

Abstract
Motivation: Defining the precise location of structural variations (SVs) at single-nucleotide breakpoint resolution is an important problem, as it is a prerequisite for classifying SVs, evaluating their functional impact and reconstructing personal genome sequences. Given approximate breakpoint locations and a bridging assembly or split read, the problem essentially reduces to finding a correct sequence alignment. Classical algorithms for alignment and their generalizations guarantee finding the optimal (in terms of scoring) global or local alignment of two sequences. However, they cannot generally be applied to finding the biologically correct alignment of genomic sequences containing SVs because of the need to simultaneously span the SV (e.g. make a large gap) and perform precise local alignments at the flanking ends.
Results: Here, we formulate the computations involved in this problem and describe a dynamic-programming algorithm for its solution. Specifically, our algorithm, called AGE for Alignment with Gap Excision, finds the optimal solution by simultaneously aligning the 5′ and 3′ ends of two given sequences and introducing a ‘large-gap jump’ between the local end alignments to maximize the total alignment score. We also describe extensions allowing the application of AGE to tandem duplications, inversions and complex events involving two large gaps. We develop a memory-efficient implementation of AGE (allowing application to long contigs) and make it available as a downloadable software package. Finally, we applied AGE for breakpoint determination and standardization in the 1000 Genomes Project by aligning locally assembled contigs to the human genome.
Availability and Implementation: AGE is freely available at http://sv.gersteinlab.org/age.
Contact: pi@gersteinlab.org
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The problem of single-nucleotide breakpoint resolution for genome structural variations (SVs) (deletions, insertions, inversions, etc.) is of great importance for a number of reasons. First, as recently demonstrated (Lam et al., 2010), single-nucleotide breakpoint resolution is absolutely necessary for SV classification and annotation. It is also important for genotyping known SVs in newly sequenced genomes (Lam et al., 2010). Second, precise breakpoints are required to evaluate the functional impact of SVs. For example, uncertainty in breakpoints in just a few bases may lead to ambiguous conclusions when an SV is close to a splice-junction and/or regulation sites or overlaps exon(s). Last, but not least, construction of personal diploid genomes (one of the ultimate long-term goals of human genome analysis) cannot be done properly without precise knowledge of SV breakpoints.
It might seem obvious, but the only plausible way to achieve single-nucleotide breakpoint resolution is to align two sequences: one without an SV (e.g. a region in the reference human genome) and another containing an SV (e.g. locally assembled contig, completely sequenced and assembled fosmid clone or long read). Most commonly used methods for SV detection provide only approximate breakpoint locations. Paired-end mapping (also called read-pair) approaches inherently have uncertainty in breakpoint resolution, due to uncertainty in the distance between sequenced ends and the possibility of read mismapping (Korbel et al., 2009; Medvedev et al., 2009). Resolution of breakpoints by array comparative genomic hybridization analysis and read-depth approaches is limited by the probe density (for array) and the genomic bin size (for read-depth) used to produce the subsequently analyzed signal (Abyzov et al., 2010; Medvedev et al., 2009; Wang et al., 2009).
While being imprecise in breakpoint resolution, the approaches mentioned above yield approximate SV locations, where a local assembly of a haplotype bridging an SV region could be accomplished. Subsequently, alignment of the assembled contig to the predicted SV region identifies precise SV breakpoints. The described strategy is employed by the 1000 Genomes Project (Durbin et al., 2010; Mills, 2010), where tens of thousands of local haplotype assemblies in the SV regions are made. Proper alignment of those contigs will and already is an important challenge that must be fulfilled precisely and computationally efficiently, given the number of expected local assemblies. Single-nucleotide resolution of SV breakpoints will allow their standardization and analysis in a single framework.
The problem of aligning two sequences containing SVs might seem to be trivial, but upon deeper consideration it is not. The major complications are due to possible repeats within aligned sequence, sequence homology/identity around breakpoints and, the often complex nature of SVs, where, for instance, a deletion/insertion is accompanied by smaller insertion/deletion. More specifically, classical algorithms, which guarantee the finding the optimal global Needleman–Wunsch (Gotoh, 1982; Needleman and Wunsch, 1970) and local SmithWaterman (Smith and Waterman, 1981) alignments, generally cannot arrive at a biologically correct solution when aligned sequences contain SVs (Fig. 1). The major problem with those algorithms is the gap penalty. A large gap penalty does not allow for the extension of alignment across an SV. Reducing the gap penalty interferes with the alignment scoring scheme and jeopardizes the construction of the proper alignment in regions flanking the SV and when the sequence(s) contains repeats. In addition, it offers only a partial solution to the problem, and cases when the SV is not a pure deletion or insertion are still not solved (Fig. 1B).
Fig. 1.

Schematics of the expected optimal alignment around a structural variation (left) and alignments produced by global Needleman–Wunsch (NW) and local Smith–Waterman (SW) algorithms (right). The structural variation, i.e. deletion, is in red. In (B), the deletion is accompanied by a small insertion (blue). Throughout the figure, alignable flanking regions are shown in green and orange. Both SW and NW algorithms generally cannot arrive at a biologically correct alignment.
A generalized global alignment (Huang and Chao, 2003) algorithm is generally also unable to solve the formulated problem. The algorithm works by introducing the concept of a ‘difference block’, e.g. large gap, and imposing a cap on the penalty for having such a block in an alignment. When a block is small, e.g. small gap, it is penalized, as it would be in the classical Needleman–Wunsch algorithm. For a large block, e.g. large gap, the penalty is constant. Therefore, it can only be applied to the alignment of sequences where the SV size is large enough for the algorithm to work in the non-classical mode. More importantly, the algorithm can be misled by sequence similarity around SV breakpoints. Specifically, when sequences around breakpoints are homologous (Fig. 2A), the algorithm has to choose between aligning with a higher sequence identity—but introducing a large gap—or aligning with a lower sequence identity and no gap (Fig. 2B). Only the former scenario is correct, but either one can be chosen by the algorithm (considered to be optimal) depending on the scoring scheme, size of deletion, length and percent of homology around breakpoints, and the lengths of aligned sequences flanking the breakpoints (longer flanking sequences allow one to resolve breakpoints within longer and higher homologous sequences). Incidentally, this problem is inherent to all algorithms employing a concave/piecewise gap penalty. It is also inherent to Needleman–Wunsch and Smith–Waterman algorithms. Therefore, the described problem may hamper the discovery and characterization of a particular class of NAHR (Lam et al., 2010) SVs that are characterized by long similar/homologous sequences around breakpoints.
Fig. 2.

Sequence similarity (see A) around SV breakpoints (shades of green) can mislead local and/or global alignment(s) to produce incorrect alignment (see B). Conceptually, to produce correct alignment one has to find an optimal jump between overlapping local alignments. However, local alignment calculation and jump finding have to be done simultaneously rather than successively to guarantee finding the optimal alignment (Supplementary Fig. S4).
The ‘sandwich dynamic programming’ algorithm, introduced (Wu and Watanabe, 2005) to align cDNAs to exons, could be useful, but even if adopted for aligning sequences containing SVs, it does not offer a general solution, as it has the same problems as the Needleman–Wunsch algorithm when handling events that are not pure deletions or insertions (Fig. 1B). Also, none of the mentioned algorithms could be applied to determine tandem duplication and inversion breakpoints.
Hence, with the aim of achieving single-nucleotide SV breakpoint resolution and standardization, we have developed an algorithm for the correct alignment of sequences containing SVs. This article first describes an algorithm for optimal sequence alignment containing only a single SV deletion or insertion. We then describe algorithm extensions to align sequences containing other SVs. To accomplish the first aim, we formulated it as a problem of finding the optimal local alignment of two sequences containing one unaligned and unpenalized region/gap (corresponding to one SV) between two aligned regions.
The rationale is that flanking regions of an SV are very similar and can be aligned collinearly (5′ end to 5′ end and 3′ end to 3′ end) using a local Smith–Waterman algorithm (Fig. 2C). To yield the final alignment, the two local ones should simply be combined. However, if the alignments of the flanking regions overlap, combining two local alignments becomes complicated, and an optimal jump from one local alignment to another must be found—that is, a gap must be introduced—to maximize the alignment score. The optimal (highest scoring) alignment may not be found if the jump is searched between already calculated local alignments because trimming a local alignment does not guarantee that it is still optimal (a simple example demonstrating the concept is shown in Supplementary Fig. S4). Therefore, the calculation of flanking sequence alignments and finding the optimal jump between the two must be done simultaneously rather than successively.
When formulated this way, the problem explicitly addresses only the issue of the largest gap in the alignment and does not require adjustment or modifications of the alignment scoring scheme. Therefore, substitution matrices and gap penalties tuned to a particular alignment purpose, e.g. contig or short/long-read alignment, can be used unchanged.
2 METHODS
2.1 Algorithm
Let us denote the lengths of two compared sequences as N and M. The algorithm starts with the construction of two [0,N + 1]x[0,M + 1] alignment scoring matrices SL and SR, a la the Smith–Waterman algorithm, for the local alignment of two sequences (Fig. 3A). Indices [1,N] and [1,M] in each matrix, respectively, are used to store alignment scores while indices 0, N + 1, and M + 1 are used for the convenience of filling the matrices and tracing back. One matrix represents a score for the alignment initiated from the 5′ ends (left flanking region of the SV), whereas the other one represents a score for the alignment initiated from the 3′ ends (right flanking regions of the SV). The maximum in each matrix defines a cell from which to start tracing back to find the best local alignment. Importantly, the maximum in the leading/trailing submatrix does so for the local alignment of sequence ends. Specifically, the maximum ML(n, m) in the leading submatrix [0,n]x[0,m] of SL, where n<=N and m<=M, anchors the best local alignment for n and m nucleotides at the 5′ ends. Similarly, the maximum MR(n+1,m + 1) in the trailing submatrix [n+1, N+1]x[m+1,M+1] of SR, anchors the best local alignment for N − n and M − m nucleotides at the 3′ ends: i.e.
| (1) |
Then, the total score of aligning n and m nucleotides at the 5′ ends and N − n and M − m nucleotides at the 3′ ends is ML(n, m)+MR(n+1, m+1). The optimal alignment has the highest score; thus it maximizes the sum: i.e.
| (2) |
where BS is the best score. In other words, one has to maximize the sum of the maxima in the paired submatrices of SL and SR (Fig. 3B). Such a maximum can be found in quadratic time. Note that
| (3) |
Using (3), one can convert matrices SL and SR to have values ML(n, m) and MR(n, m), respectively. During such conversion, one can trace from whence the value in each cell was assigned, just like when constructing an alignment score matrix. Having matrices ML and MR calculated, one can find the highest score sum (2) in one pass through the matrices. The corresponding alignment is then constructed by, first, tracing back the maximum location in each matrix and, then, tracing back alignments for the 5′ and 3′ ends (i.e. alignment is inferred from each matrix) and combining them (Fig. 3C). The unaligned region is the one between 5′ and 3′ end alignments.
Fig. 3.

Schematics of the algorithm. (A) Two alignment score matrices for the alignment of the 5′ ends (SL matrix) and the 3′ ends (SR matrix) are constructed a la the Smith–Waterman local alignment. In this example, scoring is as follows: match = 1, mismatch = −1, gap open penalty = 4 and gap extend penalty = 2. Orange arrows represent trace-back information. The best alignment maximizes the sum of the maximum in the leading submatrix (highlighted buff) in SL and the maximum in the paired trailing submatrix (highlighted cyan) in SR. (B) Matrices are converted so that each cell contains the maximum score of the leading submatrix in SL and the trailing submatrix in SR. The location of score maxima is traced (blue arrows), just like the score is traced in scoring matrices. Maximum score sum (red) can now be found in one pass through the matrices. (C) Alignment is constructed by, first, tracing back the maximum location (red arrows) in each matrix and then, tracing back alignments for the 5′ and 3′ ends (green arrow). The resulting alignment is the sum of the alignments at the 5′ and 3′ ends, with the unaligned region in-between. The maximum score can be redundant (bold rectangles). However, the resulting alignment will be the same.
The best score can be redundant (Fig. 3B). However, redundancy does not necessarily imply alternative alignments. As shown in the figure, the sum for indices (n, m) falling in the bold area is equal to the best score. However, tracing back for the maximum locations in each matrix will lead to the same cells, i.e. the very northwest and southeast cells of the bold area for matrices SL and SR, respectively. Intuitively, one can think about finding the optimal (maximum) score as a procedure of trying all possible sequence splitting into two subsequences (5′ end and 3′ end) and optimally aligning those subsequences. The splitting of one of the sequences within the SV region does not change flanking alignment, and, thus, generates the same maximum score (Supplementary Fig. S6). Thus, only different maximum locations are indicative of alternative alignments. It is a trivial computational task to check for such alternative alignments.
Another reason for a redundant maximum score sum is sequence identity around SV breakpoints (Supplementary Fig. S2). In such cases, ‘shuttling’ of one or a few pairs of aligned nucleotides in alignment from one breakpoint to another does not change the overall score. Thus, in some cases, the resolution of SV breakpoints is naturally limited to the length of sequences that are identical around breakpoints. Note, that the limitation is not methodological, but, rather, biological. Moreover, wherever the actual breakpoints within the identical sequences are, the resulting sequence, after SV excision, is the same. Therefore, breakpoint uncertainty caused by sequence identity will not affect downstream analysis. Such cases can be easily identified and described by post-processing the produced alignments.
2.2 Generalization of the algorithm
The generalizations described below allow for breakpoint inference for multiple deletions/insertions, tandem duplication, inversions and for splice sites within genes. As previously explained, approximate locations of breakpoints and read/assembly bridging SV breakpoints or splice sites are a prerequisite.
2.2.1 Inferring breakpoints when sequences contain multiple deletions/insertions
When aligned sequences contain two SVs (either insertions or deletions) an optimal alignment with two unaligned regions must be found. Thus, it is necessary to introduce two jumps between three matrices (Supplementary Fig. S5). As before, two matrices ML and MR represent the alignment of sequence 5′ and 3′ ends and one SR represents the local alignment of sequence fragments between two SVs. The optimal alignment will maximize the total score of aligning all three fragments: i.e.
| (4) |
where TSR(n, m) is a trace−back path for element (n, m) in matrix SR. The last condition states that elements (n, m) and (n′, m′) are on the same alignment path. The maximum can efficiently be found by checking all pairs of (n, m) and only those (n′, m′) pairs that can be traced back from the (n+1, m+1) element in matrix SR. Tracing back is a linear procedure and may require up to min(N, M) operations: thus, the optimal solution can be found in O(NM min(N, M)) time in the worst case.
Furthermore, the algorithm can be inductively generalized to produce alignments with any given k number of SVs. For k = 3, one needs to maximize the sum of scores for Smith–Waterman alignment of 5′ end sequences and that of 3′ end sequences with two SVs: i.e.
where BSr is the best score for aligning residual sequences at 3′ ends, i.e. [n+1, N] and [m+1, M]. Generally, for k > 2 SVs in alignment, best score can be found using the following induction
| (5) |
For each increment of k > 2, the time complexity of the algorithm increases multiplicatively by a factor of O(NM).
2.2.2 Inferring splice sites by cDNA alignment
Given a cDNA sequence containing two exons and an approximate location of an intron between the two exons, the described algorithm can be extended to determine the splice site at breakpoint resolution. Aligning such cDNA to the genome is similar to aligning sequences containing deletion/insertion SVs, but has an important difference. The complete cDNA sequence is expected to align to the genome, i.e. cases when deletion is accompanied by micro insertion (Fig. 1B) do not happen. In other words, with an excised intron the sequence should align to the genome globaly. This can be accomplished by introducing two changes to the algorithm: (i) matrices SL and SR should be calculated a la the Needelman–Wunsch algorithm; (ii) maxima should be calculated for different submatrices
| (6) |
where we assume that the second sequence is cDNA. Now ML(n, m) stores the value of the the best global alignment for the 5′ end, while MR(n, m) does so for the 3′ end. Subsequent steps (finding best score and tracking back) should be done as before.
2.2.3 Inferring breakpoints for tandem duplications
Aligning a contig or a read that spans tandem duplication breakpoints is similar to aligning sequences with deletions and insertions (Fig. 4). In particular, when a contig/read spans an insertion site around a breakpoint that is farther from the duplication original site (Fig. 4A), then the alignment procedure is exactly the same as for sequences with simple deletion or insertion. In cases when a contig/read spans an insertion site around a breakpoint that is closer to the duplication original site (Fig. 4B), then the order of the aligned fragments is different in the two sequences. Using the described methodology, but maximizing a different function (assuming the second sequence is a contig/read)
| (7) |
one can find the highest scoring split-alignment for a contig's 3′- and 5′-ends. Subsequently, post-processing can be applied to ensure that end alignments do not overlap.
Fig. 4.

Schematics for aligning sequences with tandem duplication and inversion. Color gradient reflects the directionality of sequences from the 5′-end to the 3′-end. (A) Aligning a contig/read spanning a duplication breakpoint can be no different than aligning sequences with a deletion/insertion. (B) For contigs spanning other duplication breakpoints, the order of the aligned fragments is different in the two se-quences. (C) Optimal split-alignment for sequences with inversions is calculated by aligning the 5′-end of the first sequence to the 5′-end of the second one and aligning the 3′-end of the first sequence to the 3′-end of the reverse complement of the second one.
2.2.4 Inferring breakpoints for inversions
The algorithm for the optimal alignment of inversions was described a number of years ago (Schoniger and Waterman, 1992). However, it is only applicable to cases in which an inversion is completely enclosed in either of the aligned sequences. Cases in which an inversion is incomplete, i.e. one sequence spans only the inversion breakpoint, are not handled by that algorithm, but this can be accomplished by generalizing the algorithm described above. Indeed, if we have two sequences, part of the first sequence will align to the second and another non-overlapping part will align to the reverse complement of the second sequence (see schematics in Fig. 4). Again, using the described methodology one can find split-alignment for a contig's 3′ and 5′ ends. Note that one should use a reverse complement for one of the aligned sequences to construct matrix SR together with maximizing the function in Equation (7). The same post-processing as for tandem duplication breakpoint inference can be applied to ensure that end alignments do not overlap.
2.3 Alternative recurrence
The described algorithm and generalizations can be restated using alternative but equivalent recurrence. Namely, to infer deletion/insertion breakpoints, the recurrence is
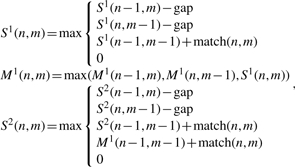 |
(8) |
and to infer splice sites, the recurrence is
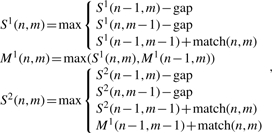 |
(9) |
where second sequence is cDNA, match(n, m) is match or mismatch between nucleotides in positions n and m, and gap is a gap function. The matrices S1 and M1 are the same as SL and ML, while the matrix S2 keeps the alignment score for the left flanking region (matrix S1) and (via M1) for the right flanking region. Generalizing to two or more SVs/introns in an alignment requires the use of additional pairs of matrices Mi and Si for each SV/intron, where the recurrence for Mi utilizes values from Si (just as M1 does from S1), and the recurrence for Si+1 utilizes values from Mi (just as S2 does from M1). Linear space alignment algorithms (Chao et al., 1994; Hirschberg, 1975) can also be applied with this recurrence.
3 RESULTS
3.1 Implementation, Alignment with Gap Excision program
We have implemented the algorithm described above in the C++ language as the Alignment with Gap Excision (AGE ) program (freely available at http://sv.gersteinlab.org/age). The current implementation is limited to aligning sequences containing only one deletion, insertion or inversion. The major challenge in implementation was to reduce memory usage, as the algorithm operates on two matrices (unlike only one, in the case of the classical Smith–Waterman algorithm) and requires additional storage for recording of traces to locations of maxima (Fig. 3B). In addition, genomics sequences containing SVs are typically thousands (at times, hundreds of thousands) of nucleotides in length requiring large matrices for alignment calculations. To reduce memory usage, we used small integers for scoring and greedy affine gap penalty calculations (Supplementary Material). Having done this, we showed that AGE is practical even when aligning long sequences of fosmid clones (Supplementary Material).
The input to the program is simple and consists of two FASTA files, e.g. reference sequence and assembled contig, specification of expected SV, e.g. option –inv for alignment with inversions, and optional specification of sequence subranges and scoring parameters. The output is comprehensive and includes breakpoint coordinates of unaligned/excised region(s) for the two best alignments, i.e. having the same maximum score (this can be used to evaluate alignment uniqueness), coordinates of aligned regions, i.e. regions flanking the SV from left and right, and lengths of identical sequence at breakpoints calculated in three different ways: around breakpoints, inside breakpoints and outside breakpoints (Supplementary Fig. S2). The reported numbers are followed by actual sequence alignment in blast-like format.
We have tested AGE to ensure that it reports accurate breakpoints. To do this, we constructed a total of 312 contigs, by merging the 500 bp flanking regions of large deletions (>1 kb), known with breakpoint resolution from sequencing and assembly by long Sanger reads (Levy et al., 2007). We then aligned these contigs to deletion regions extended by 1 kb. In 303 (97%) cases, SV breakpoints from optimal alignment(s) by AGE exactly matched those we started from. In 8 (2.5%) cases, contigs could be perfectly aligned (i.e. no mismatched and no gap) to the reference genome; thus, no breakpoints were reported by AGE. In one case, however, breakpoints were different. This may be due to the existence of more than two optimal solutions, with the solution containing actual deletion breakpoints simply not being reported. Indeed, when increasing contig length up to 5 kb we found exact breakpoints for all deletions. Thus, AGE is perfectly accurate in SV breakpoint determination.
3.2 Application to 1000 Genomes Project data
The AGE program has been used to align genomic contigs that were locally assembled by the 1000 Genome Project around predicted SV regions (mostly deletions), with the aim of confirming predictions and standardizing deletion breakpoints. Due to technical limitations, current local assembly was done for a fraction (∼50%) of predicted deletions and mostly for predictions made by read-pair and split-read approaches, with only a tiny fraction of contigs assembled for SV regions predicted by read-depth approaches. Thus, assembled regions are not representative of all predicted deletions but are still suited for demonstration purposes. The length of contigs ranged from 43 to 2472, with an average of 915 nt. A total of 38 226 contigs were aligned.
Below we will refer to predicted breakpoints, determined by a variety of approaches (read-pair, read-depth, etc.) as ‘predicted’ and to breakpoints inferred from contig alignment using AGE as ‘derived’. ‘Predicted’ breakpoints are typically a few dozen nucleotides away from the true ones. Of all SV predictions, 35 822 (94%) have regions between predicted and derived breakpoints overlapping by 80%, reciprocally. Moreover, for 36 290 (95%) predictions, we observed excellent (within 50 bases) agreement of breakpoints (Supplementary Fig. S3). The analyzed set of deletions was a union of predictions made using different data and approaches that have different breakpoint resolutions. Therefore, we use either criterion: 80% reciprocal overall or 50 bases difference at each breakpoint, to consider a prediction confirmed by assembly. A total of 36 986 (97%) deletions have been confirmed, ranging in size from 51 to 994 703 nt in length. For the confirmed deletions, we investigated sequence identity around breakpoints (see definition in Supplementary Fig. S2). We observed that 97% of sequences that are identical around breakpoints are shorter than 20 nt, but can be as long as 100 nt. Distribution of sequence identity around breakpoints has two distinct peaks (around 0 and 15 bp). The first one is likely to represent random sequence matches, while the second one is indicative of transposable elements (Lam et al., 2010), characterized by Target Site Duplication around breakpoints (∼15 nt in length). Identical sequences outside breakpoints (Supplementary Fig. S3) were almost all no longer than 30 nt and were typically shorter than 10 nt. Similarly, 98% of identical sequences inside breakpoints were shorter than 10 nt.
Most of the contigs could be aligned as pure deletions, but in 4340 (11%) cases, deletion was accompanied by micro-insertion (as on schematics in Fig. 1B). We further analyzed such cases (Supplementary Fig. S3). The distribution of insertion lengths peaks around 1 nt and decays exponentially. There is a slight elevation in the event frequency for insertions of length 12 and 15 nt. This could be further studied to determine whether this is a biological phenomenon. In one case, the inserted sequences was extremely long (1385 nt) and could be partially aligned to the opposite DNA strand in the region of deletion and to the genome in the region next to corresponding deletion. Thus, the region is a complex SV event that has a deletion, duplication and inversion. Another alternative is that the region was misassembled.
3.3 Examples
Application of other existing programs aimed at long gap alignment to the same set of contigs from the 1000 Genomes Project revealed a substantially higher SV confirmation rate when using AGE, thereby suggesting its superior performance in determining SV breakpoints (Supplementary Material). As it was pointed out in Section 1, SV breakpoints with sequences homology around them are the most challenging to determine (Fig. 2). For such SVs, successful resolution of breakpoints depends on the length and percent of homology of breakpoint sequences as well as the lengths of the aligned sequence. To demonstrate the advantage of using AGE in practice, we provide a few examples of alignments in such cases. We chose to demonstrate examples for AGE, CrossMatch (http://www.phrap.org/phredphrapconsed.html, implementing the Smith–Waterman alignment), GAP3 (Huang and Chao, 2003) (implementing a generalized global alignment with piecewise gap penalty), and Blat (Kent, 2002), a popular heuristic alignment program aimed at aligning highly similar sequences with large gaps.
Figure 5A shows an example of comparative contig-to-genome alignments made by these programs. The contig is the local assembly of the alternative (to the reference genome) haplotype around the region of predicted deletion chr20:2,969,769-2,970,056. AGE alignment clearly identifies a large unaligned region, confirms the predicted deletion and derives deletion breakpoints as chr20:2,969,756-2,970,052—in excellent agreement with the prediction. GAP3 was challenged by sequence homology around deletion breakpoints and did not introduce a large gap. Instead, it aligned the left flanking sequence with gaps and mismatches. CrossMatch aligned two regions, but contig sequence fragments in those two alignments overlap by 315 bases and additional analysis is required for breakpoints identification. Note that, as mentioned, post-processing of local overlapping alignment does not guarantee finding optimal alignment around breakpoints (see also Supplementary Fig. S4). Blat heuristically starts alignment from near-exact matches and, in fact, penalizes large gaps. Exact repeats within homologous regions flanking breakpoints misled the program into initiating alignment in the wrong regions and, as a result, produced incorrect alignment. Another example (Supplementary Fig. 5B) demonstrates the difficulty the GAP3 program has producing the correct alignment in cases when SV flanking sequences are not long enough. Note all alignment methods that utilize concave/piecewise gap penalty will face the same challenge.
Fig. 5.

Comparison of assembled contig alignments in the region of predicted deletions. The first line in each alignment is the sequence for the genomic region, while the second is for the contig sequence. Nucleotide numbering is sequential, starting from one in both compared sequences. Each alignment is accompanied by a schematic representation underneath. (A) The predicted deletion is chr20:2,969,769-2,970,056. The contig that is 614 bp in length has been aligned by the AGE, GAP3, CrossMatch and Blat programs to the predicted region of deletion, which is extended by 1 kb in each direction, i.e. from 2,968,769-2,971,056. The first sequence (genomic region) has two pairs of homologous sequences: orange to yellow and dark green to light green. AGE alignment clearly identifies a large unaligned region, confirms a predicted deletion, and derives deletion breakpoints as chr20:2,969,756-2,970,052 (coordinates are for the first and the last deleted bases). Note that the resulting breakpoints are in excellent agreement (within 13 bp) with the prediction. No other program was able to produce the correct alignment. (B) Predicted deletion is chr8:118,292,728-118,292,987. The contig of 530 bp in length has been aligned by the AGE and GAP3 programs to the predicted region of deletion, which is extended by 1 kb in each direction, i.e. from 118 291 728 to 118 293 987. AGE alignment clearly identifies a large unaligned region, confirms a predicted deletion, and derives deletion breakpoints as chr8:118,292,711-118,292,990 (coordinates are for first and last deleted bases). GAP3 is not able to align the left flanking sequence, as the penalty for a long gap outweighs the matches at the left flanking sequence. All coordinates are for human hg18 reference.
4 DISCUSSION
We have described an algorithm for the correct alignment of two nucleotide sequences containing SVs, i.e. deletion, insertion, tandem duplication or inversion, called AGE. The algorithm does not require the adjustment or modification of the alignment scoring scheme(s) that is usually tuned for a particular alignment purpose, e.g. cross-species, contig or read alignments. Thus, the algorithm can be universally applied in various biological studies relying on alignment. Its distinguishing feature is that it produces correct alignments in cases that are challenging for methods utilizing concave/piecewise gap penalty, i.e. cases with long sequence homology around breakpoints and/or a short SV region and/or short flanking sequences. The algorithm naturally handles certain cases of complex SV events, such as when deletion is accompanied by insertion.
The most straightforward application of AGE is single-nucleotide SV breakpoint resolution and standardization, as has just been demonstrated by using the algorithm implemented in AGE software. While the algorithm can be generalized to align sequences containing any number of SVs, its most practical (due to computational scalability) application is to align sequence with one SV, which are also the most common. Sequences containing more SVs are very rare, even when aligning long sequences of fosmid clones (Kidd et al., 2008). Still, AGE can also be useful in aligning such sequences. One may envision a strategy in which SV breakpoints are approximately localized (e.g. by analysis of local alignments) and then precisely identified using AGE to align only subsequences that flank SVs.
Of perhaps equal importance, the algorithm can be used to refine read alignment once a read has been heuristically mapped to a particular genomic location that is expected to contain an SV. Such read realignment has potential implications for genotyping known SVs in newly sequenced individuals, and/or discovering de novo SVs within loci that are known or expected to have a strong copy-number association with genetic diseases (McCarroll and Altshuler, 2007). Finally, the algorithm is not alphabet specific and can therefore be applied to the alignment of protein sequences.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Xinmeng Jasmine Mu and Declan Clarke for useful discussions, and Nicholas Carriero for help with algorithm implementation. We also thank the anonymous reviewers for valuable suggestions.
Funding: National Institutes of Health (NIH); the AL Williams Professorship funds; ‘Yale University Biomedical High Performance Computing Center’; its support team (in particular, Robert Bjornson and Nicholas Carriero); NIH (grant RR19895).
Conflict of Interest: None declared.
REFERENCES
- Abyzov A, et al. CNVnator: an approach to discover, genotype and characterize typical and atypical cnvs from family and population genome sequencing. Genome Res. 2011 doi: 10.1101/gr.114876.110. (submitted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao KM, et al. Recent developments in linear-space alignment methods: a survey. J. Comput. Biol. 1994;1:271–291. doi: 10.1089/cmb.1994.1.271. [DOI] [PubMed] [Google Scholar]
- Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh O. An improved algorithm for matching biological sequences. J. Mol. Biol. 1982;162:705–708. doi: 10.1016/0022-2836(82)90398-9. [DOI] [PubMed] [Google Scholar]
- Hirschberg DS. A linear space algorithm for computing maximal common subsequences. Commun. ACM. 1975;18:341–343. [Google Scholar]
- Huang X, Chao K-M. A generalized global alignment algorithm. Bioinformatics. 2003;19:228–233. doi: 10.1093/bioinformatics/19.2.228. [DOI] [PubMed] [Google Scholar]
- Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, et al. PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol. 2009;10:R23. doi: 10.1186/gb-2009-10-2-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam HY, et al. Nucleotide-resolution analysis of structural variants using BreakSeq and a breakpoint library. Nat. Biotechnol. 2010;28:47–55. doi: 10.1038/nbt.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA, Altshuler DM. Copy-number variation and association studies of human disease. Nat. Genet. 2007;39:S37–S42. doi: 10.1038/ng2080. [DOI] [PubMed] [Google Scholar]
- Medvedev P, et al. Computational methods for discovering structural variation with next-generation sequencing. Nat. Methods. 2009;6:S13–S20. doi: 10.1038/nmeth.1374. [DOI] [PubMed] [Google Scholar]
- Mills RE. Mapping structural variation at fine-scale by population genome sequencing. Nature. 2011 (in press) [Google Scholar]
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Schoniger M, Waterman MS. A local algorithm for DNA sequence alignment with inversions. Bull. Math. Biol. 1992;54:521–536. doi: 10.1007/BF02459633. [DOI] [PubMed] [Google Scholar]
- Smith TF, Waterman MS. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Wang LY, et al. MSB: a mean-shift-based approach for the analysis of structural variation in the genome. Genome Res. 2009;19:106–117. doi: 10.1101/gr.080069.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.