, after which the supernatant can be removed, the somules resuspended in ~4 ml warmed, fresh Basch’s medium, and then returned to a new 6-well plate.
GENETIC MANIPULATION OF CULTURED SCHISTOSOMES
General considerations
The recent release of draft genome sequences of two of the major human schistosomes has underscored the pressing need to develop functional genomics approaches for these significant pathogens (Schistosoma japonicum Genome Sequencing and Functional Analysis Consortium et al. 2009; Berriman et al. 2009). Progress with functional genomics approaches and tools for schistosomes have been reviewed (Beckmann et al. 2007; Brindley and Pearce, 2007; Ndegwa et al. 2007; Mann et al. 2008). In particular, RNA interference (RNAi) has been developed to target numerous genes in S. mansoni and S. japonicum (e.g. Freitas et al. 2007; Morales et al. 2008; Ndegwa et al. 2007; Rinaldi et al. 2009). RNAi using short interfering RNAs (20 to 30 nucleotides) as well as longer double stranded RNAs (100 to more than 1000 nt) has been successful. In addition, the schistosome genome sequences make feasible genome- scale investigation of transgene integration into schistosome chromosomes. We have adapted several gene therapy approaches to transform schistosomes with the long-term aim of establishment of lines of transgenic schistosomes (Mann et al. 2008). These approaches include deployment of the piggyBac transposon and pseudotyped murine leukaemia retrovirus (Kines et al. 2006, 2008; Morales et al. 2007). Retrovirus- and piggyBac-mediated transductions both offer a means to establish transgenic lines of schistosomes, to elucidate schistosome gene function and expression, and to advance functional genomics approaches for the parasites. The theory, history and methods of binary transposons systems and retroviral/lentiviral approaches for transgenesis have been reviewed, e.g. Plasterk et al. 1999; O’Brochta et al. 2003; Miskey et al. 2005; Alfa and Blesch, 2006; Pfeifer and Hofmann, 2009. Given our recent experience with both RNAi and transgenesis approaches with S. mansoni, we have included here brief methods that facilitate transfer of foreign genetic materials and reporter genes into cultured schistosomes, including the deployment of square wave electroporation.
RNA interference
RNAi for schistosomes was pioneered by Skelly et al. (2003) and subsequently employed by many workers (see Ndegwa et al. 2007). Developmental stages of schistosomes including sporocysts and adults have been probed with dsRNA or short interfering RNAs (siRNAs) in RNAi investigations (e.g. Delcroix et al. 2006). Electroporation of dsRNA/siRNA, rather than soaking alone, appears to be much more efficient for schistosomules and adult S. mansoni worms (Ndegwa et al. 2007). Recently, we developed methods to assess for the presence of a viable RNAi pathway by silencing the exogenous reporter gene, firefly luciferase. We established the method in S. mansoni and thereafter confirmed its utility in the liver fluke Fasciola hepatica. This straightforward reporter system could provide investigators with a means to test the presence of a functional RNAi pathway in other parasites that are by other means intractable (Rinaldi et al. 2008).
Double stranded RNA (dsRNA) has been introduced into schistosome eggs for RNAi analysis (Freitas et al. 2007; Rinaldi et al. 2009). After the isolation of eggs from mouse livers (above), we have cultured eggs in a small volume of DMEM complete media as described above, at 37 °C under 5% CO2 in 24 well-plates (~5000 eggs per well). For RNAi studies, in our case with investigation of proteolytic enzymes of eggs, we have soaked eggs in 20 μg/ml of dsRNA. Provided that enough dsRNA is available, we change the culture media supplemented with the dsRNA daily or every second day, with frequent microscopic examination to identify visual phenotypic effects (if relevant). We harvested eggs after seven days in culture with dsRNA, for subsequent RNA and biochemical activity analyses (Rinaldi et al. 2009). After one week in culture, eggs retain the capacity to hatch when transferred to distilled water (Figs. 1, 2) (above).
Electroporation
For electroporation of dsRNA, the schistosomula can be removed at 3 h and at 7, 9 and 11 days, or at other suitable times, after cercarial transformation. We rinse cultured developmental stages of schistosomes free of erythrocytes and/or serum by three or more washes in somule wash medium. At the time of electroporation, dsRNA is added at a final concentration 0.1 μg/μl to 3-h-old schistosomules, and at a final concentration 0.3 μg/μl for 7-, 9- and 11-day-old somules. We have accomplished electroporation using a single 20 ms square wave pulse at 125 V in 4 mm gap cuvettes (BTX, San Diego, CA) using a BTX ElectroSquarePoratorTM ECM830 with ~2000 schistosomules suspended in 100 μl of wash medium containing 10 to 30 μg of dsRNA. Immediately following electroporation, we transfer the parasites to pre-warmed Basch’s medium (Table 3) and maintain in culture, as above (Correnti and Pearce, 2004; Correnti et al. 2005; Morales et al. 2008). After electroporation, we also have used the transformed schistosomes to infect mice. For example, we have resuspended 500 to 2000 in ~200 μl Modified Eagle’s Medium and injected them into the thigh muscle or the peritoneal cavity of mice using a 22-guage needle (James and Taylor, 1976; Morales et al. 2008).
Transposons
In addition to dsRNA/siRNA, we have introduced binary transposon constructs into schistosomes by square wave electroporation (Morales et al. 2007). For electroporation in transposon studies using donor plasmids and helper (transposase) as mRNA, the schistosomules were removed from culture 15 days after cercarial transformation if expression of the reporter gene is important. Otherwise the schistosomules could be electroporated at younger age (Correnti et al. 2005; Morales et al. 2008). In brief, transposase mRNA was added to the DNA at a final concentration of 6 μg mRNA/100 μl. Electroporations were accomplished in 4 mm gap cuvettes (BTX) with ~1000–2000 parasites (somules, sporocysts or eggs) or 15–20 adults resuspended in 100 μl of wash medium containing 10–12 μg donor plasmid and 3–6 μg of helper mRNA using the BTX ElectroSquarePorator™ ECM830. We introduce either intact circular or linearized (donor) plasmids, with or without transposase mRNA, into schistosomules by square wave electroporation (125 V, 20 ms, 4 mm). Immediately after electroporation, parasites are transferred to pre-warmed Basch’s medium and cultured as described above. Subsequently, schistosomula were washed three times with pre-warmed wash medium and treated with DNase (New England Biolabs, Ipswich, MA) for 1 h at 37 °C to remove any residual donor plasmids. Parasites were then washed three more times with wash media to remove DNase, employed to infect mice (see above), or stored at −80 °C.
Retroviruses
With retrovirus virions as the transgenesis vectors, we have observed that both soaking and electroporation techniques can effectively facilitate integration of transgenes into the schistosome genome. We have exposed eggs, sporocysts, schistosomules and adult stages to Moloney murine leukaemia retrovirus (MLV) virions pseudotyped with vesicular stomatitis virus glycoprotein (VSVG) (Clontech) or lentiviral virions pseudotyped with VSVG (Invitrogen) (Kines et al. 2006, 2008; P. Brindley and others, unpublished). Both these viruses are replication-incompetent and accordingly they can be employed using Biological Safety Level 2 (BSL2) containment. To introduce the virus into the schistosomes, we soak worms in the presence of pseudotyped virions or in addition subject the parasites to square wave electroporation. With soaking alone, we reduce the volume of media in a 6-well plate to one ml, and add one hundred μl of concentrated virus (>1×106 transducing units) to the well. We disperse the virus by gentle rocking of the plate. We add polybrene (Sigma) to 6 μg/ml to facilitate virion attachment to the surface of the target parasites. (Polybrene neutralizes charge repulsion between retrovirus virions and sialic acid on the surface of cells [Davis et al. 2004]). After gentle mixing, we incubate the plate overnight at 37 °C under 5% CO2 (for sporocysts, 27 °C, 90% N2, 5%CO2, 5%O2) (Table 1). For electroporation of parasites, we remove the parasites from the 6-well plate, pellet them as above, and resuspend in 100 μl of Opti-Mem 1 (Invitrogen). We transfer them to a 4 mm gap cuvette; add 100 μl of virion preparation in Opti-Mem 1 medium and electroporate, as above. After electroporation, we transfer the schistosomes to a 6-well plate and add 800 μl of DMEM with 10% FBS and 100U of penicillin and streptomycin (Invitrogen) to the well. They are incubated overnight at 37 °C under 5% CO2 unless the life stage is the sporocyst (see above). The following day, after soaking and/or electroporation, we pellet the parasites at 800× g and wash 3 times in DPBS. Eggs tolerate this electroporation, can hatch afterwards, and the resulting miracidia will infect snails (V. Mann, unpublished). After exposure to retrovirus virions, we have returned developmental stages of schistosomes to culture in new media appropriate for the life stage (Table 1). After appropriate intervals of time in culture, virion-exposed schistosomes can be assessed for productive transduction using appropriate approaches such as anchored PCR (Morales et al. 2007).
CONCLUDING REMARKS
Because many standard tools and approaches are not yet available, and because the schistosome developmental cycle cannot be completed without animal hosts, in vitro culture methods and procedures have particular importance for genetic manipulation of schistosomes. Whereas the methods that are currently available are not optimal, they can facilitate advances in genetic manipulation which, in turn, will enhance our understanding of schistosome physiology and the host-parasite relationship. Despite the limitations of maintenance of schistosomes in vitro, new insights into fundamental helminth biology are accumulating. Geneticmanipulation approaches will gain momentum because of the newly available draft genomes and the application of RNAi and nascent transgenesis technologies. At the same time, much of our understanding of Th2-type immune responses, functions of regulatory T cells, generation of alternatively activated macrophages and the transmission dynamics of infectious agents, for example, are derived from schistosome and other parasitic helminth paradigms. Ultimately, advances in molecular and medical helminthology – including manipulation of the schistosome genome – can be expected to be translated into new interventions targeting schistosomiasis and other helminthiases, and/or therapeutics for other ailments (Hotez et al. 2008; Han et al. 2009; Hewitson et al. 2009).
Acknowledgments
We thank Kristine Kines, Tunika Okatcha and Mary Ayuk for informative discussions and methods. We obtained advice and schistosome-infected snails and mice from Dr. Fred A. Lewis, Biomedical Research Institute, Rockville, Maryland through National Institute of Allergy and Infectious Diseases contract NO155270. We gratefully acknowledge support from the NIAID, award number RO1 AI072773.
References
- Alfa RW, Blesch A. Murine and HIV-based retroviral vectors for in vitro and in vivo gene transfer. Methods in Molecular Medicine. 2006;129:241–254. doi: 10.1385/1-59745-213-0:241. [DOI] [PubMed] [Google Scholar]
- Basch PF. Cultivation of Schistosoma mansoni in vitro. I. Establishment of cultures from cercariae and development until pairing. Journal of Parasitology. 1981;67:179–185. [PubMed] [Google Scholar]
- Beckmann S, Wippersteg V, El-Bahay A, Hirzmann J, Oliveira G, Grevelding CG. Schistosoma mansoni: germ-line transformation approaches and actin-promoter analysis. Experimental Parasitology. 2007;117:292–303. doi: 10.1016/j.exppara.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Berriman M, Haas BJ, LoVerde PT, Wilson RA, Dillon GP, Cerqueira GC, Mashiyama ST, Al-Lazikani B, Andrade LF, Ashton PD, Aslett MA, Bartholomeu DC, Blandin G, Caffrey CR, Coghlan A, Coulson R, Day TA, Delcher A, DeMarco R, Djikeng A, Eyre T, Gamble JA, Ghedin E, Gu Y, Hertz-Fowler C, Hirai H, Hirai Y, Houston R, Ivens A, Johnston DA, Lacerda D, Macedo CD, McVeigh P, Ning Z, Oliveira G, Overington JP, Parkhill J, Pertea M, Pierce RJ, Protasio AV, Quail MA, Rajandream MA, Rogers J, Sajid M, Salzberg SL, Stanke M, Tivey AR, White O, Williams DL, Wortman J, Wu W, Zamanian M, Zerlotini A, Fraser-Liggett CM, Barrell BG, El-Sayed NM. The genome of the blood fluke Schistosoma mansoni. Nature. 2009;460:352–358. doi: 10.1038/nature08160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bixler LM, Lerner JP, Ivanchenko M, McCormick RS, Barnes DW, Bayne CJ. Axenic culture of Schistosoma mansoni sporocysts in low O2 environments. Journal of Parasitology. 2001;87:1167–1168. doi: 10.1645/0022-3395(2001)087[1167:ACOSMS]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, Cogliano V WHO International Agency for Research on Cancer Monograph Working Group. A review of human carcinogens – Part B: biological agents. Lancet Oncology. 2009;10:321–322. doi: 10.1016/s1470-2045(09)70096-8. [DOI] [PubMed] [Google Scholar]
- Brindley PJ, Pearce EJ. Genetic manipulation of schistosomes. International Journal for Parasitology. 2007;37:465–473. doi: 10.1016/j.ijpara. 2006.12.012. [DOI] [PubMed] [Google Scholar]
- Correnti JM, Brindley PJ, Pearce EJ. Long-term suppression of cathepsin B levels by RNA interference retards schistosome growth. Molecular and Biochemical Parasitology. 2005;143:209–215. doi: 10.1016/j.molbiopara.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Correnti JM, Jung E, Freitas TC, Pearce EJ. Transfection of Schistosoma mansoni by electroporation and the description of a new promoter sequence for transgene expression. International Journal for Parasitology. 2007;37:1107–1115. doi: 10.1016/j.ijpara.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Correnti JM, Pearce EJ. Transgene expression in Schistosoma mansoni: introduction of RNA into schistosomula by electroporation. Molecular and Biochemical Parasitology. 2004;137:75–79. doi: 10.1016/j.molbiopara.2004.04.015. [DOI] [PubMed] [Google Scholar]
- Coustau C, Yoshino TP. Flukes without snails: advances in the in vitro cultivation of intramolluscan stages of trematodes. Experimental Parasitology. 2000;94:62–66. doi: 10.1006/expr.1999.4462. [DOI] [PubMed] [Google Scholar]
- Crabb BS, Gilson PR. A new system for rapid plasmid integration in Plasmodium parasites. Trends in Microbiology. 2007;15:3–6. doi: 10.1016/j.tim.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Dalton JP, Day SR, Drew AC, Brindley PJ. A method for the isolation of schistosome eggs and miracidia free of contaminating host tissues. Parasitology. 1997;115:29–32. doi: 10.1017/s0031182097001091. [DOI] [PubMed] [Google Scholar]
- Davis HE, Rosinski M, Morgan JR, Yarmush ML. Charged polymers modulate retrovirus transduction via membrane charge neutralization and virus aggregation. Biophysical Journal. 2004;86:1234–1242. doi: 10.1016/S0006-3495(04)74197-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcroix M, Sajid M, Caffrey CR, Lim KC, Dvorak J, Hsieh I, Bahgat M, Dissous C, McKerrow JH. A multienzyme network functions in intestinal protein digestion by a platyhelminth parasite. Journal of Biological Chemistry. 2006;281:39316–39329. doi: 10.1074/jbc.M607128200. [DOI] [PubMed] [Google Scholar]
- Duvall RH, DeWitt WB. An improved perfusion technique for recovering adult schistosomes from laboratory animals. American Journal of Tropical Medicine and Hygiene. 1967;16:483–486. doi: 10.4269/ajtmh.1967.16.483. [DOI] [PubMed] [Google Scholar]
- Freitas TC, Jung E, Pearce EJ. TGF-beta signaling controls embryo development in the parasitic flatworm Schistosoma mansoni. PLoS Pathogens. 2007;3:e52. doi: 10.1371/journal.ppat.0030052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobert GN, Moertel L, Brindley PJ, McManus DP. Developmental gene expression profiles of the human pathogen Schistosoma japonicum. BMC Genomics. 2009;10:128. doi: 10.1186/1471-2164-10-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett F. The culture of Schistosoma mansoni and production of life cycle stages. Methods in Molecular Biology (Clifton, NJ) 1993;21:89–99. doi: 10.1385/0-89603-239-6:89. [DOI] [PubMed] [Google Scholar]
- Han ZG, Brindley PJ, Wang SY, Chen Z. Schistosome genetics: new perspectives on schistosome biology and host-parasite interaction. Annual Review of Genomics and Human Genetics. 2009 doi: 10.1146/annurev-genom-082908-150036. [DOI] [PubMed] [Google Scholar]
- Hewitson JP, Grainger JR, Maizels RM. Helminth immunoregulation: the role of parasite secreted proteins in modulating host immunity. Molecular and Biochemical Parasitology. 2009;167:1–11. doi: 10.1016/j.molbiopara.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Taguchi T, Saitoh Y, Kawanaka M, Sugiyama H, Habe S, Okamoto M, Hirata M, Shimada M, Tiu WU, Lai K, Upatham ES, Agatsuma T. Chromosomal differentiation of the Schistosoma japonicum complex. International Journal for Parasitology. 2000;30:441–452. doi: 10.1016/s0020-7519(99)00186-1. [DOI] [PubMed] [Google Scholar]
- Hokke CH, Fitzpatrick JM, Hoffmann KF. Integrating transcriptome, proteome and glycome analyses of Schistosoma biology. Trends in Parasitology. 2007;23:165–174. doi: 10.1016/j.pt.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Hotez PJ, Brindley PJ, Bethony JM, King CH, Pearce EJ, Jacobson J. Helminth infections: the great neglected tropical diseases. Journal of Clinical Investigation. 2008;118:1311–1321. doi: 10.1172/JCI34261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Brindley PJ, McManus DP, Feng Z, Han ZG. Schistosome transcriptomes: new insights into the parasite and schistosomiasis. Trends in Molecular Medicine. 2004;10:217–225. doi: 10.1016/j.molmed.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Hu W, Yan Q, Shen DK, Liu F, Zhu ZD, Song HD, Xu XR, Wang ZJ, Rong YP, Zeng LC, Wu J, Zhang X, Wang JJ, Xu XN, Wang SY, Fu G, Zhang XL, Wang ZQ, Brindley PJ, McManus DP, Xue CL, Feng Z, Chen Z, Han ZG. Evolutionary and biomedical implications of a Schistosoma japonicum complementary DNA resource. Nature Genetics. 2003;35:139–147. doi: 10.1038/ng1236. [DOI] [PubMed] [Google Scholar]
- Ivanchenko MG, Lerner JP, McCormick RS, Toumadje A, Allen B, Fischer K, Hedstrom O, Helmrich A, Barnes DW, Bayne CJ. Continuous in vitro propagation and differentiation of cultures of the intramolluscan stages of the human parasite Schistosoma mansoni. Proceedings of the National Academy of Sciences, USA. 1999;96:4965–4970. doi: 10.1073/pnas.96.9.4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James ER, Taylor MG. Transformation of cercariae to schistosomula: a quantitative comparison of transformation techniques and of infectivity by different injection routes of the organisms produced. Journal of Helminthology. 1976;50:223–233. doi: 10.1017/s0022149x0002664x. [DOI] [PubMed] [Google Scholar]
- Jolly ER, Chin CS, Miller S, Bahgat MM, Lim KC, DeRisi J, McKerrow JH. Gene expression patterns during adaptation of a helminth parasite to different environmental niches. Genome Biology. 2007;8:R65. doi: 10.1186/gb-2007-8-4-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapp K, Coustau C, Wippersteg V, Jourdane J, Kunz W, Grevelding CG. Transplantation of in vitro-generated Schistosoma mansoni mother sporocysts into Biomphalaria glabrata. Parasitology Research. 2003;91:482–485. doi: 10.1007/s00436-003-0951-1. [DOI] [PubMed] [Google Scholar]
- Kawanaka M, Hayashi S, Ohtomo H. A minimum essential medium for cultivation of Schistosoma japonicum eggs. Journal of Parasitology. 1983;69:991–992. [PubMed] [Google Scholar]
- Kawanaka M, Sidner RA, Carter CE. In vitro transformation of Schistosoma japonicum miracidia to young sporocysts in a culture system for egg maturation. Journal of Parasitology. 1985;71:368–370. [PubMed] [Google Scholar]
- Kines KJ, Mann VH, Morales ME, Shelby BD, Kalinna BH, Gobert GN, Chirgwin SR, Brindley PJ. Transduction of Schistosoma mansoni by vesicular stomatitis virus glycoprotein-pseudotyped Moloney murine leukemia retrovirus. Experimental Parasitology. 2006;112:209–220. doi: 10.1016/j.exppara.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Kines KJ, Morales ME, Mann VH, Gobert GN, Brindley PJ. Integration of reporter transgenes into Schistosoma mansoni chromosomes mediated by pseudotyped murine leukemia virus. FASEB Journal. 2008;22:2936–2948. doi: 10.1096/fj.08-108308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazdins JK, Stein MJ, David JR, Sher A. Schistosoma mansoni: rapid isolation and purification of schistosomula of different developmental stages by centrifugation on discontinuous density gradients of Percoll. Experimental Parasitology. 1982;53:39–44. doi: 10.1016/0014-4894(82)90090-x. [DOI] [PubMed] [Google Scholar]
- Lewis F, editor. Current Protocols in Immunology. Suppl 28. 1998. Schistosomiasis. [DOI] [PMC free article] [PubMed] [Google Scholar]; Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W, editors. Animal Models for Infectious Diseases. Wiley; New York: [Google Scholar]
- Liu F, Chen P, Cui SJ, Wang ZQ, Han ZG. SjTPdb: integrated transcriptome and proteome database and analysis platform for Schistosoma japonicum. BMC Genomics. 2008;9:304. doi: 10.1186/1471-2164-9-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Lu J, Hu W, Wang SY, Cui SJ, Chi M, Yan Q, Wang XR, Song HD, Xu XN, Wang JJ, Zhang XL, Zhang X, Wang ZQ, Xue CL, Brindley PJ, McManus DP, Yang PY, Feng Z, Chen Z, Han ZG. New perspectives on host-parasite interplay by comparative transcriptomic and proteomic analyses of Schistosoma japonicum. PLoS Pathogens. 2006;2:e29. doi: 10.1371/journal.ppat.0020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann VH, Morales ME, Kines KJ, Brindley PJ. Transgenesis of schistosomes: approaches employing mobile genetic elements. Parasitology. 2008;135:141–153. doi: 10.1017/S0031182007003824. [DOI] [PubMed] [Google Scholar]
- Miskey C, Izsvak Z, Kawakami K, Ivics Z. DNA transposons in vertebrate functional genomics. Cellular and Molecular Life Sciences. 2005;62:629–641. doi: 10.1007/s00018-004-4232-7. [DOI] [PubMed] [Google Scholar]
- Morales ME, Mann VH, Kines KJ, Gobert GN, Fraser MJ, Jr, Kalinna BH, Correnti JM, Pearce EJ, Brindley PJ. piggyBac transposon mediated transgenesis of the human blood fluke, Schistosoma mansoni. FASEB Journal. 2007;21:3479–3489. doi: 10.1096/fj.07-8726com. [DOI] [PubMed] [Google Scholar]
- Morales ME, Rinaldi G, Gobert GN, Kines KJ, Tort JF, Brindley PJ. RNA interference of Schistosoma mansoni cathepsin D, the apical enzyme of the hemoglobin proteolysis cascade. Molecular and Biochemical Parasitology. 2008;157:160–168. doi: 10.1016/j.molbiopara.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndegwa D, Krautz-Peterson G, Skelly PJ. Protocols for gene silencing in schistosomes. Experimental Parasitology. 2007;117:284–291. doi: 10.1016/j.exppara.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brochta DA, Sethuraman N, Wilson R, Hice RH, Pinkerton AC, Levesque CS, Bideshi DK, Jasinskiene N, Coates CJ, James AA, Lehane MJ, Atkinson PW. Gene vector and transposable element behavior in mosquitoes. The Journal of Experimental Biology. 2003;206:3823–3834. doi: 10.1242/jeb.00638. [DOI] [PubMed] [Google Scholar]
- Pfeifer A, Hofmann A. Lentiviral transgenesis. In: Clifton NJ, editor. Methods in Molecular Biology. Vol. 530. 2009. pp. 391–405. [DOI] [PubMed] [Google Scholar]
- Plasterk RH, Izsvak Z, Ivics Z. Resident aliens: the Tc1/mariner superfamily of transposable elements. Trends in Genetics. 1999;15:326–332. doi: 10.1016/s0168-9525(99)01777-1. [DOI] [PubMed] [Google Scholar]
- Rinaldi G, Morales ME, Alrefaei YN, Cancela M, Castillo E, Dalton JP, Tort JF, Brindley PJ. RNA interference targeting leucine aminopeptidase blocks hatching of Schistosoma mansoni eggs. Molecular and Biochemical Parasitology. 2009;167:118–126. doi: 10.1016/j.molbiopara.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi G, Morales ME, Cancela M, Castillo E, Brindley PJ, Tort JF. Development of functional genomic tools in trematodes: RNA interference and luciferase reporter gene activity in Fasciola hepatica. PLoS Neglected Tropical Diseases. 2008;2:e260. doi: 10.1371/journal.pntd.0000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki E, Suemizu H, Shimada A, Hanazawa K, Oiwa R, Kamioka M, Tomioka I, Sotomaru Y, Hirakawa R, Eto T, Shiozawa S, Maeda T, Ito M, Ito R, Kito C, Yagihashi C, Kawai K, Miyoshi H, Tanioka Y, Tamaoki N, Habu S, Okano H, Nomura T. Generation of transgenic non-human primates with germline transmission. Nature. 2009;459:523–527. doi: 10.1038/nature08090. [DOI] [PubMed] [Google Scholar]
- Schistosoma japonicum Genome Sequencing and Functional Analysis Consortium. Liu F, Zhou Y, Wang ZQ, Lu G, Zheng H, Brindley PJ, McManus DP, Blair D, Zhang QH, Zhong Y, Wang S, Han ZG, Chen Z. The Schistosoma japonicum genome reveals features of host-parasite interplay. Nature. 2009;460:345–351. doi: 10.1038/nature08140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelly PJ, Da’dara A, Harn DA. Suppression of cathepsin B expression in Schistosoma mansoni by RNA interference. International Journal for Parasitology. 2003;33:363–369. doi: 10.1016/s0020-7519(03)00030-4. [DOI] [PubMed] [Google Scholar]
- Smithers SR, Terry RJ. The infection of laboratory hosts with cercariae of Schistosoma mansoni and the recovery of the adult worms. Parasitology. 1965;55:695–700. doi: 10.1017/s0031182000086248. [DOI] [PubMed] [Google Scholar]
- Stirewalt M, Lewis FA. Schistosoma mansoni: effect of rotifers on cercarial output, motility and infectivity. International Journal for Parasitology. 1981;11:301–308. doi: 10.1016/0020-7519(81)90040-0. [DOI] [PubMed] [Google Scholar]
- Terenius O, Marinotti O, Siegla D, James AA. Molecular genetic manipulation of vector mosquitoes. Cell Host and Microbe. 2008;4:417–423. doi: 10.1016/j.chom.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzinger J, Keiser J. Schistosomiasis and soil-transmitted helminthiasis: common drugs for treatment and control. Expert Opinion on Pharmacotherapy. 2004;5:263–285. doi: 10.1517/14656566.5.2.263. [DOI] [PubMed] [Google Scholar]
- Verjovski-Almeida S, DeMarco R, Martins EA, Guimaraes PE, Ojopi EP, Paquola AC, Piazza JP, Nishiyama MY, Jr, Kitajima JP, Adamson RE, Ashton PD, Bonaldo MF, Coulson PS, Dillon GP, Farias LP, Gregorio SP, Ho PL, Leite RA, Malaquias LC, Marques RC, Miyasato PA, Nascimento AL, Ohlweiler FP, Reis EM, Ribeiro MA, Sa RG, Stukart GC, Soares MB, Gargioni C, Kawano T, Rodrigues V, Madeira AM, Wilson RA, Menck CF, Setubal JC, Leite LC, Dias-Neto E. Transcriptome analysis of the acoelomate human parasite Schistosoma mansoni. Nature Genetics. 2003;35:148–157. doi: 10.1038/ng1237. [DOI] [PubMed] [Google Scholar]