

Abstract
BACKGROUND AND PURPOSE
The binding of transmitter to specialized binding pockets leads to rearrangements in the structure of the receptor eventually resulting in channel opening. We used voltage-clamp fluorometry to investigate the pharmacological basis and biophysical processes that underlie structural changes at the transmitter binding site of the rat α1β2γ2L GABAA receptor.
EXPERIMENTAL APPROACH
Simultaneous electrophysiological and site-specific fluorescence measurements were conducted on receptors expressed in Xenopus oocytes and labelled with an environmentally-sensitive fluorophore, Alexa 546 maleimide, at the α1L127C site.
KEY RESULTS
Receptors activated by GABA demonstrate a concentration-dependent increase in fluorescence intensity, indicating that the environment surrounding the fluorophore becomes less polar upon activation. Qualitatively similar responses were observed with other GABA site ligands such as piperidine-4-sulphonic acid, muscimol, β-alanine and 4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol. Fluorescence changes were not affected by the direction of current flow. During long applications of GABA significant desensitization developed, which was not accompanied by additional changes in fluorescence. Pentobarbital was an efficacious agonist of the labelled mutant receptor but did not cause changes in fluorescence. Direct activation by etomidate or the steroid allopregnanolone also did not result in fluorescence changes. Functional potentiation of GABA-activated receptors by allopregnanolone or etomidate enhanced both the GABA-elicited functional response and the fluorescence change. In contrast, potentiation by pentobarbital was not accompanied by an enhanced fluorescence response.
CONCLUSIONS AND IMPLICATIONS
The data indicate that there is no direct correlation between current flow or position of the activation gate and the structural changes as detected by Alexa 546-labelled α1L127Cβ2γ2L GABAA receptors. Channel potentiation by pentobarbital qualitatively differs from potentiation by etomidate or allopregnanolone.
Keywords: GABAA receptor, modulation, fluorescence
Introduction
The GABAA receptor consists of five homologous subunits, each of which contains a large aminoterminal extracellular domain followed by four membrane-spanning regions and a short carboxyterminal tail. The two transmitter binding sites are located in the extracellular region, at the β-α subunit interfaces (for review, please see Kash et al., 2004). The occupation of the transmitter binding sites with GABA (or a number of structurally related compounds) leads to changes in receptor structure eventually resulting in the opening of the channel gate in the transmembrane region of the receptor. The GABAA receptor can also be directly activated by barbiturates (e.g. pentobarbital), neuroactive steroids (e.g. allopregnanolone) and general anaesthetics (e.g. etomidate), which interact with distinct sites that do not overlap with the transmitter binding site. Furthermore, barbiturates, etomidate and neuroactive steroids can also potentiate currents elicited by low concentrations of GABA.
The structural changes that arise from the occupation of the transmitter binding sites, and how the initial rearrangements in the extracellular domain lead to channel opening have been of great interest to channel biophysicists. We have used site-specific fluorescence, combined with simultaneous electrophysiological recordings, to detect structural changes at the transmitter binding site. In this approach, a residue in the region of interest is labelled with an environmentally sensitive fluorescent reporter. The reporter responds to changes in the environment, such as those happening following activation- and modulation-related conformational changes, with a change in quantum yield, which is observed and recorded as a change in the intensity of the fluorescence signal (Mannuzzu et al., 1996).
Here, we labelled ternary α1β2γ2L receptors at the α1L127C residue with Alexa Fluor 546 C5 maleimide, and probed for structural changes surrounding the labelled residue following channel activation and modulation by a wide range of neuroactive compounds. The overall goal of the study was to determine whether the structural changes at the α1L127C site are related to movement of the channel gate.
The α1L127 site is within the transmitter binding pocket, contributed by loop E of the ‘−’ interface (Figure 1). Previous work has shown that a reporter attached to this residue responds with changes in fluorescence intensity following channel activation by GABA but not pentobarbital (Muroi et al., 2006; 2009;). We have extended the previous work by probing for fluorescence changes (ΔF) in response to exposure to a variety of GABA site ligands, the actions of which range from competitive antagonism (gabazine) to full agonism (GABA, β-alanine). Furthermore, we examined direct activation and modulation of GABA-activated receptors by pentobarbital, etomidate and the neurosteroid allopregnanolone. The main conclusion of the study is that conformational changes at the α1L127C site do not directly reflect channel activation, but are more likely associated with local conformational changes associated with occupation of the transmitter binding site.
Figure 1.
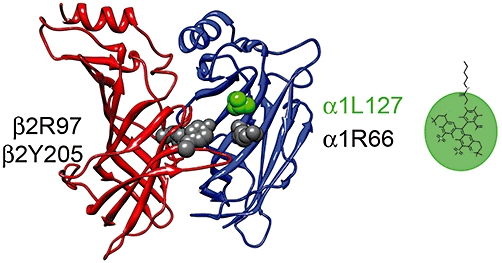
The transmitter binding site of the GABAA receptor. The site is formed at the interface between the α (blue) and β subunits (red). The β subunit contributes the so-called ‘+’ interface (with loops A, B and C), and the α subunit contributes the ‘−’ interface (loops D, E and F). The figure shows the putative transmitter binding pocket lined with the β2Y205 (Amin and Weiss, 1993), β2Y97 (Padgett et al., 2007), α1R66 (Holden and Czajkowski, 2002) and α1L127 (Muroi et al., 2006) residues. In the present work, the receptors were labelled with Alexa 546 maleimide at the α1L127C site (loop E), and electrophysiological and fluorescence measurements conducted in the presence of various agonists and modulators. The relative size of the linker and fluorophore are shown with a line and oval respectively. The length of the five-ring system is approximately 12 Å (Chem 3D, CambridgeSoft).
Methods
The site-specific fluorescence experiments were conducted on wild-type and mutant rat α1β2γ2L GABAA receptors expressed in Xenopus oocytes. The cDNAs for the receptor subunits were subcloned into the pcDNA3 expression vector in the T7 orientation. The mutations [α1L127C and the α1(Q241L + L127C) double mutant] were generated using the QuikChange site-directed mutagenesis kit (Stratagene, San Diego, CA, USA). The mutated subunits were fully sequenced to verify that only the desired mutation(s) had been produced. The cDNA was linearized by Xba I (NEB Laboratories, Ipswich, MA, USA) digestion, and the cRNA was produced using mMessage mMachine (Ambion, Austin, TX, USA). The oocytes were injected with 7–14 ng cRNA per subunit (α : β: γ 1:1:1 ratio) in a final volume of 20–60 nL, and incubated at 16°C for 3–4 days before labelling and recording.
Labelling with Alexa Fluor 546 C5 maleimide (Invitrogen, Carlsbad, CA, USA) was carried out using incubation for 45–60 min at room temperature in the dark with 20 µM Alexa Fluor 546 C5 maleimide (A5m) dissolved in OR2 (92.5 mM NaCl, 2.5 mM KCl, 1 mM MgCl2, 10 mM HEPES) at pH 7.2. The oocytes were washed in the bath solution (OR2, pH 7.5) before being transferred to the recording chamber.
We used a custom-made recording chamber (Chang and Weiss, 2002; Khatri et al., 2009; Li et al., 2010). The chamber contains two compartments, separated by a 0.8 mm aperture on which the oocyte is placed. The oocyte is impaled in the top compartment, while the agonist and modulators are applied in the lower compartment. An inverted microscope (Nikon Diaphot TMD) fitted with a Nikon 20X LWD 0.4 NA objective was used to image the fluorescence signal from the lower chamber. The microscope holds dichroic (565DCLP) and emission (D605/55m) filters (Chroma Technology Corp, Rockingham, VT, USA) for fluorescence detection. Due to a relatively tight seal between the oocyte and the aperture, there is little leakage between the two compartments, and the fluorescence and current signals are measured from the same population of receptors.
The system used for fluorescence measurements was purchased from Photon Technology International (Birmingham, NJ, USA). The system consists of a DeltaRAM monochromator, from which the light (546 nm) is passed into the microscope via a liquid light guide, a photomultiplier tube (R1527P Hamamatsu Photonics, Bridgewater, NJ, USA) mounted on the side port of the microscope, BryteBox acquisition hardware, and FeliX32 software for control of excitation. A 75-watt Xenon short arc lamp (Ushio Inc., Tokyo, Japan) served as a light source.
Standard two-electrode voltage clamp was used to record the currents. Both voltage and current electrodes were patch-clamp electrodes filled with 3 M KCl and had resistances of 0.5 to 1.5 MΩ. The oocytes were typically clamped at −60 mV. The chamber was perfused continuously at approximately 5 mL·min−1. Bath solution was perfused between all test applications. Solutions were switched by pClamp using a Warner Instruments VC-8T valve controller. Solutions were applied from glass reservoirs via metal or Teflon tubing to reduce adsorption. A typical drug application protocol was to expose an oocyte to bath solution for 10 s, followed by a 20 s drug (agonist, modulators) application, and a switch back to bath solution. The washout period between successive drug applications was 2–5 min.
The current responses were amplified with an Axoclamp 900A amplifier (Molecular Devices, Sunnyvale, CA, USA), digitized with a Digidata 1320 series digitizer (Molecular Devices) at a 100 Hz sampling rate, and stored using pClamp (Molecular Devices). Current and fluorescent transients were analysed with Clampfit (Molecular Devices). The baseline was corrected for bleaching when needed. The fluorescence response values are calculated as percentage change from baseline fluorescence with positive values indicating an increase in fluorescence. The typical responses to near saturating concentrations (1–5 mM) of GABA were 0.5–2%. The fluorescence and current responses were normalized to the response from the same cell to a high (1–5 mM) concentration of GABA. Data are presented as mean ± SD. The GABA concentration–response curves were fitted using the program nfit (The University of Texas, Medical Branch at Galveston). Statistical analyses were carried out using Excel (Microsoft, Richmond, WA, USA).
Single-channel experiments were conducted on α1L127Cβ2γ2 receptors transiently expressed in HEK 293 cells. Cell culture, receptor expression, and the details of cell-attached electrophysiological recordings and data analysis have been described in detail previously (Steinbach and Akk, 2001; Akk et al., 2008).
The drugs were purchased from Sigma-Aldrich (salts, GABA, pentobarbital, allopregnanolone, etomidate), Tocris Bioscience (gabazine) or Invitrogen (Alexa Fluor 546 C5 maleimide). Stock solutions of allopregnanolone (10 mM) and etomidate (20 mM) were made in DMSO; dilutions to test concentrations were made on the day of the experiment. The stock solution of Alexa 546 maleimide (5 mM) was made in DMSO, and stored in aliquots at −80°C until used for labelling.
Results
Channel activation by GABA results in ΔF
Exposure of oocytes clamped at −60 mV expressing rat α1β2γ2L GABAA receptors to GABA results in inward current. In receptors containing the α1L127C mutation (Figure 2A) and labelled with A5m, the inward current is accompanied by an increase in fluorescence intensity suggesting that the environment around the labelled residue reporter becomes less polar upon channel activation. The ΔF occurs rapidly, following the functional response, and is reversible, decreasing upon the termination of agonist application.
Figure 2.
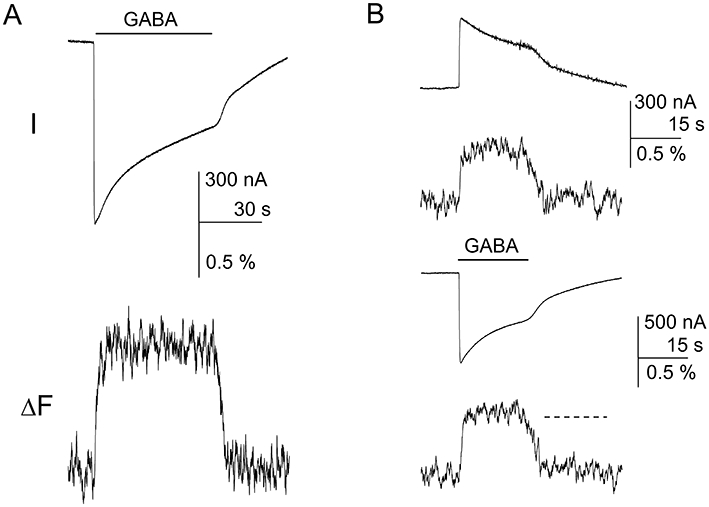
Channel desensitization and direction of current flow do not affect fluorescence changes (ΔF) at the α1L127C site. (A) The oocyte expressing α1L127Cβ2γ2L receptors labelled with Alexa Fluor 546 C5 maleimide was exposed to a 60 s pulse of 1 mM GABA. The current response shows a considerable degree of desensitization during drug application. Channel desensitization was not accompanied by changes in ΔF. The oocyte was clamped at −60 mV. (B) The oocyte was clamped at −15 mV (top) or −60 mV (bottom), resulting in outward or inward current flow respectively. Regardless of the direction of current flow, channel activation was accompanied by an increase in ΔF indicating that the fluorophore is exposed to a less polar environment. The dashed line in the fluorescence recording at −60 mV shows the mean level of ΔF at −15 mV (from top panel).
The ΔF is specific to α1L127Cβ2γ2L receptors labelled with A5m. In control experiments, no ΔF was observed when oocytes expressing wild-type α1β2γ2L receptors were incubated with A5m and then exposed to saturating concentrations of GABA. The ΔF in the presence of 300 µM GABA was −0.08 ± 0.22% (statistically not different from no response). Similarly, α1L127Cβ2γ2L receptors not exposed to a fluorophore did not demonstrate significant ΔF when challenged with 3 mM GABA (−0.10 ± 0.26%). Finally, uninjected oocytes, exposed to A5m, did not respond to applications of GABA with current or fluorescence responses.
The α1L127C mutation shifts the activation concentration–response curve to higher GABA concentrations. The midpoint of the wild-type α1β2γ2L receptor concentration–response curve was 4.3 µM. When the receptors contain the α1L127C mutation the EC50 of the concentration–response curve for activation was 20 ± 1 µM. The EC50 for activation was 117 ± 41 µM when the α1L127C mutant receptors were labelled with A5m. At higher agonist concentrations, the ΔF, similar to the current response, was enhanced. We estimated that the EC50 of the concentration–response curve for ΔF in labelled α1L127C mutant receptors is 173 ± 60 µM (Figure 3). The data are summarized in Table 1.
Figure 3.
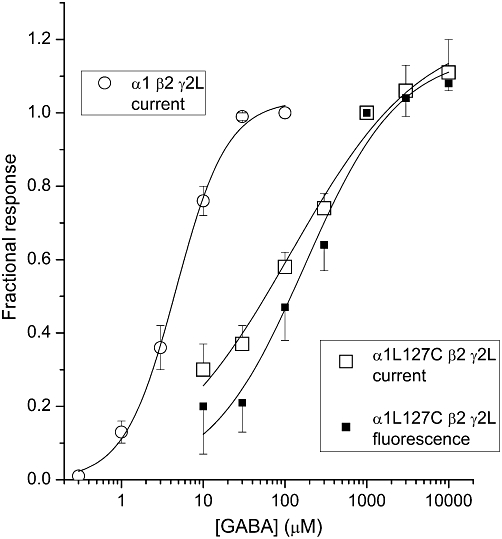
The concentration–response properties for wild-type and α1L127Cβ2γ2L mutant receptors. Current (open squares) and fluorescence change (filled squares) concentration–response curves are shown for the α1L127Cβ2γ2L receptor labelled with Alexa Fluor 546 C5 maleimide. The curves were fitted to the Hill equation yielding an EC50 of 117 ± 41 µM, nH= 0.5 ± 0.1 and maximal response = 1.2 ± 0.1, and an EC50 of 173 ± 60 µM, nH= 0.8 ± 0.2 and maximal response = 1.2 ± 0.1 for current and fluorescence change respectively. For comparison, the concentration–response curve for currents from wild-type α1β2γ2L receptors (open circles) had an EC50 of 4.3 ± 0.3 µM, and a Hill coefficient of 1.5 ± 0.1. The oocytes were clamped at −60 mV. Each data point shows mean ± SEM from 3–9 oocytes.
Table 1.
Concentration–response properties of wild-type and mutant receptors
| α1β2γ2L wild-type Unlabelled Current | ΔF | A5m Current | ΔF | α1L127Cβ2γ2L Unlabelled Current | ΔF | A5m Current | ΔF | |
|---|---|---|---|---|---|---|---|---|
| Maximal response | 1.03 ± 0.03 | nd | 1.06 ± 0.07 | nr# | 0.98 ± 0.02 | nr# | 1.24 ± 0.09 | 1.16 ± 0.09 |
| EC50 | 4.6 ± 0.4 µM | nd | 4.6 ± 0.9 µM | 20 ± 1 µM | 117 ± 41 µM | 173 ± 60 µM | ||
| nH | 1.4 ± 0.1 | nd | 1.3 ± 0.3 | 1.2 ± 0.1 | 0.5 ± 0.1 | 0.8 ± 0.2 |
The concentration–response curves were fitted to: response = maximal response/(1 + EC50/[GABA])nH, where EC50 is the concentration of GABA producing a half-maximal reponse, and nH is the Hill slope of the curve. The data were normalized to the response at 100 µM GABA (wild-type receptors) or 3 mM GABA (mutant receptors). Data are from 3–9 oocytes. ΔF, fluorescence change; A5m, Alexa Fluor 546 C5 maleimide; nd, not determined; nr#, no ΔF response was observed in the presence of GABA. For more details please see text.
During prolonged agonist exposure channels desensitize. This manifests as reduced current response. We tested the effect of desensitization on the ΔF by comparing current and fluorescence responses during a 1 min application of 1 mM GABA. The relative current level at the end of the agonist application was 49 ± 4% of peak response (n= 4 cells; P < 0.001, paired t-test). The reduction in current level was not accompanied by a change in the ΔF. During the last 5 s of GABA application the ΔF was 103 ± 23% (P > 0.82) of the ΔF measured during the first 5 s of the response. Sample current and fluorescence traces are shown in Figure 2A. These results confirm the observations made by Muroi et al. (2006) using the fluorophores A5m and tetramethylrhodamine.
We also tested the effect of voltage and direction of current flow on the ΔF at the α1L127C site. The bulk of the experiments was conducted at −60 mV resulting in inward current flow (outward movement of Cl-). To change the direction of current flow we clamped the cells at potentials between −20 mV and −15 mV. The data demonstrate that changes in voltage and/or direction of current flow have no effect on the ΔF at the α1L127C site. In three cells, the mean ΔF during inward current flow was 95 ± 12% (P > 0.56) of the fluorescence response during outward current flow. Sample recordings are shown in Figure 2B.
It has been reported that the amplitude, and even the sign, of ΔF for a probe linked at the α1L127C position can be different for receptors containing the α1, β2 and γ2 subunits as opposed to receptors consisting of only the α1 and β2 subunits (Muroi et al., 2006; 2009;). To eliminate the possibility that the receptors expressed lacked the γ2L subunit, we tested the ability of zinc ions (100 µM) to inhibit GABA-elicited currents. Receptors containing α and β subunits are strongly inhibited, whereas αβγ receptors are little affected (Draguhn et al., 1990; Smart et al., 1991). When only α1L127C and β2 subunits were expressed, the steady-state response to 1 mM GABA was reduced to 25 ± 11% of control in the presence of zinc (n= 4 cells; P < 0.001). In contrast, responses from oocytes injected with α1L127C, β2 and γ2L subunits were unaffected (96 ± 15%, n= 10 cells; P > 0.54).
Other ligands to the GABA binding site elicit ΔF
The concentration–response data for GABA indicate that activation of the receptor and the increase in fluorescence have a similar dependence on GABA concentration. To further examine the relationship between receptor activation and amplitude of fluorescence signal we compared current and fluorescence responses for selected ligands of the GABA binding site. We used relatively high concentrations of β-alanine (20 mM), muscimol (100 µM), piperidine-4-sulphonic acid (P4S, 1 mM), and 4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol (THIP, 10 mM). Each of these drugs activated the labelled mutant receptor. The average peak responses ranged from 44% (P4S) to 71% (muscimol) of that observed with 1 mM GABA in the same oocyte. Although the functional response was accompanied by an increase in fluorescence intensity there was no clear correlation between the peak current and ΔF. For example, the peak current in the presence of P4S was less than half; however, the ΔF was 215 ± 69% (n= 5 cells; P < 0.05) of that observed with GABA. Besides P4S, the current and fluorescence responses to THIP showed no linear correlation with those to GABA (current: 55 ± 16%; ΔF: 258 ± 102%). In contrast, exposure to muscimol and β-alanine resulted in smaller electrophysiological and fluorescence responses in a similar ratio to GABA. The data are summarized in Figure 4.
Figure 4.
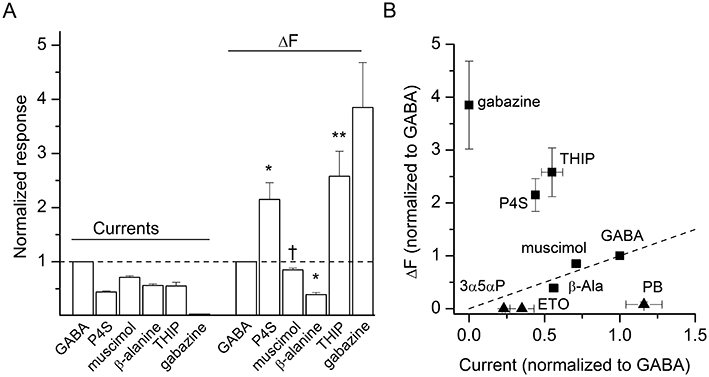
Occupation of the GABA site results in fluorescence changes (ΔF). (A) Comparison of current and ΔF from oocytes exposed to 1 mM GABA, 1 mM piperidine-4-sulphonic acid (P4S), 100 µM muscimol, 20 mM β-alanine, 10 mM 4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol (THIP), or 50 µM gabazine. The responses are normalized to those to GABA. Gabazine elicited no current response, but exhibited the largest ΔF. Statistical analysis was conducted on a parameter R (calculated as relative ΔF/relative current). Significance levels apply to comparison with the data obtained in the presence of GABA. *P < 0.05; **P < 0.01; †not significant. For gabazine, due to lack of currents R could not be calculated. (B) The normalized ΔF is plotted as a function of normalized current response. For comparison, data for non-GABA site ligands pentobarbital (PB, 1 mM), allopregnanolone (3α5αP, 10 µM) and etomidate (ETO, 5 µM) are shown with triangles. The dashed line goes from no response to the GABA point (slope of 1).
We also tested the competitive antagonist of GABA binding, gabazine (SR-95531). As expected, the application of 50 µM gabazine elicited no functional response. However, we observed an increase in fluorescence intensity during exposure to gabazine. The mean ΔF was 385 ± 144% (n= 3 cells) of the response to GABA. A previous study in which the receptors were labelled with tetramethylrhodamine at the α1L127C site showed that while the application of GABA results in an increase in fluorescence intensity, exposure to gabazine decreases fluorescence (Muroi et al., 2006). We have confirmed the reduction of fluorescence intensity in oocytes labelled with tetramethylrhodamine exposed to gabazine (data not shown). Accordingly, the two fluorophores give different signs for the ΔF in response to gabazine.
These results indicate that the fluorescence increase at the α1L127C site is associated with occupation of the transmitter binding site, but does not directly correlate with channel activation.
Direct activation by non-GABA site ligands pentobarbital, etomidate or the neurosteroid allopregnanolone does not result in ΔF at the α1L127C site
We next tested whether channel activation by anaesthetic ligands pentobarbital, etomidate or the neurosteroid allopregnanolone cause ΔF at the transmitter binding site. The oocytes were exposed to 10–2000 µM pentobarbital, 1–10 µM allopregnanolone, or 5 µM etomidate, and the current and fluorescence responses were recorded. Each egg was additionally exposed to 1–5 mM GABA to obtain a control current and fluorescence response. All results are expressed as % of the response to GABA.
The electrophysiological recordings show that pentobarbital is an efficacious activator of the labelled α1L127C mutant receptor. The mean response to 1 mM pentobarbital was 116 ± 23% (n= 4 cells) of the response to 1 mM GABA. However, the ΔF was only 8 ± 10% (statistically not different from no response; P > 0.2) of that in the presence of GABA. When 2 mM pentobarbital was used to activate the receptors the current responses were slightly diminished (72 ± 17% of the response to GABA), probably due to channel block (Akaike et al., 1987). The current response was not accompanied by a ΔF (relative response 17 ± 22% of the response to GABA; P > 0.2). The sample currents and a summary of the data are given in Figure 5A,D.
Figure 5.
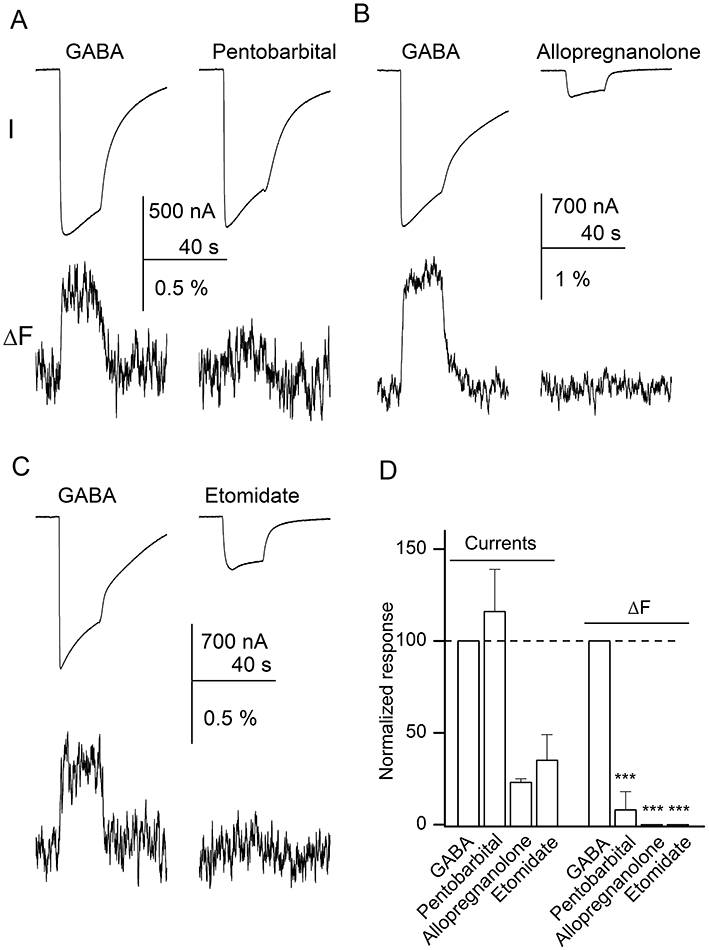
Direct activation by pentobarbital, allopregnanolone or etomidate does not result in fluorescence changes (ΔF). The oocytes were activated by 1 mM pentobarbital (A), 10 µM allopregnanolone (B), or 5 µM etomidate (C). For comparison, each cell was additionally exposed to 1–5 mM GABA. Current (I) and ΔF are shown. (D) Summary of the current and fluorescence responses. The signal amplitudes are normalized to the response in the presence of GABA (100%). The data indicate that direct activation by allopregnanolone, etomidate or pentobarbital did not result in ΔF. Statistical analysis was conducted on a parameter R (calculated as relative ΔF/relative current). Significance levels apply to comparison with the data obtained in the presence of GABA. ***P < 0.001.
We also recorded current and fluorescence responses in the presence of the neurosteroid allopregnanolone. Allopregnanolone was a relatively inefficacious activator. The current response to 1 µM allopregnanolone was only 4 ± 1% (n= 3 cells) of that to 5 mM GABA. When 10 µM steroid was used to activate the receptors, the mean current response was 23 ± 2% of the response to GABA. In either case there was no ΔF accompanying channel activation (Figure 5B,D).
The application of 5 µM etomidate resulted in an inward current with an average peak response of 35 ± 14% (n= 3 cells) of the peak current observed in the presence of 5 mM GABA. Channel activation was not accompanied by changes in fluorescence intensity (Figure 5C,D).
These experiments demonstrate that channel activation per se is not sufficient to elicit structural changes at the α1L127C site.
Potentiation by allopregnanolone and etomidate results in increased ΔF
A simple allosteric model for receptor activation associates an increase in affinity for agonist, at the agonist binding site, with the open state of the receptor (Colquhoun, 1998). This model predicts that a potentiating drug which increases the activation of the channel at a given agonist concentration would also increase ΔF. We tested whether potentiation of channel function is associated with enhanced ΔF as detected by A5m linked to the α1L127C site by examining the effects of etomidate and the neurosteroid allopregnanolone on currents and ΔF.
Co-application of 10 µM allopregnanolone with 100 µM GABA enhanced the current response from 45 ± 4% to 93 ± 8% (n= 4 cells; P < 0.01). The increase in functional response was accompanied by an enhanced ΔF that was increased from 29 ± 23% to 117 ± 39% (P < 0.05). All values are expressed as % of the response to 1 mM GABA. Sample traces are shown in Figure 6.
Figure 6.
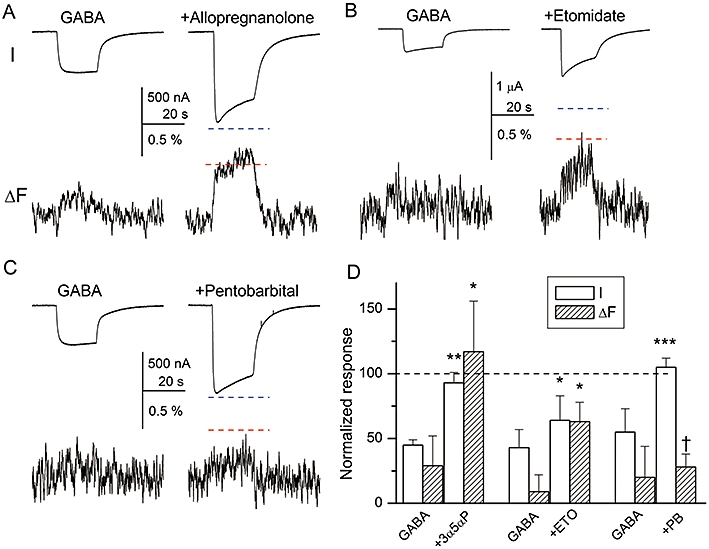
Potentiation by allopregnanolone or etomidate, but not pentobarbital is accompanied by increased fluorescence change (ΔF). The oocytes were exposed to a low concentration of GABA (30–100 µM, eliciting an approximately half-maximal control response) in the absence and presence of 10 µM allopregnanolone (A), 5 µM etomidate (B) or 100 µM pentobarbital (C). Current (I) and ΔF are shown. The blue and red dashed lines show the levels of the peak response and ΔF respectively, in the presence of 1 mM GABA. (D) Summary of the current and fluorescence responses. The signal amplitudes are normalized to the response in the presence of 1 mM GABA. Allopregnanolone (3α5αP) is a strong potentiator, which enhances the peak current and ΔF to the level seen in the presence of 1 mM GABA. In the presence of etomidate (ETO) both peak current and ΔF are enhanced. In contrast, pentobarbital (PB) enhances the current response but is without effect on ΔF. Significance levels apply to comparison between control condition and in the presence of modulator. *P < 0.05; **P < 0.01; ***P < 0.001; †not significant.
To verify that the effect of allopregnanolone is mediated by interactions with the classic binding site mediating the effects of potentiating steroids, we tested the effect of the α1Q241L mutation, previously shown to abolish channel potentiation by allopregnanolone (Hosie et al., 2006; Akk et al., 2008) on ΔF. In this experiment, the α1(Q241L + L127C) double mutant was coexpressed with wild-type β2 and γ2L subunits. The drug application protocol was analogous to the one used in experiments described above: each egg was exposed to a high concentration of GABA (5 mM), a low concentration of GABA (100 µM) and a low concentration of GABA + 10 µM allopregnanolone. The application of 100 µM GABA elicited a current response that was 36 ± 19% and a ΔF that was 58 ± 20% (n= 3 cells) of that in the presence of 5 mM GABA. As expected, the co-application of allopregnanolone with 100 µM GABA did not potentiate the current response, which was 92 ± 14% (P > 0.39) of the response in the presence of 100 µM GABA alone. Also, the ΔF was not modulated in the presence of this steroid (96 ± 18% of control, P > 0.76). These data thus suggest that the actions of allopregnanolone on ΔF are mediated by steroid interactions with the classic steroid interaction site.
Co-application of 5 µM etomidate with 100 µM GABA enhanced the peak response from 43 ± 14% to 64 ± 19% (n= 4 cells; P < 0.05). Functional potentiation was accompanied by enhanced ΔF. The ΔF was 9 ± 13% in the presence of 100 µM GABA, and 63 ± 15% in the presence of GABA + etomidate (P < 0.05). The values are expressed as % of the response in the presence of 1 mM GABA. Sample traces are shown in Figure 6.
Thus, these experiments demonstrate that potentiation of the electrophysiological response by allopregnanolone or etomidate is accompanied by an enhanced fluorescence response at the transmitter binding site.
Modulation by pentobarbital does not lead to increased ΔF
We also tested the potentiating effect of pentobarbital on currents and ΔF. In the first experiment, we co-applied 100 µM pentobarbital with 30 µM GABA. The application of pentobarbital increased the peak current from 55 ± 18% to 105 ± 7% (n= 5 cells; P < 0.001) of the response seen in the presence of 1 mM GABA. The increase in the functional response was not accompanied by an increased ΔF. The ΔF in the presence of 30 µM GABA was 20 ± 24% of that in the presence of 1 mM GABA. When pentobarbital was co-applied with GABA the ΔF was 28 ± 10% (P > 0.54). Sample traces and a summary are shown in Figure 6.
We confirmed the dissociation between the effects of pentobarbital on current and ΔF by testing the effect of pentobarbital on receptors activated by 0.5 mM P4S. The average peak current and ΔF in the presence of P4S alone were 35 ± 13% and 75 ± 11% (n= 3 cells) of the response to 1 mM GABA respectively. Co-application of 50 µM pentobarbital with P4S enhanced the peak current response to 155 ± 10% (P < 0.01) of control, but was without effect on the ΔF (100 ± 8% of control; P > 0.95).
We also probed for the effect of pentobarbital on the ΔF produced by the inhibitor gabazine, which acts as an allosteric inhibitor of pentobarbital activation. This experiment allowed us to independently verify the interactions of both drugs with the receptor: the binding of pentobarbital is confirmed by the current response, and the binding of gabazine to the same receptors is verified by the reduction of the current amplitude. The data show that in the presence of 10 µM gabazine the electrophysiological response to 1 mM pentobarbital was reduced to 58 ± 22% of control (n= 3 cells). However, functional inhibition was not accompanied by changes in fluorescence elicited by gabazine (106 ± 8% of control, P > 0.35).
These results indicate that the effect of pentobarbital on channel open probability is not accompanied by structural changes at the α1L127C site as detected from changes in fluorescence intensity of the A5m fluorophore.
Does pentobarbital potentiate the α1L127Cβ2γ2L mutant receptor?
The data presented above show that co-application of pentobarbital with low concentrations of GABA or P4S enhances the functional response but not ΔF. To exclude the possibility that the functional response in the presence of GABA + pentobarbital is an independent sum of electrophysiological responses to GABA and pentobarbital, we verified functional potentiation of mutant receptors by pentobarbital using single-channel patch clamp. The α1L127C, β2 and γ2L subunits were transiently expressed in HEK 293 cells, and single-channel activity in the cell-attached configuration recorded in the presence of GABA, pentobarbital and GABA + pentobarbital. We focused on channel open durations. A previous study investigating the mechanism of action of pentobarbital found that the drug potentiates the wild-type α1β2γ2L GABAA receptor by increasing the mean duration and prevalence of long-lived openings (Steinbach and Akk, 2001). Accordingly, we reasoned that potentiation would be evident as increased mean open duration. In contrast, independent direct activation by the two drugs would be expected to reduce the mean closed time duration but not necessarily affect the mean open duration.
In the presence of 100 µM GABA, the open time histograms, similar to those for wild-type receptors, were best described by the sum of three exponentials (Table 2). The addition of 100 µM pentobarbital significantly increased the mean open duration, as a result of increases in the mean duration and prevalence of the longest-lived open state. Pentobarbital alone elicited openings as well, which showed only two open time components and lacked the long-duration component. Sample currents are shown in Figure 7. Thus, the data indicate that pentobarbital potentiates receptors containing the α1L127C mutation similarly to the wild-type α1β2γ2L receptor.
Table 2.
Potentiation of single-channel currents by pentobarbital
| [GABA] | [Pentobarbital] | Mean OT (ms) | OT1 (ms) | Fraction OT1 | OT2 (ms) | Fraction OT2 | OT3 (ms) | Fraction OT3 | n patches |
|---|---|---|---|---|---|---|---|---|---|
| 100 µM | – | 1.8 ± 0.3 | 0.18 ± 0.11 | 0.26 ± 0.07 | 1.7 ± 0.2 | 0.59 ± 0.15 | 3.6 ± 0.5 | 0.15 ± 0.09 | 3 |
| 100 µM | 100 µM | 6.0 ± 1.5** | 0.26 ± 0.15† | 0.44 ± 0.13† | 2.9 ± 1.5† | 0.22 ± 0.09** | 12.7 ± 2.9*** | 0.35 ± 0.09* | 6 |
| – | 100 µM | 2.1 ± 0.3 | 0.56 ± 0.24 | 0.49 ± 0.11 | 3.4 ± 1.0 | 0.51 ± 0.11 | – | – | 3 |
Results from single-channel patch clamp experiments on HEK cells transiently expressing α1L127C, β2 and γ2L subunits. The open time histograms were fitted to sums of two (pentobarbital) or three (GABA, GABA + pentobarbital) exponentials. The Table gives the mean open duration, and the mean durations and fractions (relative frequency) of the individual open time components. The data indicate that the application of pentobarbital potentiates the mutant receptor by increasing the mean duration and prevalence of OT3. For GABA + pentobarbital, the significance level applies to comparison to data obtained in the presence of GABA alone.
P < 0.05;
P < 0.01;
P < 0.001;
not significant. OT, open time.
Figure 7.
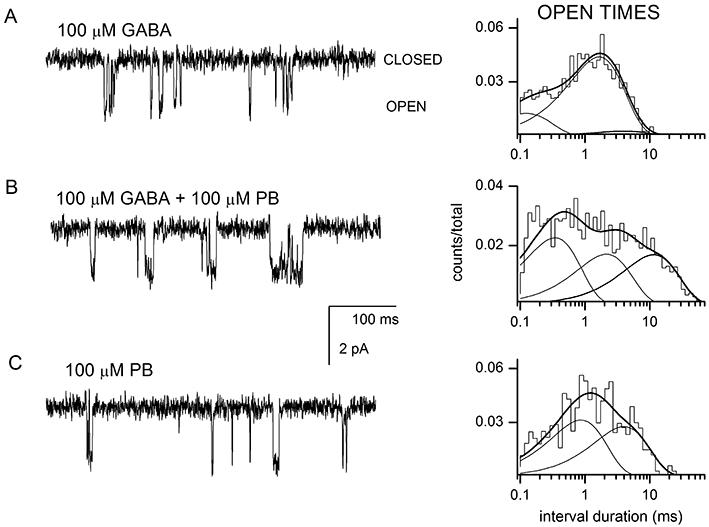
Pentobarbital potentiates open times from receptors containing the α1L127C mutation. Sample currents obtained from receptors activated by 100 µM GABA (A), 100 µM GABA + 100 µM pentobarbital (PB) (B) or 100 µM pentobarbital (C). Channel openings are shown as downward deflections. Open time histograms from the respective patches are shown next to the data traces. For receptors activated by GABA, the open times were 0.11 ms (21%), 1.5 ms (74%) and 3.8 ms (4%). For receptors activated by GABA + PB, the open times were 0.32 ms (41%), 2.0 ms (30%) and 10.5 ms (29%). For receptors activated by PB, the open times were 0.8 ms (53%) and 3.7 ms (47%). The averaged data from multiple patches are given in Table 2.
Discussion and conclusions
In this study, we have used simultaneous electrophysiology and site-directed fluorometry to investigate the pharmacological basis for structural changes near the GABAA receptor transmitter binding site. We labelled the α1L127C residue with Alexa Fluor 546 C5 maleimide. This residue is in the vicinity of the GABA binding pocket contributed by loop E of the ‘−’ interface. Previous work has shown that mutations in this region affect agonist binding and channel gating by 5-iodo-3-[2(S)-2-azetidinyl-methoxy] pyridine (5I-A-85380) in the related nicotinic acetylcholine receptor (Hamouda et al., 2009). The present study was aimed at determining which component of channel function (transmitter binding, channel gating, current flow) leads to structural changes, as determined from ΔF, at the α1L127C site. We also examined how three potentiators of GABAA receptor function affect currents and ΔF elicited by ligands for the GABA binding site.
The data demonstrate that receptor exposure to GABA leads to a concentration-dependent increase in fluorescence intensity, indicative of the local environment surrounding the fluorophore becoming less polar. The effect is mimicked by other ligands to the transmitter binding sites such as muscimol, β-alanine, THIP and P4S. Interestingly, there was no clear correlation between the ability of the agonist to elicit current (i.e. peak response) and cause structural change (amplitude of ΔF). The peak responses to saturating concentrations of P4S and THIP were smaller but the ΔF was larger than in the presence of GABA. Furthermore, the competitive antagonist gabazine caused a strong increase in ΔF but was ineffective at producing a functional response.
It should be noted that a previous study in which the α1L127C was labelled with tetramethylrhodamine found that GABA and the competitive antagonist gabazine elicited ΔF of opposite direction (Muroi et al., 2006). It was proposed that the two ligands cause distinct types of structural changes in the transmitter binding sites, and that changes associated with the binding of GABA, but not gabazine, lead to channel opening. Our data demonstrate that receptors labelled with A5m exhibit ΔF of the same direction in the presence of GABA and gabazine. The exact cause for differences between the data obtained with A5m versus tetramethylrhodamine is unclear, but differences in linker lengths (5 Å for tetramethylrhodamine vs. 15 Å for A5m) positioning the fluorophores in slightly different locations where they report dissimilar changes in environment is a possibility. As the underlying structural motions in the presence of gabazine are probably the same regardless of the nature of the bound fluorophore, the findings suggest that results pertaining to the direction of the ΔF can be less informative than previously considered, at least when different fluorophores are used.
During long agonist applications channel desensitization develops. In the related nicotinic receptor, slow desensitization has been explained by a conformational change in the receptor, which is distinct from the closing of the activation gate (Auerbach and Akk, 1998; Purohit and Grosman, 2006). In the GABAA receptor labelled at the α1L127C site, functional desensitization did not affect the ΔF. These data confirm the observations by Muroi et al. (2006) that entry into the slow-desensitized state does not affect the fluorescence signal. In addition we found that ΔF was insensitive to voltage and direction of the current flow. We infer that the structural change at the α1L127C site does not correlate with current flow.
Overall, these results for ligands of the GABA binding site indicate that the fluorescence increase is more closely associated with occupancy of the binding site than with receptor activation. However, the fact that different ligands can elicit ΔF of differing amplitude indicates that occupancy per se is not the sole determinant, and that some additional aspects of the interaction between ligand and receptor influence the signal.
Channel activation by ligands which do not interact with the transmitter binding site did not result in ΔF at the α1L127C site. Pentobarbital is a strong activator of the mutant receptor; however, no significant ΔF was observed in the presence of 10–2000 µM pentobarbital. This finding confirms a previous report by Muroi et al. (2009). A previous study of the rate of reaction of the α1L127C residue with MTS-biotin found that pentobarbital does not significantly change the rate, although both GABA and gabazine reduced the rate of reaction (Kloda and Czajkowski, 2007). We also found that etomidate and the neurosteroid allopregnanolone activate the labelled mutant receptor but elicit no ΔF. However, we note that the low current levels during direct activation by etomidate and allopregnanolone may contribute to the lack of detection of ΔF. We also point out that lack of ΔF does not exclude the possibility that a structural change near the fluorophore has taken place. If the local environments surrounding the fluorophore are similar in the inactive and active states, ΔF would not be observed.
Pentobarbital, etomidate and allopregnanolone, in addition to acting as direct activators of the GABAA receptor, potentiate the response to low concentrations of GABA. In the simplest model for allosteric potentiation, the increased response will be associated with an increased occupancy of the GABA binding site by GABA. This is because the open-channel state of the receptor has a higher affinity for GABA, so at a constant concentration of GABA the occupancy will be higher when the potentiator increases the fraction of receptors with open channels. Qualitatively, potentiation with either allopregnanolone or etomidate conformed with this simple idea. In the presence of potentiator, both the current and ΔF elicited by GABA were increased.
In contrast, pentobarbital increased the current but did not increase the ΔF elicited by GABA. However, examination of single-channel currents indicated that pentobarbital was able to potentiate GABA-elicited currents. We also examined potentiation of responses to a second agonist, P4S, with similar results. We then tested the interaction between gabazine and pentobarbital. Gabazine binds to the GABA binding site, to competitively inhibit activation by GABA, but additionally acts as a non-competitive antagonist of activation by other agonists including pentobarbital and neuroactive steroids (Ueno et al., 1997). Co-application of gabazine with pentobarbital reduces the peak current, most probably by stabilizing an inactive state of the receptor, that is, gabazine has a higher affinity for an inactive receptor than an active one. For an allosteric interaction between an activator (pentobarbital) and an inhibitor (gabazine) it would be expected that the presence of the activator would shift the receptor population towards states with a low affinity for inhibitor and hence reduce the fraction of sites with inhibitor bound. The observation that gabazine reduced the response to pentobarbital supports the idea that the two drugs interact with the same population of receptors. However, the ΔF elicited by gabazine is unaffected by a concentration of pentobarbital which elicits a maximal current. This result is consistent with the observations using pentobarbital and GABA-site agonists. It also indicates that quenching of the fluorescence signal by pentobarbital is unlikely to be an explanation for the observations. In the cases of GABA and P4S, potentiation by pentobarbital would be predicted to increase the fluorescence while in the case of gabazine to decrease the signal, but in both cases no significant change was produced.
The full interpretation of these observations is not yet possible, because the exact structural bases of the change in fluorescence intensity are not known. A similar change could result from somewhat different changes in receptor structure, so long as the changes in the environment around the probe were similar, and the absence of a change does not conclusively demonstrate that no structural change occurred in the receptor. However, the qualitative differences between the results obtained with allopregnanolone and etomidate, on the one hand, and pentobarbital, on the other, suggest that the mechanisms of potentiation differ in the two cases. The results also suggest that the structural changes in the receptor during activation may be more complex than expected from simple models. If there can be a change in the probability that a receptor is active without an associated change in the affinity of the GABA binding site for GABA, it would suggest that coupling between the gate region and the agonist binding site is not as tight as is often thought.
Acknowledgments
We thank Chuck Zorumski and Steve Mennerick for providing Xenopus laevis oocytes. This work was supported by the National Institutes of Health Grant GM47969. JHS is the Russell and Mary Shelden Professor of Anesthesiology.
Glossary
Abbreviations
- ΔF
fluorescence change
- P4S
piperidine-4-sulphonic acid
- THIP
4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol
Conflicts of interest
The authors state no conflict of interest.
Supplemental material
References
- Akaike N, Maruyama T, Tokutomi N. Kinetic properties of the pentobarbitone-gated chloride current in frog sensory neurones. J Physiol. 1987;394:85–98. doi: 10.1113/jphysiol.1987.sp016861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Li P, Bracamontes J, Reichert DE, Covey DF, Steinbach JH. Mutations of the GABAA receptor alpha1 subunit M1 domain reveal unexpected complexity for modulation by neuroactive steroids. Mol Pharmacol. 2008;74:614–627. doi: 10.1124/mol.108.048520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin J, Weiss DS. GABAA receptor needs two homologous domains of the β-subunit for activation by GABA but not by pentobarbital. Nature. 1993;366:565–569. doi: 10.1038/366565a0. [DOI] [PubMed] [Google Scholar]
- Auerbach A, Akk G. Desensitization of mouse nicotinic acetylcholine receptor channels. A two-gate mechanism. J Gen Physiol. 1998;112:181–197. doi: 10.1085/jgp.112.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Weiss DS. Site-specific fluorescence reveals distinct structural changes with GABA receptor activation and antagonism. Nat Neurosci. 2002;5:1163–1168. doi: 10.1038/nn926. [DOI] [PubMed] [Google Scholar]
- Colquhoun D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol. 1998;125:924–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draguhn A, Verdorn TA, Ewert M, Seeburg PH, Sakmann B. Functional and molecular distinction between recombinant rat GABAA receptor subtypes by Zn2+ Neuron. 1990;5:781–788. doi: 10.1016/0896-6273(90)90337-f. [DOI] [PubMed] [Google Scholar]
- Hamouda AK, Jin X, Sanghvi M, Srivastava S, Pandhare A, Duddempudi PK, et al. Photoaffinity labeling the agonist binding domain of α4β4 and α4β2 neuronal nicotinic acetylcholine receptors with [(125)I]epibatidine and 5[(125)I]A-85380. Biochim Biophys Acta. 2009;1788:1987–1995. doi: 10.1016/j.bbamem.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden JH, Czajkowski C. Different residues in the GABAA receptor α1T60-α1K70 region mediate GABA and SR-95531 actions. J Biol Chem. 2002;277:18785–18792. doi: 10.1074/jbc.M111778200. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva HM, Smart TG. Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature. 2006;444:486–489. doi: 10.1038/nature05324. [DOI] [PubMed] [Google Scholar]
- Kash TL, Trudell JR, Harrison NL. Structural elements involved in activation of the γ-aminobutyric acid type A (GABAA) receptor. Biochem Soc Trans. 2004;32:540–546. doi: 10.1042/BST0320540. [DOI] [PubMed] [Google Scholar]
- Khatri A, Sedelnikova A, Weiss DS. Structural rearrangements in loop F of the GABA receptor signal ligand binding, not channel activation. Biophys J. 2009;96:45–55. doi: 10.1016/j.bpj.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloda JH, Czajkowski C. Agonist-, antagonist-, and benzodiazepine-induced structural changes in the α1 Met113-Leu132 region of the GABAA receptor. Mol Pharmacol. 2007;71:483–493. doi: 10.1124/mol.106.028662. [DOI] [PubMed] [Google Scholar]
- Li P, Khatri A, Bracamontes J, Weiss DS, Steinbach JH, Akk G. Site-specific fluorescence reveals distinct structural changes induced in the human ρ1 GABA receptor by inhibitory neurosteroids. Mol Pharmacol. 2010;77:539–546. doi: 10.1124/mol.109.062885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannuzzu LM, Moronne MM, Isacoff EY. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 1996;271:213–216. doi: 10.1126/science.271.5246.213. [DOI] [PubMed] [Google Scholar]
- Muroi Y, Czajkowski C, Jackson MB. Local and global ligand-induced changes in the structure of the GABAA receptor. Biochemistry. 2006;45:7013–7022. doi: 10.1021/bi060222v. [DOI] [PubMed] [Google Scholar]
- Muroi Y, Theusch CM, Czajkowski C, Jackson MB. Distinct structural changes in the GABAA receptor elicited by pentobarbital and GABA. Biophys J. 2009;96:499–509. doi: 10.1016/j.bpj.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padgett CL, Hanek AP, Lester HA, Dougherty DA, Lummis SC. Unnatural amino acid mutagenesis of the GABAA receptor binding site residues reveals a novel cation-π interaction between GABA and β2Tyr97. J Neurosci. 2007;27:886–892. doi: 10.1523/JNEUROSCI.4791-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit Y, Grosman C. Block of muscle nicotinic receptors by choline suggests that the activation and desensitization gates act as distinct molecular entities. J Gen Physiol. 2006;127:703–717. doi: 10.1085/jgp.200509437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart TG, Moss SJ, Xie X, Huganir RL. GABAA receptors are differentially sensitive to zinc: dependence on subunit composition. Br J Pharmacol. 1991;103:1837–1839. doi: 10.1111/j.1476-5381.1991.tb12337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach JH, Akk G. Modulation of GABAA receptor channel gating by pentobarbital. J Physiol. 2001;537:715–733. doi: 10.1111/j.1469-7793.2001.00715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Bracamontes J, Zorumski C, Weiss DS, Steinbach JH. Bicuculline and gabazine are allosteric inhibitors of channel opening of the GABAA receptor. J Neurosci. 1997;17:625–634. doi: 10.1523/JNEUROSCI.17-02-00625.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.