

Abstract
Motivation: Admixed populations offer a unique opportunity for mapping diseases that have large disease allele frequency differences between ancestral populations. However, association analysis in such populations is challenging because population stratification may lead to association with loci unlinked to the disease locus.
Methods and results: We show that local ancestry at a test single nucleotide polymorphism (SNP) may confound with the association signal and ignoring it can lead to spurious association. We demonstrate theoretically that adjustment for local ancestry at the test SNP is sufficient to remove the spurious association regardless of the mechanism of population stratification, whether due to local or global ancestry differences among study subjects; however, global ancestry adjustment procedures may not be effective. We further develop two novel association tests that adjust for local ancestry. Our first test is based on a conditional likelihood framework which models the distribution of the test SNP given disease status and flanking marker genotypes. A key advantage of this test lies in its ability to incorporate different directions of association in the ancestral populations. Our second test, which is computationally simpler, is based on logistic regression, with adjustment for local ancestry proportion. We conducted extensive simulations and found that the Type I error rates of our tests are under control; however, the global adjustment procedures yielded inflated Type I error rates when stratification is due to local ancestry difference.
Contact: mingyao@upenn.edu; chun.li@vanderbilt.edu.
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
African Americans and Hispanic Americans represent the two largest racial minority groups in the USA, comprising ∼28% of the US population. Both groups are recently admixed and have inherited ancestry from more than one ancestral population. Such admixed populations offer a unique opportunity for mapping disease genes that have large allele frequency differences between ancestral populations. Recently, admixture mapping has become one of the main approaches for gene mapping studies in admixed populations (Hoggart et al., 2004; McKeigue, 1998; Montana and Pritchard, 2004; Patterson et al., 2004; Zhang et al., 2004; Zhu et al., 2004). However, traditional admixture mapping has a substantially lower resolution than association analysis (Smith et al., 2005; Zhu et al., 2008). Moreover, admixture mapping cannot identify disease loci that have similar allele frequencies or disease prevalences in the ancestral populations.
With the increasing availability of large volumes of high-density SNP genotyping data generated in genome-wide association studies (GWAS), the analysis of admixed populations is now moving toward SNP association. In admixed populations, the proportion of admixture may vary across individuals. This variation can lead to associations of the disease with loci unlinked to the disease locus, a phenomenon well known as ‘population stratification’, which can produce both false-positive and false-negative association signals if not appropriately controlled.
Over the past decade, various methods have been developed to deal with this population stratification effect (Price et al., 2010; Sillanpaa, 2010). In general, these methods can be classified into three categories: (i) genomic control (Devlin et al., 1999), (ii) structured association (Pritchard et al., 2000; Satten et al., 2001) and (iii) principal components-based methods (Epstein et al., 2007; Li et al., 2010; Price et al., 2006; Zhang et al., 2003; Zhu et al., 2002). The genomic control approach attempts to correct for population stratification by adjusting association statistics with a single overall inflation factor obtained from a set of random markers that are not associated with the phenotype of interest. In contrast, structured association methods assign the study subjects to estimated discrete subpopulations and then aggregates evidence of association within each subpopulation. The current state-of-the-art approach is the principal component-based approach implemented in EIGENSTRAT (Price et al., 2006), which computes coefficients of principal components using SNPs across the genome to control for population structure.
The central idea of all the above-mentioned methods is to use markers across the genome to capture the global population structure within the study subjects. Variation in global population structure is driven mainly by demographic histories such as migration and genetic random drift. However, for admixed populations, a person's genome is a mosaic of ancestral chromosomes. This introduces considerable amount of variation in local ancestry at certain genomic regions, and the local ancestry may have little correlation with global ancestry. Local ancestry has been useful in localizing disease susceptibility genes in admixture mapping studies (Hoggart et al., 2004; McKeigue, 1998; Montana and Pritchard, 2004; Patterson et al., 2004; Zhang et al., 2004; Zhu et al., 2004). In this article, we show that local ancestry may confound with the association signal at the test SNP, and if ignored, can lead to spurious association in genetic association analysis. Methods that adjust for global ancestry may fail to remove this spurious association because the global ancestry information obtained from all markers across the genome may not accurately reflect the amount of ancestry variation in local regions.
There are two types of mechanisms that may lead to population stratification: (i) stratification due to local ancestry difference driven by natural selection at certain genomic regions and (ii) stratification due to global ancestry difference driven by the demographic history of a population or genetic random drift due to finite population size. The existing methods for population stratification correction are appropriate when the stratification is due to global ancestry difference; however, they may be ineffective in removing the effect of population stratification when the stratification is induced by natural selection that occurs only in certain genomic regions. Population stratification due to natural selection is not uncommon. A well-known example is the lactase gene, which has been shown to be under recent positive selection (Bersaglieri et al., 2004). Since both the lactase gene ancestry and height track with northwest versus southeast European ancestry, naïve association analysis between the lactase gene and height will lead to a spurious association (Campbell et al., 2005). Another example of recent natural selection was reported in admixed Puerto Ricans (Tang et al., 2007). In this study, three chromosomal regions, including the human leukocyte antigen region on 6p, 8q and 11q, were reported to exhibit deficiencies in the European-ancestry proportion as compared with the genome-wide European-ancestry. With the increased evidence of recent positive selection in many regions of the genome (Pickrell et al., 2009; Voight et al., 2006), we anticipate that unrecognized population stratification due to natural selection might be present for many other phenotypes.
In the Appendix (Supplementary Material), we show theoretically that to remove the confounding effect of local ancestry, it is sufficient to condition on local ancestry at the test SNP, whereas conditioning on global ancestry may be ineffective. To our knowledge, it is the first time that local ancestry adjustment is shown to be sufficient in eliminating spurious association in the analysis of admixed populations. We further present two novel association tests that correct for population stratification by adjusting for local ancestry at the test SNP. Our first test is a likelihood ratio test (denoted by LRT), which is based on a conditional likelihood framework which models the distribution of a test SNP given disease status and genotypes of flanking markers. This conditional likelihood allows us to model local ancestry difference among study subjects explicitly and thus eliminates the effect of population stratification at the test SNP. A key advantage of this test lies in its ability to incorporate different directions of association in the ancestral populations, a ‘flip-flop’ phenomenon due to either population differences in allele frequencies, or to multi-locus effects and variation in inter-locus correlations (Lin et al., 2007). Our second test, denoted as Logistic-Local, is based on a simple logistic regression in which the test SNP is a predictor and the estimated local ancestry proportion at the test SNP is included as a covariate in the analysis. Compared with the first test, this test imposes additional assumptions on the disease risk model but is computationally simpler. Our procedures are developed as general tests of genetic association for admixed populations and can be applied in the GWAS setting. In contrast to a requirement in admixture mapping, neither of our tests requires the allele frequencies or the disease prevalences to be different in the ancestral populations, and thus they can identify disease loci that may be missed by admixture mapping.
We conducted extensive simulations to evaluate the performance of the proposed tests. Our results indicate that regardless of the mechanism of population stratification—whether due to local or global ancestry differences—the Type I error rates of our procedures were always under control; however, global ancestry adjustment procedures such as EIGENSTRAT (Price et al., 2006) may fail to control Type I error rates when population stratification is due to local ancestry differences.
2 METHODS
2.1 Notation
We assume that admixture has occurred between two ancestral populations, denoted by X and Y, in a recently admixed population. Assume a set of markers with known genetic locations is available for estimating ancestry. Consider a test SNP with alleles A and a (with frequencies pX, qX and pY, qY, in populations X and Y, respectively). Let π be the probability that a randomly selected allele at the test SNP comes from population X. For each individual, let Z be the disease status (1 = affected; 0 = unaffected), ISNP (= 0, 1, 2) denote the number of alleles at the test SNP that come from population X, GSNP(= 0, 1, 2) denote the number of allele A at the test SNP and GANC denote the flanking marker genotypes that are used to infer the local ancestry at the test SNP.
2.2 Confounding due to local ancestry
We now show that local ancestry at the test SNP can confound with the association signal in genetic association analysis. The local ancestry at the test SNP can be considered as a ‘diallelic marker’ with alleles ‘X’ and ‘Y’. There are four marker-ancestry haplotypes, AX, aX, AY and aY, with respective frequencies
 |
Based on these haplotype frequencies, we can calculate the square of the correlation coefficient between the test SNP and the local ancestry as
Therefore, r2 = 0 when π = 0 or 1, or pX = pY and r2 > 0 when 0 < π < 1 and pX ≠ pY. In an admixed population, π is always between 0 and 1. Thus, there is linkage disequilibrium (LD) between the test SNP and the local ancestry at the SNP as long as the allele frequencies are different in the two ancestral populations. In this situation, if the local ancestry is correlated with disease risk, the test SNP will appear to be disease associated, even when the SNP is not close to the disease locus.
To help understand this phenomenon, consider a typical admixture mapping scan with unrelated cases and controls. In the traditional admixture mapping analysis, one may identify a region that shows association evidence between a local ancestry estimate and disease status; however, due to the shared ancestry among many markers, this region may extend over several megabases. For a marker that falls in the region, the local ancestry at the marker will correlate with both the marker (due to the marker-ancestry LD described above) and the disease status (due to the admixture mapping signal). However, most markers in the region will be far from the disease-causing variant and will not be in LD with the variant in any of the ancestral populations. Naïvely testing for association between a marker and disease status while ignoring the confounding effect due to the local ancestry may lead to a spurious association. Below we describe statistical procedures that can remove this confounding effect. In the Appendix (Supplementary Material), we show that such local ancestry adjustment is sufficient and that adjustment for global ancestry may not be enough. Our tests do not require the disease allele frequencies or the disease prevalences to be different in the ancestral populations.
2.3 Conditional likelihood for unrelated cases and controls
The conditional likelihood of GSNP, the genotype at the test SNP, given disease status Z and flanking marker genotypes GANC, is
 |
(1) |
where
 |
(2) |
Given a set of unrelated cases and controls, the overall likelihood of the observed data is L = ∏P(GSNP|Z, GANC), where the product is taken over all individuals. To estimate the expression in (1) and (2), we need to estimate: (i) P(Z|GSNP, ISNP), the probability that an individual is affected or unaffected given his genotype and ancestry state at the test SNP, (ii) P(GSNP|ISNP), the genotype frequency given the ancestry state at the test SNP and (iii) P(ISNP|GANC), the probability of ancestry state at the test SNP given flanking marker genotypes. Below we describe how to estimate each of the three probabilities.
2.4 Model for disease risk
To calculate P(Z|GSNP, ISNP), we need to have a model that relates disease risk to the SNP genotype and the underlying latent ancestry state. It is generally believed that disease risks are the same in the ancestral populations at the true disease locus (Kaplan et al., 1998). However, in genetic association studies, it is more likely that the test SNP is not the true disease locus but instead is in LD with it. Since the degrees of LD may differ in the ancestral populations, the penetrances manifested at the test SNP may be different. Therefore, two sets of penetrances are needed to reflect the fact that the probability of being affected depends not only on the individual's genotype at the test SNP but also on the underlying ancestry state. Taking this into consideration, we propose to model the penetrances at the test SNP as shown in Table 1.
Table 1.
Model for disease risk P(Z = 1|GSNP, ISNP)
| GSNP | ISNP = 0 | ISNP = 2 | ISNP = 1 | ||
|---|---|---|---|---|---|
| Additive | Dominant | Recessive |
|||
| 0 | f0,Y | f0,X | f0,X + f0,Y)/2 | f0,X | f0,Y |
| 1 | f1,Y | f1,X | f1,X + f1,Y)/2 | f1,X | f1,Y |
| 2 | f2,Y | f2,X | f2,X + f2,Y)/2 | f2,X | f2,Y |
Here (f0,X, f1,X, f2,X) and (f0,Y, f1,Y, f2,Y) denote the penetrances of genotypes aa, Aa and AA in ancestral populations X and Y, respectively. For individuals carrying one allele from population X and one allele from population Y, we allow the ancestry risk (defined as the risk of having disease due to ancestry state at the test SNP) to be additive, recessive or dominant (Table 1). We note that imposing different penetrances in different ancestral populations has been previously considered by Pritchard et al. (2000). The key advantage of using two sets of penetrances is that this allows us to incorporate different directions of association in the ancestral populations, a ‘flip-flop’ phenomenon due either to population differences or multi-locus effects and variation in inter-locus correlations (Lin et al., 2007).
2.5 Genotype frequencies given local ancestry state
Assuming Hardy–Weinberg equilibrium (HWE) in both ancestral populations, the genotype frequencies at the test SNP can be easily calculated. For example, when ISNP = 2, i.e. both alleles come from population X, the frequency for genotype aa is qX2. Table 2 shows the genotype frequencies for all three scenarios of ISNP (Table 2).
Table 2.
Genotype frequency given ancestry state at the test SNP
| GSNP | ISNP = 2 | ISNP = 1 | ISNP = 0 |
|---|---|---|---|
| 0 | qX2 | qXqY | qY2 |
| 1 | 2pXqX | pXqY +qXpY | 2pYqY |
| 2 | pX2 | pXpY | pY2 |
2.6 Estimation of ancestry state at the test SNP
The conditional probability distribution of the ancestry state at the test SNP given flanking marker genotypes can be obtained from external programs. Several software packages are available for estimating local ancestry, including ANCESTRYMAP (Patterson et al., 2004), MALDSOFT (Montana and Pritchard, 2004), ADMIXPROGRAM (Zhu et al., 2006), SABER (Tang et al., 2006), LAMP (Sankararaman et al., 2008), HAPAA (Sundquist et al., 2008) and HAPMIX (Price et al., 2009). The choice of which program to use will depend on the nature of the data. For studies where the flanking markers are not densely spaced, programs such as ANCESTRYMAP and ADMIXPROGRAM would be sufficient. For markers from GWAS, programs such as SABER, HAPAA, LAMP and HAPMIX would be more appropriate. The current state-of-the-art method is HAPMIX, which for GWAS data can yield an estimated ancestry that has as high as 98% correlation with the true ancestry (Price et al., 2009).
2.7 Tests of association
With the previously developed likelihood framework, we can evaluate whether the test SNP is associated with the disease of interest. Under the null hypothesis that the test SNP is not associated with the disease, the penetrances at the test SNP should be the same for different SNP genotypes for both populations X and Y. This indicates that we can test for association by testing H0: f0,X=f1,X=f2,X and f0,Y=f1,Y=f2,Y. Our method requires parameter estimation, including {pX, pY, f0,X, f1,X, f2,X, f0,Y, f1,Y, f2,Y}. When only case–control data are available, these parameters are not all identifiable. To estimate these parameters, we therefore need to add parameter constraints. We choose to fix the disease prevalences in the ancestral populations, as they can often be obtained from external sources. Similar strategies have been employed elsewhere (Zollner et al., 2007). To maximize the likelihood, we use a simplex algorithm (Nelder et al., 1965), an optimization method that does not require calculation of derivatives. To address the issue of local maxima, we try multiple sets of starting values so that the procedure will converge to the point of maximum likelihood.
To assess the evidence of association, we propose to use a likelihood ratio test,  , where
, where 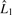 and
and  are the likelihood maximized under the general and null models, respectively. Under the null hypothesis of no association, LRT is asymptotically distributed as a χ2 with four degrees of freedom (d.f.). We could further reduce the d.f. by assuming a more restrictive risk model (e.g. additive, dominant or recessive) for the SNP genotype in the two ancestral populations. For example, with an additive model, we have the constraints 2f1,X = f0,X+f2,X and 2f1,Y = f0,Y+f2,Y on the parameters, and the corresponding test statistic will have two d.f.
are the likelihood maximized under the general and null models, respectively. Under the null hypothesis of no association, LRT is asymptotically distributed as a χ2 with four degrees of freedom (d.f.). We could further reduce the d.f. by assuming a more restrictive risk model (e.g. additive, dominant or recessive) for the SNP genotype in the two ancestral populations. For example, with an additive model, we have the constraints 2f1,X = f0,X+f2,X and 2f1,Y = f0,Y+f2,Y on the parameters, and the corresponding test statistic will have two d.f.
The above procedure explicitly models the ancestry penetrances at the test SNP. An alternative and simpler approach is to conduct a logistic regression
where propSNP = 0.5P(ISNP = 1|GANC)+P(ISNP = 2|GANC) is the local ancestry proportion at the test SNP. Association with the SNP can be assessed by testing H0 : β1 = β2 = 0 using a two d.f. likelihood ratio test. We denote this test as ‘Logistic-Local’. This approach assumes that the ancestry risk is additive on the logit scale and the direction of SNP association is the same irrespective of ancestry, and thus may not be as flexible as the LRT approach proposed earlier; however, it may be more powerful than the LRT approach due to its reduced number of d.f.
We note that neither of our tests requires the disease allele frequencies or disease prevalences to be different in the ancestral populations. Therefore they are able to identify signals that can be missed by the traditional admixture mapping approach. In the Appendix (Supplementary Material), we show that local ancestry adjustment is sufficient for controlling for population stratification in case–control data, whereas spurious association may occur with an adjustment only for global ancestry.
2.8 Simulation setup
To evaluate the performance of the proposed tests, we conducted extensive simulations. The allele frequencies of the 2774 ancestry informative markers (AIMs) in Smith et al. (2004) were used as marker allele frequencies in X and Y, respectively. We assumed HWE and linkage equilibrium between adjacent AIMs in each population. Samples were simulated according to the continuous gene flow model. In brief, in Generation 0, the marker genotypes for the AIMs of 50 000 unrelated individuals in population X were simulated. An admixed population was generated by forming inter-population marriages in subsequent generations. Specifically, in each generation, we took a proportion λ randomly selected individuals to marry individuals generated according to the marker allele frequencies in population Y, and let the remaining proportion, 1 − λ, mate randomly among themselves. The number of children in each marriage was assumed to follow a Poisson distribution with mean size two. The number of crossovers between two adjacent markers was determined by the genetic distance between them. This process was repeated six times to reach the current generation. We chose the value of λ such that an average individual in the current generation has ∼80% X ancestry and 20% Y ancestry, similar to that in African Americans.
To evaluate Type I error rates, we considered two mechanisms of population stratification: (i) stratification due to local ancestry difference at the test SNP between cases and controls and (ii) stratification due to global ancestry difference between cases and controls. In the first scenario, the true disease variant and the test SNP may be in the same region with shared ancestry but they are far from each other. When stratification was due to local ancestry difference, the case–control status was assigned according to the local ancestry state. Specifically, for individuals with two copies of population X alleles, the probability of being affected was 0.3; for individuals with two copies of population Y alleles, the probability of being affected was 0.1; for individuals with one copy of population X allele and one copy of population Y allele, the probability of being affected was either 0.2 (additive ancestry risk), 0.1 (recessive ancestry risk) or 0.3 (dominant ancestry risk). When stratification was due to global ancestry difference, the probability of being affected was equal to the global ancestry proportion of the individual, calculated as the average of the ancestry proportions across all markers in the genome. For power evaluation, we simulated case–control data following disease models specified in Table 7. As noted earlier, an advantage of our tests is that they do not require the disease allele frequencies or disease prevalences to be different in the ancestral populations. To evaluate the performance of our tests in this setting, we simulated data assuming the disease allele frequency was 0.4, disease prevalence was 0.3, and genotype relative risk (GRR) was 1.2 in both ancestral populations.
Table 7.
Comparison of power (%)
| GRR | Ancestry Risk | LRT-A | LRT-R | LRT-D | LL |
|---|---|---|---|---|---|
| GRRX = 1.2 | Add | 67.3 | 68.1 | 67.6 | 77.0 |
| GRRY = 1.1 | Rec | 61.2 | 62.0 | 59.4 | 68.2 |
| Dom | 75.9 | 74.9 | 75.7 | 84.8 | |
| GRRX = 1.2 | Add | 63.5 | 62.0 | 61.5 | 72.8 |
| GRRY = 1.0 | Rec | 56.3 | 60.5 | 47.1 | 57.9 |
| Dom | 75.0 | 74.8 | 75.9 | 84.9 | |
| GRRX = 1.2 | Add | 66.5 | 58.3 | 57.3 | 53.4 |
| GRRY = 0.7 | Rec | 81.1 | 96.8 | 20.9 | 14.8 |
| Dom | 75.2 | 69.6 | 80.0 | 80.6 |
The prevalence of population X is 0.3 and the prevalence of population Y is 0.1. pX = 0.6 and pY = 0.2. GRRX is the genotype relative risk for population X at the test marker. GRRY is the genotype relative risk for population Y at the test marker.
Type I error rates were estimated based on 10 000 replicate datasets, and power was estimated based on 1000 replicates. Each replicate dataset consisted of 1000 unrelated cases and 1000 unrelated controls. We analyzed each simulated dataset using the following tests: (i) LRT, (ii) Logistic-Local, (iii) Logistic-Global, a logistic regression procedure with the global ancestry proportion (calculated as the average of local ancestry proportions across all markers in the genome) as a covariate and (iv) EIGENSTRAT. We assumed additive model for the test SNP genotype in all tests. For the LRT approach, we conducted the test assuming the ancestry risk model is additive, dominant or recessive. For EIGENSTRAT, we included the first 10 principal components in the analysis.
2.9 Simulation of a synthetic GWAS dataset
To test the performance of our tests in GWAS settings, we simulated a synthetic GWAS dataset based on ancestry characteristics in the Maywood study (Kang et al., 2010). In this study, 701 unrelated African Americans were collected from Maywood, IL, USA. All study subjects were genotyped using Affymetrix 6.0 SNP array. Due to its small sample size and the lack of disease phenotypes, the Maywood dataset is not appropriate for testing the performance of different tests. However, we can use it to simulate GWAS dataset with realistic admixture patterns. Specifically, we used ADMIXPROGRAM (Zhu et al., 2006) to infer each individual's ancestry using 2606 selected ancestry informative SNPs and obtained the distribution of ancestry proportion. Based on this distribution, we then simulated an admixed population with average 20% Caucasian and 80% African ancestries. For each individual with average ancestry proportion wi (sampled from the ancestry distribution estimated from Maywood), we simulated SNP genotypes for 22 autosomal chromosomes using data generated by HapMap, which includes 1 969 739 SNPs with complete haplotype information for CEU (Utah residents with ancestry from northern and western Europe) and YRI (Yruba in Ibadan, Nigeria) samples. For each chromosome, we simulated the number of crossover points s from a Poisson distribution with mean μ = l × g × 10−7, where l is the length of the chromosome, g is the generation since the individual began admixture and was randomly sampled from 1 to 10. We then randomly sampled haplotype segments from CEU or YRI haplotypes between two crossovers independently according to the average ancestry proportion. Next, we introduced selection to a 5 Mb region centered at rs6576848 (87 053 359 bp) on Chromosome 1 by simulating haplotype segments with 40% Caucasian ancestry and 60% African ancestry. The size of the selection region is similar to that reported by Tang et al. (2007). Similar simulation strategies have been employed by Qin et al. (2010). We assigned disease status according to local ancestry at rs6576848: the probabilities of being affected are 0.1, 0.2 and 0.3, respectively, if the numbers of YRI alleles at rs6576848 are 0, 1 and 2. Applying this simulation procedure, we simulated 1000 cases and 1000 controls. We then thinned the data to Affymetrix 6.0 SNP density, leaving ∼800 000 autosomal SNPs in the analysis. In this simulated dataset, there is no allelic association between any SNPs and the disease status. The association between the SNPs and disease status is simply induced by shared ancestry due to natural selection.
3 RESULTS
3.1 Comparison of Type I error rates
Table 3 shows the estimated Type I error rates of different tests when population stratification is due to local ancestry difference at the test SNP between cases and controls. Not surprisingly, tests that adjust for global ancestry cannot effectively remove the effect of population stratification. For example, when the allele frequencies in populations X and Y were 0.4 and 0.2 and the ancestry risk model was additive, the Type I error rates of Logistic-Global and EIGENSTRAT were 12.44 and 7.62%, respectively. This Type I error rate inflation increased further when the allele frequency difference between populations X and Y was increased. In contrast, the Type I error rates of the LRT and Logistic-Local approaches were under control. In particular, for the LRT approach, its Type I error rates were less than the nominal level even when the ancestry risk model was misspecified in the analysis. When population stratification was due to global ancestry difference among study subjects, the Type I error rates of all tests were under control (Table 4). Our results indicate that regardless of the mechanism of population stratification, whether due to local or global ancestry differences, it is sufficient to adjust for local ancestry at the test SNP. This is consistent with the theoretical result in the Appendix (Supplementary Material). However, global adjustment procedures such as Logistic-Global and EIGENSTRAT may fail to control Type I error rates when population stratification is due to local ancestry difference. To investigate this further, we calculated the correlation coefficient between global ancestry and the local ancestry at the test SNP, and found the degree of correlation is generally <0.3. This explains why global ancestry adjustment may fail to control population stratification that is due to local but not global ancestry.
Table 3.
Comparison of Type I error rates (%) when population stratification is due to local ancestry difference between cases and controls
| Allele Freq | Ancestry Risk | LRT-A | LRT-R | LRT-D | LL | LG | ES |
|---|---|---|---|---|---|---|---|
| pX = 0.4 | Add | 3.31 | 3.50 | 3.35 | 3.20 | 12.44 | 7.62 |
| pY = 0.2 | Rec | 3.67 | 3.25 | 3.81 | 3.17 | 32.92 | 16.47 |
| Dom | 2.69 | 2.71 | 2.68 | 2.43 | 4.12 | 3.29 | |
| pX = 0.6 | Add | 2.83 | 3.06 | 3.59 | 2.65 | 40.89 | 34.67 |
| pY = 0.2 | Rec | 3.17 | 3.25 | 3.50 | 2.95 | 84.70 | 53.36 |
| Dom | 2.66 | 2.58 | 3.11 | 2.40 | 9.03 | 5.92 | |
| pX = 0.8 | Add | 2.88 | 2.86 | 3.48 | 2.87 | 79.47 | 74.28 |
| pY = 0.2 | Rec | 3.45 | 3.25 | 3.58 | 3.09 | 99.40 | 91.08 |
| Dom | 3.01 | 2.87 | 3.16 | 2.77 | 20.32 | 12.07 |
Significance was assessed at the 5% level. LRT-A, LRT assuming ancestry risk is additive; LRT-R, LRT assuming ancestry risk is recessive; LRT-D, LRT assuming ancestry risk is dominant; LL, Logistic-Local; LG, Logistic-Global; ES, EIGENSTRAT.
Table 4.
Comparison of Type I error rates (%) when population stratification is due to global ancestry difference between cases and controls
| pX | pY | LRT-A | LRT-R | LRT-D | LL | LG | ES |
|---|---|---|---|---|---|---|---|
| 0.4 | 0.2 | 3.25 | 3.15 | 3.88 | 3.14 | 2.98 | 3.08 |
| 0.6 | 0.2 | 3.20 | 3.11 | 3.59 | 2.82 | 3.48 | 3.36 |
| 0.8 | 0.2 | 3.28 | 3.18 | 3.18 | 3.28 | 3.69 | 4.04 |
Significance was assessed at the 5% level.
In the above simulations, we assumed the ancestry state at the test SNP was known. We also assessed the performance of our tests when there is uncertainty in ancestry probability estimation. Ideally, one should use programs such as HAPMIX to estimate ancestry probabilities. However, these programs are computationally intensive and it is not feasible to run these programs on all simulated datasets. To circumvent this difficulty, we added uncertainties to the ancestry states according to the error model specified in Table 5, which creates similar patterns and magnitude of uncertainty as those in the estimated ancestry from HAPMIX and LAMP based on our analysis of testing datasets. In our error model, we set the uncertainty parameter for population X to be less than that for population Y. The rationale is that for the data we simulated (mimicking African Americans), 80% of the genome came from population X and only 20% came from population Y. Thus, the effective sample size for population X was larger, and this led to more accurate ancestry probability estimates than for population Y. Table 6 shows the Type I error rates of the LRT and Logistic-Local approaches when there are uncertainties in ancestry estimation. Our results indicate that the Logistic-Local approach is robust to uncertainties in ancestry estimation. Its Type I error rates were below the nominal level under all the settings we considered. For the LRT approach, its Type I error rates were under control when assuming additive or dominant model for the ancestry risk, and slightly inflated when the assumed ancestry risk model was recessive and the degree of uncertainty was high. We note that when GWAS data are available, with the current state-of-the-art program such as HAPMIX, ancestry probabilities can be reliably estimated for an admixed population such as African American population. Therefore, we anticipate that the amount of uncertainty in practical GWAS may not be a concern.
Table 5.
Uncertainty model for ancestry probability estimation
| Ancestry probability | True probability | Probability with uncertainty |
|---|---|---|
| P(ISNP = 0|GANC | 0 | εX2 |
| P(ISNP = 1|GANC | 0 | 2εX(1 − εX) |
| P(ISNP = 2|GANC) | 1 | (1 − εX)2 |
| P(ISNP = 0|GANC) | 0 | εX(1 − εY) |
| P(ISNP = 1|GANC) | 1 | εXεY+(1 − εX)(1 − εY) |
| P(ISNP = 2|GANC) | 0 | (1 − εX)εY |
| P(ISNP = 0|GANC) | 1 | (1 − εY)2 |
| P(ISNP = 1|GANC) | 0 | 2εY(1 − εY) |
| P(ISNP = 2|GANC) | 0 | εY2 |
εX is randomly generated from uniform (0, EX), and εY is generated from uniform (0, EY), where EX and EY are uncertainty parameters.
Table 6.
Type I error rates (%) of LRT and Logistic-Local when there are uncertainties in ancestry probabilities
| Ancestry Risk | EX | EY | LRT-A | LRT-R | LRT-D | LL |
|---|---|---|---|---|---|---|
| Add | 0.01 | 0.05 | 2.81 | 3.86 | 2.99 | 2.63 |
| 0.01 | 0.10 | 3.18 | 5.37 | 2.81 | 2.67 | |
| 0.03 | 0.05 | 3.04 | 4.39 | 2.92 | 2.70 | |
| Rec | 0.01 | 0.05 | 3.20 | 3.86 | 4.19 | 2.95 |
| 0.01 | 0.10 | 3.45 | 4.86 | 3.03 | 2.98 | |
| 0.03 | 0.05 | 3.51 | 4.37 | 4.22 | 2.95 | |
| Dom | 0.01 | 0.05 | 2.90 | 3.44 | 2.72 | 2.52 |
| 0.01 | 0.10 | 3.22 | 5.45 | 2.48 | 2.50 | |
| 0.03 | 0.05 | 3.12 | 4.09 | 2.63 | 2.53 |
The reference allele frequency for population X at the test marker is 0.6 and 0.2 for population Y. EX and EY are uncertainty parameters.
3.2 Comparison of power
Next, we compared the power of the LRT and Logistic-Local approaches using data simulated under disease models specified in Table 7. We did not compare with the Logistic-Global and EIGENSTRAT approaches as they have inflated Type I error rates when local ancestry is the main factor for stratification. We considered three scenarios for power evaluation: (i) the test SNP was associated with the disease in the same direction in populations X and Y (GRRX = 1.2, GRRY = 1.1), (ii) the test SNP was associated with the disease only in population X (GRRX = 1.2, GRRY = 1.0) and (iii) the test SNP was associated with the disease in opposite directions in populations X and Y (GRRX = 1.2, GRRY = 0.7). Table 7 shows the power comparison results. For the first two scenarios, the power of the Logistic-Local approach was slightly higher than LRT, especially when the true ancestry risk model was additive or dominant. However, when the test SNP was associated with the disease in opposite directions in the ancestral populations, the power of the Logistic-Local approach can be substantially lower than LRT. For example, in the third scenario, the power of the LRT approach assuming recessive ancestry risk was 96.8% when the true ancestry risk model was recessive, but the power of the Logistic-Local approach was only 14.8%. The power of the LRT approach can be higher than Logistic-Local even when the ancestry risk model is misspecified. Such a significant drop in power for the Logistic-Local approach is due to: (i) its inherent assumption on the additivity of the ancestry risk on the logit scale and (ii) its inability to model opposite directions of association in the ancestral populations. The power of this test can be greatly attenuated when these assumptions are not satisfied. In contrast, the LRT approach uses two sets of penetrances to model disease risk for the two ancestral populations, and thus is more flexible.
For the situation where the disease allele frequency and disease prevalence are equal in the two ancestral populations, we compared the power of the LRT and Logistic-Local approaches with the traditional admixture mapping method, which tests for the correlation between disease status and local ancestry proportion at the test SNP. When the disease allele frequency was 0.4, disease prevalence was 0.3 and GRR was 1.2 in both ancestral populations, the power of the traditional admixture mapping method was close to the 5% nominal level, whereas the power of the LRT and Logistic-Local approaches were 82% and 89%, respectively. Therefore, our tests can identify signals that are missed by the traditional admixture method and be used as a general tool for genetic association analysis.
3.3 Analysis of the synthetic GWAS dataset
We analyzed the synthetic GWAS dataset simulated based on the Maywood study (Kang et al., 2010) using five approaches, including LRT, Logistic-Local, Logistic-Global, EIGENSTRAT and unadjusted trend test. For the LRT, we assumed the disease prevalences in Africans and Caucasians to be 0.3 and 0.1, respectively, and analyzed the data assuming additive, dominant and recessive models for ancestry risk. Figure 1 shows the results from different tests for the region on Chromosome 1 that undergoes selection. Using local adjustment tests, we did not observe any evidence of association; however, all the other tests yield highly significant results, with many SNPs reaching genome-wide significance (P-values < 5 × 10−8). The global ancestry adjustment procedures such as Logistic-Global and EIGENSTRAT performed similarly as the unadjusted trend test, suggesting that global ancestry cannot sufficiently capture the local ancestry variation for regions with natural selection. We note that the significant results from Logistic-Global, EIGENSTRAT and trend tests are not due to allelic association between the SNPs and disease status, but are rather driven by the association between the disease status and local ancestry in the region. Our results demonstrate that global ancestry adjustment procedures may identify a wrong region if local ancestry in the region happens to be associated with disease status. As an example, it has been previously shown that EIGENSTRAT cannot completely remove the effect of population stratification at the lactase gene (Epstein et al., 2007; Li et al., 2010; Price et al., 2006). We suspect that this failure is probably because global ancestry cannot fully reflect the ancestry variation at the lactase gene.
Fig. 1.

P-values from various tests for the analysis of the synthetic GWAS dataset. Shown are the results for the region on Chromosome 1 that undergoes natural selection but with no allelic association between the SNPs and disease status. Results from LRT-Rec and LRT-Dom are similar to LRT-Add. The gray horizontal line corresponds to P-value=5 × 10−8.
We also note that admixture mapping searches for correlation between local ancestry and disease status. Our results indicate that even if there is a causal SNP in the region, adjusting for global ancestry will result in a wide region with association signals (∼5 Mb or even larger depending on the size of the ancestry block) due to inflated signals induced by association with local ancestry. Therefore, the global ancestry adjustment methods may point to the right region that harbors causal SNPs, but this region might be too wide and may lead to following up of wrong SNPs/genes. In contrast, the local ancestry adjustment procedures will appropriately control the background signals induced by association with local ancestry, and thus can pinpoint the correct SNPs/genes for follow-up study. This makes the local adjustments procedures a useful tool for fine mapping of regions identified from admixture mapping studies.
4 DISCUSSION
We showed that in genetic association analysis of admixed populations, local ancestry difference between study subjects can confound with association signal and lead to spurious associations if not appropriately controlled. We also showed theoretically that to remove the confounding effect of local ancestry, it is sufficient to condition on local ancestry at the test SNP, whereas conditioning on global ancestry may be ineffective. To remove the spurious associations due to the confounding effect of local ancestry, we further proposed two novel association tests that adjust for local ancestry at the test SNP. We conducted extensive simulations and evaluated the performance of different tests under various settings. Our simulation results indicate that regardless of the mechanism of population stratification, whether due to local or global ancestry difference, it is sufficient to control population stratification by conditioning on local ancestry at the test marker. In contrast, global ancestry adjustment procedures such as EIGENSTRAT and Logistic-Global cannot completely remove the effect of population stratification induced by local ancestry difference. The reason is that global ancestry information as represented by a global ancestry proportion or a few principal components obtained from all markers across the genome cannot accurately reflect the amount of ancestry variation in local regions.
We proposed two tests for association analysis in admixed populations. Our first test, LRT, is based on a conditional likelihood framework which models the distribution of a test SNP given disease status and flanking marker genotypes. This conditional likelihood allows us to explicitly model local ancestry differences among study subjects and thus it eliminates the effect of population stratification at the test SNP. Our second test, Logistic-Local, is based on a logistic regression model that adjusts for local ancestry proportion at the test SNP. Although the LRT approach is computationally more involved than Logistic-Local, it is more flexible and is particularly useful when the directions of association are different in the ancestral populations. Another advantage of the LRT approach is that, in addition to testing the null hypothesis as described in Section 2, we can test two other hypotheses H0: no disease–SNP association in population X, and H0: no disease–SNP association in population Y, separately. This can be achieved by testing H0:f0,X=f1,X=f2,X and H0:f0,Y=f1,Y=f2,Y, respectively. These tests allow a cross-ethnicity replication since evidence of disease association in the two ancestral populations can be directly compared (Risch et al., 2006).
Our tests are different from admixture mapping in that we directly assess the correlation between a phenotype and SNP genotypes. In contrast, admixture mapping examines the association between a phenotype and local ancestry without fully using the actual genotypes at each SNP. Therefore, SNPs falling within the same ancestry block will share similar admixture mapping signal, which explains why admixture mapping has substantially lower resolution than direct SNP association analysis. Since the association tests we proposed directly compare the allele frequencies between cases and controls, they can serve as a fine-mapping tool for regions identified from admixture mapping studies. As shown in our simulations, another advantage of our tests is that, unlike admixture mapping, which may miss disease variants with similar allele frequencies in the ancestral populations, our tests are able to identify such variants since the allele frequencies between cases and controls are directly compared. Therefore, our tests can be used as a general tool for genetic association analysis.
Our tests rely on the estimates of local ancestry probabilities. It is computationally intensive to estimate these probabilities for GWAS datasets. In addition, our LRT test requires the estimation of several parameters, which is computationally more involved than logistic regression; for example, with 1000 cases and 1000 controls, it took the LRT test about 3 days to finish the analysis of 500 000 SNPs using a single CPU. However, the computations can be parallelized across chromosomes, and thus the computation is tractable even for very large datasets if a computing cluster is available.
Our methods treat estimated ancestry as plug-in estimates in the likelihood calculation. Although simple, such a two-step procedure is not as efficient as approaches that estimate ancestry and disease model parameters simultaneously. Another problem is that the disease phenotypes are not used when estimating ancestry. However, near a disease locus, cases are more related in terms of their shared ancestry than controls. Ignoring disease status essentially assumes that all individuals are no more related to one another than would be expected by chance, and thus may lead to ancestry estimates that are biased towards the null. How to jointly model local ancestry and disease status would merit further research.
Our methods share similarity with a recent paper published by Qin et al. (2010), who proposed to correct for population stratification using local principal components (PCs). In principle, this method can be applied to admixed populations as well. However, local PCs can only approximate the local ancestry and require a predefined window size, whereas for admixed populations such as African Americans, locus-specific ancestry can be inferred accurately. As noted by Qin et al. (2010), the local PC-based approach will be more appropriate for a population whose substructure is subtle, due to either the lack of information on the ancestral population or when admixture has occurred within similar populations, for example, European Americans.
In summary, we have proposed two novel association tests for admixed populations. We showed that dependence between local ancestry and disease phenotype can lead to spurious associations. Our results indicate that it is better to adjust for local ancestry than for global ancestry to appropriately control for population stratification. The method in this article is implemented in a C program and can be obtained by contacting the last author.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Hongzhe Li for helpful discussions.
Funding: National Institutes of Health (grants R01HG004517 to M.L. and C.L., R01HG005854 to M.L. and R01HL074166 and R01 HG003054 to X.Z.).
Conflict of Interest: none declared.
REFERENCES
- Bersaglieri T, et al. Genetic signatures of strong recent positive selection at the lactase gene. Am. J. Hum. Genet. 2004;74:1111–1120. doi: 10.1086/421051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell CD, et al. Demonstrating stratification in an European American population. Nat. Genet. 2005;37:868–872. doi: 10.1038/ng1607. [DOI] [PubMed] [Google Scholar]
- Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- Epstein MP, et al. A simple and improved correction for population stratification in case-control studies. Am. J. Hum. Genet. 2007;80:921–930. doi: 10.1086/516842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggart CJ, et al. Design and analysis of admixture mapping studies. Am. J. Hum. Genet. 2004;74:965–978. doi: 10.1086/420855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SJ, et al. Genome-wide association of anthropometric traits in African- and African-derived populations. Hum. Mol. Genet. 2010;19:2725–2738. doi: 10.1093/hmg/ddq154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan NL, et al. Marker selection for the transmission/disequilibrium test, in recently admixed populations. Am. J. Hum. Genet. 1998;62:703–712. doi: 10.1086/301760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, et al. Correcting population stratification in genetic association studies using a phylogenetic approach. Bioinformatics. 2010;26:798–806. doi: 10.1093/bioinformatics/btq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P-I, et al. No gene is an island: the flip-flop phenomenon. Am. J. Hum. Genet. 2007;80:531–538. doi: 10.1086/512133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeigue PM. Mapping genes that underlie ethnic differences in disease risk: methods for detecting linkage in admixed populations. Am. J. Hum. Genet. 1998;63:241–251. doi: 10.1086/301908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montana G, Pritchard JK. Statistical tests for admixture mapping with case-control and cases-only data. Am. J. Hum. Genet. 2004;75:771–789. doi: 10.1086/425281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelder JA, Mead R. A simplex method for function minimization. Comput. J. 1965;7:308–313. [Google Scholar]
- Patterson N, et al. Methods for high-density admixture mapping of disease genes. Am. J. Hum. Genet. 2004;74:979–1000. doi: 10.1086/420871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Price AL, et al. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS. Genet. 2009;5:e1000519. doi: 10.1371/journal.pgen.1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, et al. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19:826–837. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, et al. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 2010;11:459–463. doi: 10.1038/nrg2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, et al. Association mapping in structured populations. Am. J. Hum. Genet. 2000;67:170–181. doi: 10.1086/302959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H, et al. Integrating local population structure for fine mapping in genome-wide association studies. Bioinformatics. 2010;26:2961–2968. doi: 10.1093/bioinformatics/btq560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N. Linkage strategies for genetically complex traits. I. Multilocus models. Am. J. Hum. Genet. 1990;46:222–228. [PMC free article] [PubMed] [Google Scholar]
- Risch N, Tang H. Whole genome association studies in admixed populations. Am. J. Hum. Genet. 2006;S79:254. [Google Scholar]
- Sankararaman S, et al. Estimating local ancestry in admixed populations. Am. J. Hum. Genet. 2008;82:290–303. doi: 10.1016/j.ajhg.2007.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satten GA, et al. Accounting for unmeasured population substructure in case-control studies of genetic association using a novel latent-class model. Am. J. Hum. Genet. 2001;68:466–477. doi: 10.1086/318195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillanpaa MJ. Overview of techniques to account for confounding due to population stratification and cryptic relatedness in genomic data association analyses. Heredity. 2010 doi: 10.1038/hdy.2010.91. Availabel at http://www.ncbi.nlm.nih.gov/pubmed/20628415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MW, et al. A high-density admixture map for disease gene discovery in African Americans. Am. J. Hum. Genet. 2004;74:1001–1013. doi: 10.1086/420856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundquist A, et al. Effect of genetic divergence in identifying ancestral origin using HAPAA. Genome Res. 2008;18:676–682. doi: 10.1101/gr.072850.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, et al. Reconstructing genetic ancestry blocks in admixed individuals. Am. J. Hum. Genet. 2006;79:1–12. doi: 10.1086/504302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, et al. Recent genetic selection in the ancestral admixture of Puerto Ricans. Am. J. Hum. Genet. 2007;81:626–633. doi: 10.1086/520769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, et al. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, et al. A hidden Markov modeling approach for admixture mapping based on case-control data. Genet. Epidemiol. 2004;27:225–239. doi: 10.1002/gepi.20021. [DOI] [PubMed] [Google Scholar]
- Zhang S, et al. On a semi parametric test to detect associations between quantitative traits and candidate genes using unrelated individuals. Genet. Epidemiol. 2003;24:44–56. doi: 10.1002/gepi.10196. [DOI] [PubMed] [Google Scholar]
- Zhu X, et al. Association mapping, using a mixture model for complex traits. Genet. Epidemiol. 2002;23:181–196. doi: 10.1002/gepi.210. [DOI] [PubMed] [Google Scholar]
- Zhu X, et al. Linkage analysis of a complex disease through use of admixed populations. Am. J. Hum. Genet. 2004;74:1136–1153. doi: 10.1086/421329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, et al. A classical likelihood based approach for admixture mapping using EM algorithm. Hum. Genet. 2006;120:431–445. doi: 10.1007/s00439-006-0224-z. [DOI] [PubMed] [Google Scholar]
- Zhu X, et al. Admixture mapping and the role of population structure for localizing disease genes. Adv. Genet. 2008;60:547–569. doi: 10.1016/S0065-2660(07)00419-1. [DOI] [PubMed] [Google Scholar]
- Zollner S, Pritchard K. Overcoming the winner's curse: estimating penetrance parameters from case-control data. Am. J. Hum. Genet. 2007;80:605–615. doi: 10.1086/512821. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.