

Abstract
Motivation: In genome-wide association studies (GWAS) of complex diseases, genetic variants having real but weak associations often fail to be detected at the stringent genome-wide significance level. Pathway analysis, which tests disease association with combined association signals from a group of variants in the same pathway, has become increasingly popular. However, because of the complexities in genetic data and the large sample sizes in typical GWAS, pathway analysis remains to be challenging. We propose a new statistical model for pathway analysis of GWAS. This model includes a fixed effects component that models mean disease association for a group of genes, and a random effects component that models how each gene's association with disease varies about the gene group mean, thus belongs to the class of mixed effects models.
Results: The proposed model is computationally efficient and uses only summary statistics. In addition, it corrects for the presence of overlapping genes and linkage disequilibrium (LD). Via simulated and real GWAS data, we showed our model improved power over currently available pathway analysis methods while preserving type I error rate. Furthermore, using the WTCCC Type 1 Diabetes (T1D) dataset, we demonstrated mixed model analysis identified meaningful biological processes that agreed well with previous reports on T1D. Therefore, the proposed methodology provides an efficient statistical modeling framework for systems analysis of GWAS.
Availability: The software code for mixed models analysis is freely available at http://biostat.mc.vanderbilt.edu/LilyWang.
Contact: lily.wang@vanderbilt.edu; zhongming.zhao@vanderbilt.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Genome-wide association studies (GWAS) have become a main approach for identifying disease genes. These studies examine large numbers of genetic polymorphisms across the genome in hundreds or thousands of samples at a time. So far, GWAS have identified many genetic variants statistically associated with diseases or traits (Hindorff et al., 2009; Manolio et al., 2008). However, for complex diseases, typically individual genetic variants only have weak marginal effects on disease risk, even for real associations. Therefore, after multiple comparison adjustment for the large number of tests performed in GWAS, real but weak associations are likely to be missed by the conventional strategy of considering only the most significant hits (McCarthy et al., 2008).
To help increase power and to better understand the disease mechanisms underlying complex diseases, several recent studies (Chasman, 2008; Holmans et al., 2009; Torkamani et al., 2008; Wang et al., 2007) considered GWAS Pathway Analysis (GWASPA) (Cantor et al., 2010). These approaches test for effects of groups of genetic variants that belong to the same biological pathway such as those defined in kyoto encyclopedia of genes and genomes (KEGG; Ogata et al., 1999) or Gene Ontology (Ashburner et al., 2000) databases. The underlying assumption is that complex diseases are likely to be caused by changes in activities in biological pathways, where a number of mutations in different genes each contributes a modest amount to disease predisposition but works together to cause major disruption in normal biological processes. Higher power in pathway-based analysis is achieved by combining weak signals from a number of individual genetic variants in the pathway.
Recently, a number of GWASPA approaches (Chen et al., 2010a, b; De la Cruz et al., 2009; Ruano et al. 2010; Wu et al., 2010; Yu et al., 2009 ), such as the modified gene set enrichment analysis (GSEA) algorithm (Mootha et al., 2003; Subramanian et al., 2005; Wang et al., 2007) have been proposed. Typically, to preserve correlation patterns between genes, P-values are estimated by permuting sample labels. However, for a typical GWAS, the recalculation of test statistics for half a million or more single nucleotide polymorphism (SNPs) with hundreds or even thousands of samples for each permutation is extremely computationally intensive. In addition, a recent review (Cantor et al., 2010) comments: ‘the permutation studies that are currently being conducted require raw genotype data, which are not always available. An important methodological improvement would make the P-values alone, and not raw data, the basis of analysis in GWASPA’.
When raw genotype data are not available, a simple and popular approach for pathway analysis is an over-representation analysis based on Fisher's exact test. This method condenses test statistics for multiple SNPs within a gene into a single value (usually by choosing the most significant P-value), classifies each gene as significant or not based on a pre-specified significance threshold, and then compares the proportion of genes significantly associated with disease in a pathway versus the rest of the genome. The Fisher's exact test and its extensions have been implemented in a number of software programs such as the Ingenuity Pathway Analysis (IPA, http://www.ingenuity.com/), SNPtoGO (Schwarz et al., 2008), PRP (Chen et al., 2009), ALIGATOR (Holmans et al., 2009) and JMP Genomics (http://www.jmp.com/genomics). However, when the threshold for declaring significant genes changes, the results of pathway analysis based on Fisher's test can differ. In addition, because genes in the same pathway are expected to regulate or interact with each other, the assumption that these genes are independent may not be tenable.
A further difficulty for GWASPA is that it is not immediately clear what strategy should be used for the SNP information reduction for each gene. When each gene locus is represented by a single SNP, the potential effects of multiple association signals for the gene would be missed. Moreover, for overlapping genes, bias may be introduced when SNPs mapped to multiple genes are highly significant, because these SNPs would be included multiple times in the dataset and pathway significance may be driven by only a few of these SNPs.
In this article, to address these analytical challenges, we propose an efficient, threshold-free, hierarchical generalized linear mixed model (GLMM) for GWASPA. The proposed model has several desirable properties: (i) It is computationally efficient: only summary statistics are needed; raw genotype data, which may not be available due to confidentiality concerns (Homer et al., 2008), are not required. (ii) We model all the genes and SNPs within a pathway in a hierarchical fashion using random effects, which provide the ability to borrow information across genes in the same pathway. Our method corrects for the presence of overlapping genes and linkage disequilibrium (LD) between SNPs. (iii) The proposed model improves power over currently available pathway analysis methods while preserving type I error rate. (iv) In addition to identifying pathways associated with diseases, genes that contribute most to pathway association with disease can be identified using shrinkage estimates of best linear unbiased prediction (BLUP; McCulloch and Searle, 2001). (v) Covariate information, environmental effects and other complex design factors can also be accommodated using fixed and random effects in the mixed model.
In Section 2, we will discuss a few mixed models, including our proposals for modeling overlapping genes and LD patterns across genes and SNPs within a pathway. In Section 3, we will use both simulated and real GWAS dataset to show our proposed model has an increased power over currently available methods while preserving type I error rate for identifying disease associated pathways. Finally, using GWAS dataset from the WTCCC T1D study, we will show that mixed model analysis identifies meaningful biological processes and genes that agree well with previous reports on T1D.
2 METHODS
2.1 From SNPs to pathways
There are several pre-processing steps for pathway analysis of GWAS datasets:
Determine the pathway database to be used. For the analysis of the real GWAS dataset in Section 3.2, we used gene sets from the C2-CP collection and C5-BP collection of the Molecular Signature Database (MSigDB; Subramanian et al., 2005). The C2-CP gene sets are biological processes compiled by domain experts and the C5-BP gene sets are derived from controlled vocabulary of the Gene Ontology project (Ashburner et al., 2000).
Assign SNPs to genes. For the analysis in Section 3.2, we assigned SNPs to a gene if they were located within 5 kb upstream of the first exon or downstream of the last exon.
Assign genes to pathways. We linked genes to pathways based on HUGO Gene symbols. After this step, we obtained a dataset with SNP identifiers linked to genes in each pathway for subsequent statistical analysis.
2.2 General structure of the proposed mixed models
We propose a new method for GWASPA using a class of statistical models called mixed effects models. These models include a systematic or fixed effects component that models the mean disease association for a group of genes, and a random component that models how each gene's association with disease varies about the gene group mean, thus called the mixed effects model. In the analysis of gene expression data, mixed models have been successfully applied to both single gene and pathway analysis (Chu et al., 2002; Wang et al., 2008; Wolfinger et al., 2001). For single marker analysis in GWAS, mixed models have been successfully applied to account for population structure (Yu et al., 2006; Zhang et al., 2010) and to incorporate prior biological information (Chen and Witte, 2007; Conti and Witte, 2003).
More specifically, to assess pathway association with disease, we propose two mixed models (see details in Supplementary Fig. 1) with the following general structure: let yij be the chi-squarestatistic for SNP j on gene i, based on the Cochran–Armitage trend test for a single SNP, we assume yij follows a chi-squaredistribution with mean λij, denoted as yij ∼ χ2(λij). Assuming SNP j on gene i is not associated with disease, we have λij=1 or Log (λij)=0. Next, for each pathway, pooling all SNPs mapped to genes in the pathway, we formulate the following model:
where g is the number of genes in the pathway; si is the number of SNPs on gene i; β is a fixed (intercept) effect; and u1,…, ug ∼ N(0, G) are random gene effects and G is gene–gene covariance matrix.
Fig. 1.

Design matrix for model (A), for a hypothetical gene set with three genes and three SNPs: SNP A is on gene 1, SNP B is on genes 1 and 2, SNP C is on genes 2 and 3. The outcome variable for the mixed model is the chi-square statistic for Cochran–Armitage trend test for single SNP.
To test for pathway association with disease, we test the null hypothesis.
Because the random gene effects have mean 0, statistical significance (of departure from 0) for β would indicate an overall association with disease for pathway SNPs. Under H0, the test statistic 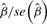 follows t distribution with n – rank[X Z] degrees of freedom (where n is the number of SNPs in the pathway and X and Z are design matrices for fixed and random effects, respectively). When the number of SNPs (n) is large, this is approximately the standard normal distribution; therefore, the null distribution for test statistics in mixed model does not depend on gene set size for pathways with a large number of SNPs.
follows t distribution with n – rank[X Z] degrees of freedom (where n is the number of SNPs in the pathway and X and Z are design matrices for fixed and random effects, respectively). When the number of SNPs (n) is large, this is approximately the standard normal distribution; therefore, the null distribution for test statistics in mixed model does not depend on gene set size for pathways with a large number of SNPs.
In the above model, the dependent variable yij is assumed to follow the chi-square distribution which belongs to the exponential family of distributions. Therefore, this model is a generalized linear model. Furthermore, since both fixed and random effects are included, it is a GLMM.
2.3 Detailed description of the mixed models
When devising the mixed models, we considered two important and challenging issues in pathway analysis: (i) accommodating overlapping genes, where a single SNP is mapped to more than one gene and (ii) accounting for the correlations between SNPs due to LD.
For the first mixed model (A), we assume the random effects u1,…, ug ∼ N(0, σ2I). Figure 1 shows a coding scheme for the independent variables, where the fixed effect β is an intercept and the random effects u1,…, ug are indicator variables for each gene. For issue (i), note that each SNP appears only once and thus contributes only once to the overall pathway association test statistic. To address issue (ii), using matrix algebra (McCulloch and Searle, 2001) it can be shown that for SNPs j and j′ on the same gene i, cov(uij,…, uij′) = σ2, j ≠ j′ so the random effects u1,…, ug account for a homogeneous covariance pattern between the SNPs in the same gene. To account for any additional variability due to unstructured covariance between SNPs on different genes, using the fact that chi-square distribution is a special case of gamma distribution, we increase the flexibility of the model by assuming yij ∼ gamma(λij, ϕ) where λ ij is the mean parameter and phi is the scale parameter. The inclusion of an additional scale parameter is a common mechanism to account for over-dispersion in generalized linear models (McCulloch and Searle, 2001).
Supplementary Figure 1 shows the details for these models. Model (B) is similar to Model (A), except that in Model (B), we propose modeling the covariance between genes in the gene set based on physical locations of the genes.
Let ui,…, ui′ be random gene effects for genes i and i′, respectively. The spatial model (B) assumes cov[ui,…, ui′] = σ2 e−dii′/α where the distance measure dii′ is calculated based on Euclidean distance between physical locations of genes i and i′ (averaged over all SNPs mapped to the gene). The parameters σ2 and α can be estimated automatically by maximizing the pseudo (restricted) maximum likelihood using PROC GLIMMIX of SAS software (Version 9.1, SAS Institute, Inc., Cary, NC, USA).
2.4 Empirical null distribution estimation
To further improve the accuracy of significance testing, rather than relying on asymptotic approximations, we pooled the estimated t-statistics corresponding to  for all gene sets, and estimated gene set P-values based on the empirical null distribution (Efron, 2010), which is a normal distribution with empirically estimated mean
for all gene sets, and estimated gene set P-values based on the empirical null distribution (Efron, 2010), which is a normal distribution with empirically estimated mean  and standard deviation
and standard deviation  . Efron (2008, 2010) showed that in large-scale simultaneous testing situations (e.g. when many gene sets are tested simultaneously in a study), serious defects in the theoretical null distribution may become obvious, while empirical Bayes methods can provide much more realistic null distributions. The empirical null distribution can be estimated using the locfdr package in R statistical software. Note that for each study, the empirical null distribution only needs to be estimated once, and can be accomplished in a few seconds, so this step adds little computational complexity to the proposed algorithm.
. Efron (2008, 2010) showed that in large-scale simultaneous testing situations (e.g. when many gene sets are tested simultaneously in a study), serious defects in the theoretical null distribution may become obvious, while empirical Bayes methods can provide much more realistic null distributions. The empirical null distribution can be estimated using the locfdr package in R statistical software. Note that for each study, the empirical null distribution only needs to be estimated once, and can be accomplished in a few seconds, so this step adds little computational complexity to the proposed algorithm.
In summary, we followed the following five steps for significance testing of gene sets:
For each gene set, fit an appropriate mixed model and obtain its t-statistic corresponding to
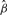 .
.Convert the t-statistic to a z-score. For example, let ti be the t-statistic for gene set i, the corresponding z-score can be obtained by zi = Φ−1(Fd(ti)) where Φ and Fd are distribution functions for standard normal distribution and t distribution with d degrees of freedom.
Pool the z-scores for all gene sets tested in a study and calculate their median value (m). Subtract m from the z-scores so that they have median of 0.
Given the median-centered z-scores, use the locfdr package, estimate location (δ) and scale (σ) parameters of the empirical null distribution.
Calculate standardized z-scores,
 , and compute the P-value for each gene set as pi=1−Φ(si).
, and compute the P-value for each gene set as pi=1−Φ(si).
2.5 Ranking of individual gene contribution to the gene set signal
As gene sets are defined based on existing knowledge in biological processes without considerations for any specific disease, typically only a subset of the genes in the pathway have genetic variations associated with disease susceptibility. Therefore, for significant gene sets, it is helpful to identify the subset of genes contributing to the gene set significance. Toward this end, we define the influential subset of genes, which are those genes contributing most to the gene set signal, to be those genes with estimated mean 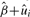 higher than the estimated overall mean
higher than the estimated overall mean 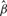 for the gene set. Recall in the general mixed model (Section 2.2), ui models the deviation of each gene's average chi-square statistic (on a log scale) from the gene set mean β. In other words, the influential subset includes all genes with
for the gene set. Recall in the general mixed model (Section 2.2), ui models the deviation of each gene's average chi-square statistic (on a log scale) from the gene set mean β. In other words, the influential subset includes all genes with  or equivalently
or equivalently  . Furthermore, we can rank these influential genes by their estimated individual gene estimate
. Furthermore, we can rank these influential genes by their estimated individual gene estimate 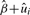 . Under the mixed model framework, these estimates are called empirical BLUP; they are shrinkage estimates that borrow information across all genes in the gene set and naturally fall into the hierarchical empirical Bayes framework. We illustrate ranking and selection of the influential genes in the pathways using a GWAS dataset in Section 3.2.
. Under the mixed model framework, these estimates are called empirical BLUP; they are shrinkage estimates that borrow information across all genes in the gene set and naturally fall into the hierarchical empirical Bayes framework. We illustrate ranking and selection of the influential genes in the pathways using a GWAS dataset in Section 3.2.
2.6 Design of a simulation experiment
To study the properties of the proposed mixed models, we simulated both null gene sets, for which the disease status of the samples were generated randomly, and causal gene sets, for which the disease status were generated based on a genetic model.
For the null gene sets, we used a genotype dataset of the genetic association information network (GAIN) GWAS for schizophrenia (Group et al., 2007). The details of data preparation were provided in our previous work (Jia et al., 2010). It includes 1158 schizophrenia cases and 1378 normal controls of European ancestry. After quality control and mapping SNPs to the pathways in the C2-CP collection of MSigDB (http://www.broadinstitute.org/gsea/msigdb), we obtained 596 gene sets with size ranging from 3 to 200 genes. For each pathway, we generated random disease status for the samples from a Bernoulli distribution (with parameter P = 0.5), so that by the experiment's design, SNPs in these pathways were not associated with the disease. This process was repeated twice, so that we had two sets of random outcomes for each pathway for a total of 1192 (596 × 2) null gene sets.
Next, for the causal gene sets, we generated genotype data for 12 000 samples representing an entire population of patient samples, and for each causal gene set, we sampled 500 cases and 500 control samples based on disease prevalence of 0.05. Since genotype data for many more samples were needed, instead of using an existing GWA dataset, we used the HAP-SAMPLE software (Wright et al., 2007) to simulate a genotype dataset with realistic LD patterns. HAP-SAMPLE simulates genotype datasets by re-sampling chromosome-length haplotypes from existing phased datasets, such as the HapMap dataset, thus preserving realistic genetic data structure. Given simulated genotype data, the samples' disease statuses were then simulated based on a genetic model with various parameters modeling the strengths of associations between pathway SNPs and the disease. More specifically, we followed these steps to generate the causal gene sets:
Simulate genotype data: the median number of genes for pathways in the C2-CP collection of MSigDB is 23. Therefore, for this simulation study, we selected the ATM pathway with 23 genes. The SNP IDs of the 262 SNPs (corresponding to the 23 genes in the ATM pathway) were entered into HAP-SAMPLE and the Caucasian cohort (CEU) from HapMap Phase II Project was used as the source data. We next used HAP-SAMPLE to generate genotype data for a total of 12 000 samples, representing the entire population of samples.
- Simulate disease status for the samples: after fixing the genotype data, we next simulated case–control status for each sample based on the following genetic model:
where gi = 0, 1, 2 represents the number of copies of the minor allele for SNP i(i = 1,…, D) and f = Probability (affected | g1,…, gD) is the penetrance for genotype {g1,…, gD}.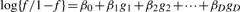
The number of causal SNPs (D) associated with disease in the pathway were set at an average of t = 0.5, 1 and 1.5 per gene, so that D = t × ngene. The first D SNPs with the lowest RSID numbers (dbSNP assigned reference SNP ID), which mapped to several different genes, were selected to be the causal SNPs. We generated βi(i = 1, D) from N(μ, τ2) where the mean log odds ratio μ = log(1.1) and τ2 = 0.3 and 0.5. Under this setup, βi can be positive or negative, therefore each simulated pathway included causal SNPs with minor alleles increasing or decreasing risk of disease.
Next, given disease prevalence K(=0.05) and βi(i = 1,…, D), β0 were estimated by maximizing the equation
Finally, given values for β0, {β1,…, βD}, and genotype data {g1,…, gD}, we computed f and generated disease status for each sample using the genetic model above. From the pool of 12 000 samples [step (1) in this section], the genotype data for the first 500 cases and 500 controls were then selected.
In Table 2, for each simulation dataset, we included 100 causal gene sets [generated by repeating step (2) in this section 100 times] and the same set of 1192 null gene sets (generated by adding random sample labels to gene sets based on GAIN schizophrenia genotype data). The type I error rate and power were estimated by the proportion of mixed model with P < 0.05 for the null gene sets or the causal gene sets, respectively.
Table 2.
Power comparisons (α = 0.05)
| Dataset | OR | τ2 | t | Mixed model |
Fisher's exact test |
|||
|---|---|---|---|---|---|---|---|---|
| A | B | Fisher0_01 | Fisher0_05 | Fisher0_1 | ||||
| 1 | 1.1 | 0.3 | 0.5 | 0.62 | 0.75 | 0.41 | 0.12 | 0.12 |
| 2 | 1.1 | 0.3 | 1.0 | 0.88 | 0.90 | 0.86 | 0.29 | 0.07 |
| 3 | 1.1 | 0.3 | 1.5 | 1.00 | 0.99 | 0.98 | 0.70 | 0.27 |
| 4 | 1.1 | 0.5 | 0.5 | 0.66 | 0.80 | 0.50 | 0.20 | 0.09 |
| 5 | 1.1 | 0.5 | 1.0 | 0.94 | 0.95 | 0.92 | 0.38 | 0.13 |
| 6 | 1.1 | 0.5 | 1.5 | 1.00 | 1.00 | 0.98 | 0.78 | 0.32 |
For Fisher's test, a Cochran-Armitage Trend test P-value was computed for each SNP, and the most significant SNP was selected to represent each gene. To classify each gene as significant or not, we used P-value cutoffs of 0.01 (Fisher0_01), 0.05 (Fisher0_05) and 0.1 (Fisher0_1).
3 RESULTS
3.1 Results of simulation study
First, we assessed the accuracy of the estimated empirical null distributions for each mixed model. In Table 1, each simulation dataset consisted of 100 causal gene sets and 1192 null gene sets. For the mixed models, given the t-statistics corresponding to 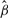 from models (A) and (B), as discussed in Section 2.4 [following the Steps (2)–(4)], we estimated the empirical null distributions using the locfdr package in R software. Since the same 1192 null gene sets were included in each simulation dataset, the estimated parameters
from models (A) and (B), as discussed in Section 2.4 [following the Steps (2)–(4)], we estimated the empirical null distributions using the locfdr package in R software. Since the same 1192 null gene sets were included in each simulation dataset, the estimated parameters 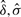 and
and 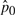 for the empirical null distribution are expected to be the same across all datasets. Among all models, the parameter estimates were most consistent for model (B), indicating the estimated empirical null distribution was most accurate for this model.
for the empirical null distribution are expected to be the same across all datasets. Among all models, the parameter estimates were most consistent for model (B), indicating the estimated empirical null distribution was most accurate for this model.
Table 1.
Type I error rate for mixed models (α = 0.05)
| Dataset | OR | τ2 | t | Mixed model |  |
 |
 |
Type I error |
|---|---|---|---|---|---|---|---|---|
| 1 | 1.1 | 0.3 | 0.5 | A | −0.01 | 1.31 | 0.98 | 0.0344 |
| 2 | 1.1 | 0.3 | 1 | A | −0.07 | 1.27 | 0.95 | 0.0453 |
| 3 | 1.1 | 0.3 | 1.5 | A | −0.16 | 1.14 | 0.90 | 0.0646 |
| 4 | 1.1 | 0.5 | 0.5 | A | −0.01 | 1.33 | 0.98 | 0.0344 |
| 5 | 1.1 | 0.5 | 1 | A | −0.11 | 1.21 | 0.93 | 0.0545 |
| 6 | 1.1 | 0.5 | 1.5 | A | −0.16 | 1.13 | 0.89 | 0.0654 |
| 1 | 1.1 | 0.3 | 0.5 | B | −0.11 | 1.54 | 0.94 | 0.0403 |
| 2 | 1.1 | 0.3 | 1 | B | −0.15 | 1.53 | 0.92 | 0.0403 |
| 3 | 1.1 | 0.3 | 1.5 | B | −0.19 | 1.49 | 0.91 | 0.0487 |
| 4 | 1.1 | 0.5 | 0.5 | B | −0.13 | 1.53 | 0.93 | 0.0403 |
| 5 | 1.1 | 0.5 | 1 | B | −0.16 | 1.50 | 0.91 | 0.0461 |
| 6 | 1.1 | 0.5 | 1.5 | B | −0.19 | 1.49 | 0.91 | 0.0495 |
OR, mean odds ratio for causal SNPs in each causal gene set; τ2, variance of causal SNPs odds ratios; t, average number of causal SNPs per gene in each causal gene set; δ (location parameter), σ (scale parameter) and p0 (proportion of null test scores in the dataset) are parameters for the empirical null distribution and were estimated by the locfdr package.
Next, for each simulation dataset, we estimated the type I error rate for each model. Under the null hypothesis, we expect the P-values to follow a uniform distribution, so a model with type I error rate equal to or less than the significance level of 0.05 is desirable. Among all models, again, Model (B) had type I error rate closest to the expected error rate of 0.05. Model (A) also had reasonable type I error rate.
Finally, we assessed the power of the proposed models. Table 2 shows the estimated power (based on 100 causal gene sets in each simulation dataset) for the mixed models and Fisher's exact tests. For Fisher's test, a Cochran–Armitage trend test P-value was computed for each SNP, and the most significant SNP was selected to represent each gene. To classify each gene as significant or not, we used P-value cutoffs of 0.01, 0.05 and 0.1 for Fisher0_01, Fisher0_05 and Fisher0_1 in Table 2, respectively. When the number of causal SNPs in the gene set was moderate (t = 1, where t is the average number of causal SNPs per gene = 1) or high (t = 1.5), all models except Fisher0_05 and Fisher0_1 performed well. When the number of causal SNPs in the gene set was low (t = 0.5), model (B) performed best, followed by model (A). As expected, the results of Fisher's exact test varied when different thresholds were used to classify each gene as significant or not, with Fisher0_01 and Fisher0_1 having the most and least power, respectively.
Note that in these simulation datasets, disease outcome for each sample was generated randomly or based on a genetic model for the null and causal gene sets, respectively, it was difficult to evaluate performance of permutation-based pathway analysis methods such as GSEA, where only a single set of disease outcomes is allowed for all genesets in a dataset. Therefore, we next analyzed the GAIN schizophrenia dataset with real disease outcomes, to compare the power of mixed model (B) with GSEA, permutation-based Fisher's exact test and one other recently proposed method—ALIGATOR (Holmans et al., 2009). Previously, we analyzed this dataset using GSEA (Wang et al., 2007) and permutation-based Fisher's exact test (or hypergeometric test) for a set of 511 curated pathways (Jia et al., 2010). Using the exact same dataset, we next performed analysis with mixed model (B). Figure 2 shows that model (B) identified more significant gene sets than the other methods. For example, the number of significant gene sets with P < 0.05 identified by mixed models, Fisher's exact test, GSEA and ALIGATOR were 26, 10, 5 and 8, respectively. Note that these comparisons based on real disease outcomes are meaningful since as shown in Table 1, type I error rate for mixed model (B) was preserved for the same GAIN genotype dataset with random outcomes. Therefore, model (B) improved power over currently available GWASPA methods, while controlling false positive rate.
Fig. 2.

Number of significant gene sets. Mixed model = model (B) described in Section 2.3, Fisher's exact test = hypergeometric test in Jia et al. (2010). P-value of 0.01 was used to classify each gene as significant or not in both Fisher's exact test and ALIGATOR.
3.2 Application to Wellcome Trust Case Control Consortium T1D GWAS dataset
To further validate the proposed methodology, we next applied mixed model (B), which performed best in the simulation study, to the GWAS dataset from the Wellcome Trust Case Control Consortium (WTCCC) Type 1 diabetes study. Similar to the analysis of GAIN dataset, we collected and tested gene sets defined by the canonical pathway (C2-CP) and biological process (C5-BP) collections of the MSigDB. We replaced the KEGG gene sets in C2-CP with the May 2010 version of KEGG gene sets. To reduce the amount of multiple testing and to avoid testing gene sets for which biological annotations are too broad, we used gene sets with size from 3 to 200 genes. This resulted in a total of 1273 gene sets for our testing. For each pathway, in order to assign SNPs to genes, we followed the procedures outlined in Section 2.1. On a Linux computer with a 3.00 GHz processor and 8 GB memory, computing time was 4 h and 33 min for this mixed model analysis of 1273 gene sets. This indicates that mixed model (B) can be run efficiently for large GWAS dataset.
Table 3 and Supplementary Table 1 show the results based on model (B). After correcting for multiple comparisons, model (B) identified 16 gene sets that are significantly associated (false discovery rate <0.2) with T1D. These pathways included antigen processing and presentation, interleukin-1 secretion and T-cell signal transduction and duplicate longstanding associations between these biological processes and T1D. Antigen processing and presentation was demonstrated to be associated with T1D more than 35 years ago (Nerup et al., 1974) and our findings of an association between SNPs in HLA-C, HLA-C and HLA-G of the major histocompatibility region and T1D indicate that the proposed mixed model could effectively detect biologically significant results (WTCCC, 2007). The identification of interleukin-1 pathway is another important replication because IL-1 was one of the first cytokines studied and has been shown to cause toxicity to islet cells, impairing their ability to produce insulin (Zawalich and Diaz, 1986). Finally, association of SNPs in genes involved in T-cell signal transduction mimics knowledge that lymphocytes are the effector cells of T1D, shown just years after the major histocompatibility complex (MHC) discovery when T1D was passively transferred from man to mouse through a lymphocyte transfusion (Buschard et al., 1978).
Table 3.
Results of mixed model analysis of the WTCCC T1D dataset
| Collection | ID | Pathway | Ngenes | Nsnps | Raw_P | FDR_P |
|---|---|---|---|---|---|---|
| C2CP | HSA04940 | Genes involved in type I diabetes mellitus | 37 | 507 | < 1 × 10−15 | < 1 × 10−15 |
| C2CP | HSA05322 | Systemic lupus erythematosus | 70 | 748 | < 1 × 10−15 | < 1 × 10−15 |
| C2CP | HSA05320 | Autoimmune thyroid disease | 43 | 432 | 1.55 × 10−13 | 5.96 × 10−11 |
| C2CP | HSA05330 | Allograft rejection | 31 | 255 | 2.01 × 10−12 | 5.80 × 10−10 |
| C2CP | HSA04612 | Genes involved in antigen processing and presentation | 63 | 359 | 3.10 × 10−9 | 5.97 × 10−7 |
| C2CP | HSA04630 | Genes involved in Jak-STAT signaling pathway | 131 | 1156 | 1.07 × 10−6 | 0.0002 |
| C5BP | GO:0007498 | Mesoderm development | 16 | 246 | 2.26 × 10−6 | 0.0003 |
| C5BP | GO:0009410 | Response to xenobiotic | 10 | 124 | 0.000165 | 0.0191 |
| C5BP | GO:0006805 | Xenobiotic metabolic process | 9 | 123 | 0.000161 | 0.0191 |
| C5BP | GO:0050708 | Regulation of protein secretion | 16 | 297 | 0.000231 | 0.0243 |
| C5BP | GO:0006626 | Protein targeting to mitochondrion | 10 | 81 | 0.000815 | 0.0786 |
| C2CP | HSA00790 | Genes involved in folate biosynthesis | 10 | 54 | 0.000972 | 0.0865 |
| C5BP | GO:0000080 | G1 phase of mitotic cell cycle | 11 | 71 | 0.001344 | 0.1111 |
| C5BP | GO:0006665 | Sphingolipid metabolic process | 26 | 485 | 0.001491 | 0.1150 |
| C5BP | GO:0050701 | Interleukin-1 secretion | 8 | 203 | 0.002051 | 0.1460 |
| C2CP | T-cell signal transduction | 40 | 493 | 0.002460 | 0.1581 |
Collection, the collection of gene sets in the MSigDB database; Ngenes, number of genes; Nsnps, number of SNPs; Raw_P, estimated unadjusted P-value; FDR_P, estimated false discovery rate.
Additionally, the recent study by Eleftherohorinou et al. (2009) also found that SNPs located in genes in the Jak-STAT signaling pathway were associated with T1D, a finding duplicated by this method. In addition, SNPs located in or near the genes CTLA4, IL2 and IL2RA have been repeatedly shown to be associated with T1D and were also found significant in this analysis (Barrett et al., 2009; Consortium, 2007). CTLA4 is a member of the immunoglobulin superfamily that sends an inhibitory signal to T cells, while IL2 and IL2RA may both be involved in the decreased synthesis of IL-2 in lymphocytes from patients with T1D (Zier et al., 1984).
4 DISCUSSION
In summary, we have described several GLMMs for pathway-based analysis of GWAS. This flexible, unified and practical approach can be implemented in common statistical packages. Our method makes several improvements over currently available algorithms for pathway testing.
First, the proposed methodology represents our attempts at rigorous statistical modeling of disease association with biological pathways in GWAS without resorting to individual-level genotype data, which are often unavailable to non-owner investigators. In particular, this approach should make large-scale meta-analysis much more convenient and practical, since currently meta-analysis often requires numerous multi-institution effort to share and coordinate genotyping data. Even when raw genotype data are available, the re-calculation of test statistics for thousands of samples in GWAS for each permutation can be computationally demanding. In contrast, maximum likelihood estimation and testing for the proposed mixed model using PROC GLIMMIX in SAS software for a typical gene set can be achieved in seconds.
Second, in our proposed mixed model, all the genes in a pathway and all the variants across each gene locus were carefully modeled in a hierarchical fashion. In addition, the correlations between SNPs (due to LD) were modeled based on the spatial distances between genes. Furthermore, for SNPs mapped to overlapping genes in the same pathway, the proposed mixed model include each SNP once and only once. In contrast, methods that select a most significant SNP to represent each gene may have difficulties, because significant SNPs mapped to multiple genes would be included multiple times and pathway significance may be driven by only a few of these SNPs. This kind of false discoveries has been frequently encountered in our analysis. For example, in the GAIN schizophrenia dataset, the ‘starch and sucrose metabolism pathway (HSA00500)’ included several genes located closely on the chromosome (e.g. UGT1A1, UGT1A3, UGT1A4, UGT1A5, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10 and others). When the most significant SNP was used to represent each gene, most of the genes in the cluster were represented by the same SNP. Therefore, if this SNP has a small P-value, the pathway would very likely be identified as a significant pathway, while in fact, the result was driven by one highly significant SNP located on multiple genes.
Third, using real and simulated GWAS dataset with realistic LD patterns, we have shown that the proposed mixed model improved power over currently available methods while preserving type I error rate. Unlike traditional pathway analysis method such as Fisher's exact test where each gene is classified as significant or not based on an arbitrary significance threshold, the proposed model uses continuous information in P-values to help improve power.
Fourth, the proposed model represents a flexible methodology that operates within a well-established statistical framework. In addition to identifying pathways associated with disease, as explained in Section 2.5, this model can also identify genes contributing most to the pathway association. Moreover, it can be easily extended to handle more complex designs with multiple sources of variations such as covariate information and environmental effects, when it is difficult to justify the exchangeability assumption behind permutation tests.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank the Wellcome Trust Case Control Consortium and the Genetic Association Information Network for sharing their data and Dr Chun Li for critical reading and discussions of the manuscript.
Funding: National Institutes of Health (5P30 HD015052-25 and 1P50 MH078028-01A1 to L.W.; RO1 AI44924, R42 DK065388 and R42 AI053984 to T.M.A.; R21AA017437, P50CA95103 and P30CA68485 to Z.Z.; 5P30CA068485-13 to X.C.; TL1 RR024978-03 and T32 GM07347 to B.L.G., partially) and a 2009 NARSAD Maltz Investigator Award to Z.Z.
Conflict of Interest: none declared.
REFERENCES
- Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschard K, et al. Passive transfer of diabetes mellitus from man to mouse. Lancet. 1978;1:908–910. doi: 10.1016/s0140-6736(78)90684-0. [DOI] [PubMed] [Google Scholar]
- Cantor RM, et al. Prioritizing GWAS results: a review of statistical methods and recommendations for their application. Am. J. Hum. Genet. 2010;86:6–22. doi: 10.1016/j.ajhg.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chasman DI. On the utility of gene set methods in genomewide association studies of quantitative traits. Genet. Epidemiol. 2008;32:658–668. doi: 10.1002/gepi.20334. [DOI] [PubMed] [Google Scholar]
- Chen GK, Witte JS. Enriching the analysis of genomewide association studies with hierarchical modeling. Am. J. Hum. Genet. 2007;81:397–404. doi: 10.1086/519794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, et al. Prioritizing risk pathways: a novel association approach to searching for disease pathways fusing SNPs and pathways. Bioinformatics. 2009;25:237–242. doi: 10.1093/bioinformatics/btn613. [DOI] [PubMed] [Google Scholar]
- Chen LS, et al. Insights into colon cancer etiology via a regularized approach to gene set analysis of GWAS Data. Am. J. Hum. Genet. 2010a;86:960–871. doi: 10.1016/j.ajhg.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, et al. Pathway-based analysis for genome-wide association studies using supervised principal components. Genet. Epidemiol. 2010b;34:716–724. doi: 10.1002/gepi.20532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu TM, et al. A systematic statistical linear modeling approach to oligonucleotide array experiments. Math. Biosci. 2002;176:35–51. doi: 10.1016/s0025-5564(01)00107-9. [DOI] [PubMed] [Google Scholar]
- Conti DV, Witte JS. Hierarchical modeling of linkage disequilibrium: genetic structure and spatial relations. Am. J. Hum. Genet. 2003;72:351–363. doi: 10.1086/346117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Cruz O, et al. Gene, region and pathway level analyses in whole-genome studies. Genet. Epidemiol. 2009;34:222–231. doi: 10.1002/gepi.20452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B. Microarrays, empirical Bayes, and the two-groups model. Statist. Sci. 2008;23:1–47. [Google Scholar]
- Efron B. Correlated z-values and the accuracy of large-scale statistical estimates. J. Am. Statist. Assoc. 2010;105:1042–1055. doi: 10.1198/jasa.2010.tm09129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eleftherohorinou H, et al. Pathway analysis of GWAS provides new insights into genetic susceptibility to 3 inflammatory diseases. PLoS One. 2009;4:e8068. doi: 10.1371/journal.pone.0008068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl Acad. Sci. USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmans P, et al. Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am. J. Hum. Genet. 2009;85:13–24. doi: 10.1016/j.ajhg.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homer N, et al. Resolving individuals contributing trace amounts of DNA to highly complex mixtures using high-density SNP genotyping microarrays. PLoS Genet. 2008;4:e1000167. doi: 10.1371/journal.pgen.1000167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia P, et al. Common variants conferring risk of schizophrenia: a pathway analysis of GWAS data. Schizophr. Res. 2010;122:38–42. doi: 10.1016/j.schres.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, et al. New models of collaboration in genome-wide association studies: the Genetic Association Information Network. Nat. Genet. 2007;39:1045–1051. doi: 10.1038/ng2127. [DOI] [PubMed] [Google Scholar]
- Manolio TA, et al. A HapMap harvest of insights into the genetics of common disease. J. Clin. Invest. 2008;118:1590–1605. doi: 10.1172/JCI34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MI, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat. Rev. Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- McCulloch CE, Searle SR. Generalized, Linear and Mixed Models. New York, NY: John Wiley & Sons, Inc.; 2001. [Google Scholar]
- Mootha VK, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Nerup J, et al. HL-A antigens and diabetes mellitus. Lancet. 1974;2:864–866. doi: 10.1016/s0140-6736(74)91201-x. [DOI] [PubMed] [Google Scholar]
- Ogata H, et al. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruano D, et al. Functional gene group analysis reveals a role of synaptic heterotrimeric G proteins in cognitive ability. Am. J. Hum. Genet. 2010;86:113–125. doi: 10.1016/j.ajhg.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz DF, et al. SNPtoGO: characterizing SNPs by enriched GO terms. Bioinformatics. 2008;24:146–148. doi: 10.1093/bioinformatics/btm551. [DOI] [PubMed] [Google Scholar]
- Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Wellcome Trust Case Control Consortium. Genome-wide association study of 14 000 cases of seven common diseases and 3000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torkamani A, et al. Pathway analysis of seven common diseases assessed by genome-wide association. Genomics. 2008;92:265–272. doi: 10.1016/j.ygeno.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, et al. Pathway-Based approaches for analysis of genomewide association studies. Am. J. Hum. Genet. 2007;81:1278–1283. doi: 10.1086/522374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, et al. An integrated approach for the analysis of biological pathways using mixed models. PLoS Genet. 2008;4:e1000115. doi: 10.1371/journal.pgen.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfinger RD, et al. Assessing gene significance from cDNA microarray expression data via mixed models. J. Comput. Biol. 2001;8:625–637. doi: 10.1089/106652701753307520. [DOI] [PubMed] [Google Scholar]
- Wright FA, et al. Simulating association studies: a data-based resampling method for candidate regions or whole genome scans. Bioinformatics. 2007;23:2581–2588. doi: 10.1093/bioinformatics/btm386. [DOI] [PubMed] [Google Scholar]
- Wu MC, et al. Powerful SNP-Set Analysis for Case-Control Genome-wide Association Studies. Am. J. Hum. Genet. 2010;86:929–942. doi: 10.1016/j.ajhg.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006;38:203–208. doi: 10.1038/ng1702. [DOI] [PubMed] [Google Scholar]
- Yu K, et al. Pathway analysis by adaptive combination of P-values. Genet. Epidemiol. 2009;33:700–709. doi: 10.1002/gepi.20422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawalich WS, Diaz VA. Interleukin 1 inhibits insulin secretion from isolated perifused rat islets. Diabetes. 1986;35:1119–1123. doi: 10.2337/diab.35.10.1119. [DOI] [PubMed] [Google Scholar]
- Zhang Z, et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 2010;42:355–360. doi: 10.1038/ng.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zier KS, et al. Decreased synthesis of interleukin-2 (IL-2) in insulin-dependent diabetes mellitus. Diabetes. 1984;33:552–555. doi: 10.2337/diab.33.6.552. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.