

Abstract
BACKGROUND AND PURPOSE
The highly lipophilic acyl-sulphonamides L-798106 and L-826266 showed surprisingly slow antagonism of the prostanoid EP3 receptor system in guinea-pig aorta. Roles of affinity and lipophilicity in the onset kinetics of these and other prostanoid ligands were investigated.
EXPERIMENTAL APPROACH
Antagonist selectivity was assessed using a panel of human recombinant prostanoid receptor-fluorimetric imaging plate reader assays. Potencies/affinities and onset half-times of agonists and antagonists were obtained on guinea-pig-isolated aorta and vas deferens. n-Octanol-water partition coefficients were predicted.
KEY RESULTS
L-798106, L-826266 and the less lipophilic congener (DG)-3ap appear to behave as selective, competitive-reversible EP3 antagonists. For ligands of low to moderate lipophilicity, potency increments for EP3 and TP (thromboxane-like) agonism on guinea-pig aorta (above pEC50 of 8.0) were associated with progressively longer onset half-times; similar trends were found for TP and histamine H1 antagonism above a pA2 limit of 8.0. In contrast, L-798106 (EP3), L-826266 (EP3, TP) and the lipophilic H1 antagonists astemizole and terfenadine exhibited very slow onset rates despite their moderate affinities; (DG)-3ap (EP3) had a faster onset. Agonism and antagonism on the vas deferens EP3 system were overall much faster, although trends were similar.
CONCLUSIONS AND IMPLICATIONS
High affinity and high liphophilicity may contribute to the slow onsets of prostanoid ligands in some isolated smooth muscle preparations. Both relationships are explicable by tissue disposition under the limited diffusion model. EP3 antagonists used as research tools should have moderate lipophilicity. The influence of lipophilicity on the potential clinical use of EP3 antagonists is discussed.
Keywords: isolated smooth muscle preparation, FLIPR assay, antagonist inhibition-curve protocol, prostanoid EP3 receptor, prostanoid TP receptor, L-798106, L-826266, BMS-180291, lipophilicity, limited diffusion model, plasmalemmal diffusion microkinetic model
Introduction
Products of fatty acid cyclooxygenase, such as prostaglandin E2 (PGE2) (Figure 1) and thromboxane A2, activate specific prostanoid receptors to subserve a variety of physiological and pathological functions (Smyth et al., 2009). The gradual emergence of these G-protein-coupled receptors, of which there are nine (Alexander et al., 2009), stimulated searches for selective antagonists in expectation of therapeutic use (Jones et al., 2009). In our investigations of the utility of these agents as research tools, high-affinity antagonists, for example the TP (thromboxane) antagonist BMS-180291 (pA2= 9.8, Zhang et al., 1996) characteristically showed slower onset of antagonism than less potent compounds on isolated smooth muscle preparations. We were therefore surprised to find that L-798106 (Gallant et al., 2002; Belley et al., 2005), an EP3 antagonist with pA2 of 7.5–8.0, slowly inhibited established contraction of guinea-pig aorta to the PGE2 analogue sulprostone; the related antagonist L-826266 (Schlemper et al., 2005) had an even slower onset. L-798106 and L-826266 are acyl-sulphonamides (Figure 1) with considerably higher lipophilicity than the majority of prostanoid ligands. We hypothesized that this high lipophilicity underpins the slow EP3 receptor antagonism and that the association is in harmony with the ‘limited diffusion model’ (Rang, 1966; Colquhoun et al., 1972; Colquhoun and Ritchie, 1972). This model (Figure 9, upper right box) proposes that ligand sequestration processes in the plasma membranes of cells comprising a solid tissue can dramatically retard diffusion of a ligand through the extracellular fluid (e.c.f.), thereby slowing its rate of equilibration with the acceptor pool; partitioning into the lipid core of the cell membrane is one such process.
Figure 1.
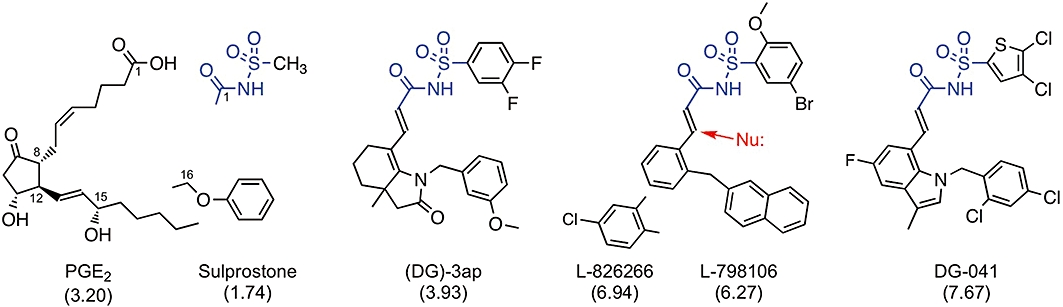
Structures of EP3 receptor ligands: prostaglandin E2 (PGE2) and its analogue sulprostone are agonists; the other compounds are antagonists. All compounds are weak acids: proton loss from carboxylate group or acyl-sulphonamide unit (blue). Predicted n-octanol-water partition coefficients (AlogP98) are shown in parentheses; differences between the antagonists are mainly due to the (lower) acyl moieties. The red arrow indicates potential covalent attack on the α, β-unsaturated amide group by a nucleophile (Nu:).
Figure 9.
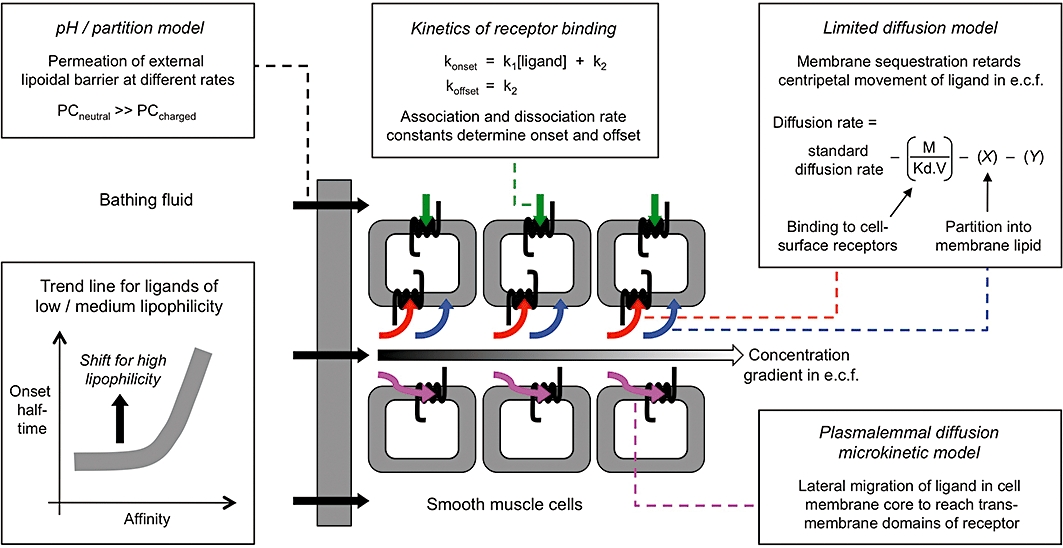
Processes potentially contributing to the kinetic profile of a receptor ligand in a multiple cell-layer tissue. Clockwise from upper left, boxes depict: (i) Passage of ligand through a lipoidal barrier surrounding the tissue mass (PC, permeability coefficient; see Pratt, 1990). (ii) Kinetics of ligand – receptor interaction. (iii) Cellular sequestration retarding movement of ligand through the extracellular fluid (e.c.f.) (limited diffusion model). For binding to cell-surface receptors, M, receptor density; Kd, equilibrium dissociation constant of ligand; V, volume of biophase. X represents a factor for partition into cell membrane lipid and Y a factor for any other sequestration process (e.g. active uptake). (iv) Access of ligand to the transmembrane domains of the receptor via lateral diffusion through the lipid core of the cell membrane (plasmalemmal diffusion microkinetic model). Processes (iii) and (iv) both tend to slow the build-up of ligand concentration in the centre of the tissue. The lower left box schematically shows the influence of ligand affinity and lipophilicity on onset rate based on data in Figure 8.
Experiments were therefore conducted with sets of agonists and antagonists on guinea-pig aorta to probe the relationships between onset rate (half-time) and potency (pEC50 or pA2) and onset rate and lipophilicity (n-octanol-water partition coefficient). Agonists were included in the study because we had observed that suprostone's action on guinea-pig aorta was slower than those of less potent EP3 agonists, which accorded with an earlier finding that highly potent TP agonists (e.g. EP-171) exhibited particularly slow onsets on vascular and respiratory smooth muscle preparations (Jones et al., 1989). Use of guinea-pig aorta allowed several receptors (EP3, TP, histamine H1) located on smooth cells to be compared and also ensured accurate measurement of slow agonism or slow antagonism as contractions were not subject to fade. The EP3 assay involved contractile synergism between EP3 agonist and the α1-adrenoceptor agonist phenylephrine (PE) (Jones and Woodward, 2010). We also performed a parallel investigation on the EP3 system in guinea-pig vas deferens, where the kinetics of both agonists and antagonists was much faster. Activation of EP3 receptors on sympathetic varicosities in the vas deferens suppresses transmitter release and this is detected as an inhibition of the twitch response induced by electrical field stimulation (EFS) (Ito and Tajima, 1979; Coleman et al., 1987; Lawrence et al., 1992).
It was deemed important to examine an acyl-sulphonamide EP3 antagonist of lower lipophilicity; (DG)-3ap (Figure 1) was synthesized for this purpose based on structure – activity relationships in O'Connell et al. (2009). In view of the limited information on the pharmacological properties of the EP3 antagonists used, their specificity and competitive behaviour were examined using Schild protocols in Ca2+-flux [fluorimetric imaging plate reader (FLIPR)] assays involving human recombinant (rc-) prostanoid receptors (Woodward et al., 2003; Matias et al., 2004) and in the guinea-pig aorta and vas deferens assays.
Our study shows that the slow onsets of particular prostanoid receptor agonists/antagonists observed in a multiple cell-layer tissue may be due to either their high affinity for the receptor or their high lipophilicity. The data trends may be explained by membrane sequestration arising from receptor binding and lipid partition, respectively, under the limited diffusion model. However, other models describing tissue disposition of a receptor ligand may be relevant, for example, the plasmalemmal diffusion microkinetic model as applied to slow-acting β2-adrenoceptor agonists (Anderson, 1993). The work emphasizes the need for caution in using highly lipophilic prostanoid antagonists as research tools because pA2 values may be distorted and receptor identification compromised. In addition, there are important implications for the clinical utility of prostanoid antagonists given the difficulties in development typically associated with lipophilic as opposed to more water-soluble analogues.
Methods
Recombinant prostanoid receptor-Ca2+ flux (FLIPR) assays
Recombinant prostanoid receptor-Ca2+ flux (FLIPR) assays were performed as described previously (Woodward et al., 2003; 2007; Matias et al., 2004). The human receptor and appropriate chimeric G protein (Gqs, Gqi) were stably transfected into HEK-293 EBNA cells, allowing activation of Gs-coupled (DP1, EP2, EP4, IP) and Gi-coupled (EP1, EP3, FP, TP) receptors to be measured as a Ca2+ signal.
Isolated smooth muscle assays
Descending thoracic aorta and vas deferens were dissected from male Dunkin-Hartley guinea-pigs (400–500 g) after they had been killed by exposure to CO2 in compliance with Schedule 1 of the UK Animals (Scientific Procedures) Act 1986. Aorta preparations (contiguous rings 4 mm in length, n= 4, intact endothelium) were suspended between L-shaped stainless steel wire holders in conventional 10 mL tissue baths; vas deferens preparations (25 mm proximal sections from left and right sides) were suspended using cotton thread. Isometric tension was recorded with Grass FT03 transducers linked to an AD Instruments PowerLab pre-amplifier-digitizer/Dell computer system. The bathing fluid was Krebs–Henseleit solution (118 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 1.18 mM KH2PO4, 25 mM NaHCO3, 10 mM glucose) aerated with 95% O2/5% CO2, maintained at 37°C, and containing 1 µM indomethacin to inhibit fatty acid cyclooxygenase activity. Resting tension was adjusted to 1.2 g for aorta and 0.5 g for vas deferens. EFS trains (50–60 V, 0.5 ms duration, 10 Hz for 1 s) were applied to vas deferens preparations every 20 or 40 s via platinum ring electrodes; EFS trains at 20 s intervals were stopped for 3–4 min during a 10 min rest period to prevent run-down of the EFS response.
Experimental protocols
Antagonists in FLIPR assays
Antagonist/vehicle (50 µL) was added to the well contents (100 µL), followed 4.5 min later by agonist (50 µL). The agonist concentration range was 0.01 nM to 10 µM, except for the TP receptor assay, which was 10-fold lower. The peak increase in fluorescence intensity was measured for each well. In each of three experiments, concentration–response data (triplicate wells) were obtained for either the standard agonist and two/three test agonists or the standard agonist in the presence of vehicle and antagonist.
Agonists in isolated smooth muscle assays
Initially, each smooth muscle preparation was treated cumulatively with the following agents: aorta: U-46619 (10–30 nM), histamine (1–5 µM) and PE (1–5 µM) for TP, H1 and α1 ligand studies respectively; PE (1–5 µM) followed by PGE2 (1–5 nM) primed by PE for EP3 agonist/EP3 antagonist studies; PE (1–5 µM) followed by U-46619 (10–30 nM) for TP agonist/EP3 antagonist studies; vas deferens: PGE2 (1–10 nM).
A cumulative log concentration–response curve was obtained for each agonist in the first instance. For a particularly slow-acting agonist, a single + maximum dose protocol (Figure 2A; Jones et al., 1989) was used to define the mid-range of the log concentration–response curve. Individual preparations were challenged with a single concentration of agonist until steady-state had been reached followed by a high concentration of standard agonist to establish the maximum response (E100). T50, the onset half-time corresponding to 50% maximal agonist response (E50), was estimated for each agonist using single concentrations inducing E30–E70. EP3 agonist activity on guinea-pig aorta was measured under priming with PE (∼E20); in each experiment PE concentration was reduced slightly (e.g. 1.0 to 0.85 µM) on all preparations in the successive sequence to maintain a consistent response over time. In addition, a log concentration–response curve for the standard agonist 17-phenyl PGE2 was obtained in the first and second sequences (S1 and S2) on separate aorta preparations in each experiment. On guinea-pig vas deferens, agonist was added exactly 5 s before an EFS train.
Figure 2.
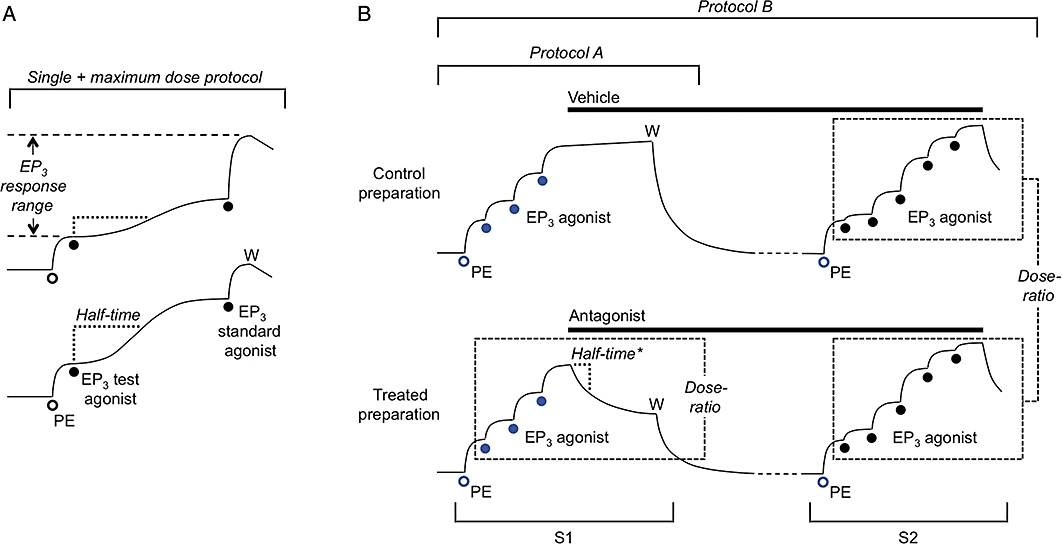
Cartoon showing protocols for isolated smooth muscle assays: in each case for guinea-pig aorta EP3 assay involving priming with phenylephrine (PE, open circles) in each cumulative agonist sequence. (A) Single + maximum dose protocol used for a slow-acting agonist. pEC50 for test agonist was derived from the mid-portion of its log concentration–response curve, which was obtained by applying single doses to individual preparations followed by a maximal dose of the standard agonist. Individual half-times were used to calculate an onset half-time corresponding to E50 (T50). (B) Antagonist protocols. Protocol A is a type of inhibition-curve protocol: the steady-state inhibition was converted into a dose-ratio (and corresponding pA2 value) using the three (EP3) agonist responses in sequence 1 (S1); a corresponding onset half-time (half-time*) was calculated from a dose-ratio – time plot. Protocol B is a Schild protocol: following washout of S1 primer/agonist (W), vehicle and antagonist treatments were continued before construction of agonist curves in S2; between-preparations dose-ratios were obtained for Schild analysis. Two additional preparations were usually treated with different concentrations of antagonist. The blue symbols indicate responses used in repeated-measures 2-factor anova (aorta EP3 assay only) to investigate matching of preparations in S1 and the effect of antagonist treatment on the priming response in S2.
Antagonists in isolated smooth muscle assays
Initial agonist sequences and standard agonists employed in the agonist studies were also used in corresponding antagonist experiments. All antagonists were examined under protocol A, a type of inhibition-curve protocol (Craig, 1993; Lazareno and Birdsall, 1993; Leff and Dougall, 1993), which is illustrated for the EP3 assay on guinea-pig aorta in Figure 2B. Three doses of standard agonist were applied cumulatively to each preparation with the highest inducing ∼E80; a single concentration of antagonist was then applied until steady-state inhibition was reached. Each antagonist was studied over a narrow range of concentration chosen to afford final dose-ratios between 2.5 and 10. On guinea-pig vas deferens, antagonist was added 10 s before the EFS train (40 s intervals).
Protocol B, an extension of protocol A (Figure 2B), involved treatment of preparations with vehicle or antagonist before construction of agonist concentration–response curves (Schild protocol). Antagonist treatment was extended beyond S1 up to a total of 180 min (with replacement at 15–25 min intervals); cumulative agonist sequences were applied in S2. Protocol B was used to estimate the pA2 value for a very slow-onset antagonist and in some cases to compare the pA2 value with that obtained under protocol A.
Prediction of physicochemical parameters
The Pipeline Pilot program (Accelrys Inc., San Diego, CA, USA) was used to predict the pKa for the most acidic/basic species of a molecule and the n-octanol-water partition coefficient for its unionized species (AlogP98) (see review by Mannhold et al., 2008).
Data analysis
Fluorimetric imaging plate reader responses were normalized to the response to 1 µM of the standard agonist (100 nM for the TP receptor) in each experiment. Contractile responses of aorta were measured from the resting level. Inhibitory (EP3) responses on vas deferens were expressed as a percentage of the two preceding control EFS twitch responses. Log concentration–response data were fitted by a 4-parameter (variable-slope) sigmoidal curve with constraint of the low-concentration asymptote to the resting, priming or established contraction level as appropriate (GraphPad Prism software, La Jolla, CA, USA).
Agonists
pEC50 values were obtained from sigmoidal fitting of log concentration–response curves. Linear regression of half-time versus % maximal response (8–12 data points) afforded a half-time corresponding to E50 (T50).
Antagonists
Protocol A
Inhibition (%) caused by each concentration of antagonist was measured at regular intervals and corrected for any change in the vehicle-treated preparation. These values were converted to dose-ratios using a sigmoidal curve fitted to the three S1 agonist data points. An onset half-time was then estimated from a dose-ratio – time plot. Linear regression of a half-time – dose-ratio plot for the set of antagonist concentrations afforded an inhibition half-time corresponding to a dose-ratio of 4 (TDR4). A mean pA2 value was calculated from the corresponding set of antagonist concentrations/steady-state dose-ratios using the Gaddum–Schild equation, pA2= log(dose-ratio – 1) + log[antagonist]. A predicted inhibition curve for (DG)-3ap on guinea-pig aorta was derived from the modified Cheng–Prussof equation (Leff and Dougall, 1993) using a pA2 of 7.92 obtained under protocol B (Table 3) and nH of 0.85 for 17-phenyl PGE2 (mean value in the set of experiments).
Table 3.
Affinities and onset rates of receptor antagonists on isolated smooth muscle preparations with corresponding physicochemical data
| pA2 | ||||||
|---|---|---|---|---|---|---|
| Antagonist | Protocol A | Protocol B | TDR4 (min) | pKa | AlogP98 | MW |
| Guinea-pig aorta | ||||||
| EP3 receptor | ||||||
| (DG)-3ap | (6.73–8.04)a | 7.92 ± 0.07 | 13.0 | 4.21b | 3.93 | 516 |
| L-798106 | Not obtainable | 7.99 ± 0.14 | See Figure 8 | 4.21b | 6.27 | 535 |
| L-826266 | Not obtainable | 7.71 ± 0.07 | See Figure 8 | 4.21b | 6.94 | 571 |
| TP receptor | ||||||
| ICI-192605 | Not obtainable | 10.24 ± 0.03c | See Figure 8 | 4.71 | 4.63 | 403 |
| BMS-180291 | 9.77 ± 0.05 | 9.98 ± 0.07d | 63 | 4.66 | 3.89 | 440 |
| I-SAP | 9.15 ± 0.04 | – | 38 | 4.84 | 4.65 | 531 |
| SQ-29548 | 8.50 ± 0.10 | – | 13.5 | 4.24 | 2.28 | 387 |
| EP-092 | 8.38 ± 0.03 | – | 13.6 | 4.84 | 5.46 | 413 |
| EP-045 | 7.54 ± 0.08 | – | 11.9 | 4.84 | 3.89 | 383 |
| MK-0524e | 6.68 ± 0.04 | – | 12.0 | 5.44 | 4.88 | 436 |
| BW-A868Ce | 4.97 ± 0.05 | – | 13.3 | 4.71 | 4.03 | 458 |
| L-826266 | Not obtainable | 7.92 ± 0.06f | See Figure 8 | 4.21b | 6.94 | 571 |
| H1 receptor | ||||||
| Doxepin | 9.64 ± 0.05 | 9.78 ± 0.05f,g | 43 | 8.98 | 3.91 | 279 |
| (+)-Chlorpheniramine | 9.05 ± 0.10 | – | 16.3 | 9.25 | 3.70 | 275 |
| Diphenhydramine | 8.21 ± 0.06 | – | 5.5 | 8.98 | 3.38 | 255 |
| Terfenadine | Not obtainable | 8.17 ± 0.09f,h | See Figure 8 | 8.59 | 6.50 | 472 |
| Astemizole | Not obtainable | 7.94 ± 0.12f,h | See Figure 8 | 6.71 | 5.21 | 458 |
| BMY-7378i | 5.95 ± 0.04 | – | 3.5 | 8.52 | 3.19 | 385 |
| Atropine | 5.38 ± 0.03 | – | 3.3 | 9.47 | 1.72 | 289 |
| Guinea-pig vas deferens | ||||||
| EP3 receptor | ||||||
| (DG)-3ap | 7.99 ± 0.11 | 7.84 ± 0.05 | 1.1 | 4.21b | 3.93 | 516 |
| L-798106 | Not obtainable | 7.53 ± 0.10f,j | See Figure 8 | 4.21b | 6.27 | 535 |
| L-826266 | Not obtainable | 7.25 ± 0.04f | See Figure 8 | 4.21b | 6.94 | 571 |
Measurement of pA2 values (±SEM): protocol A involves an inhibition-curve design with application of single antagonist concentrations to individual preparations (n= 6–12); protocol B involves antagonist pretreatment/Schild analysis (n= 4–5). TDR4 is the half-time corresponding to a dose-ratio of 4 (protocol A); some values were not obtainable owing to very slow rate of antagonism. Standard agonist: aorta/EP3, 17-phenyl PGE2 under PE priming (300 nM BMS-180291 routinely present); aorta/TP, U-46619; aorta/H1, histamine; aorta/α1, PE; vas deferens/EP3, PGE2.
Acidity constants (pKa) and n-octanol/water partition coefficients for the unionized species (AlogP98) were calculated using Pipeline Pilot (version 7.5.2) software. H1 and α1 antagonists are all amine bases. Values of AlogP98 > 5.0 and MW > 450 are shown in bold.
Range for 30–3000 nM.
Acyl-sulphonamide; other prostanoid antagonists are C1-carboxylic acids.
pA2= 9.2/9.1 on human umilical uterus/vein (Senchyna and Crankshaw, 1996; Daray et al., 2003).
pA2= 9.8 on guinea-pig aorta (Zhang et al., 1996).
Potent DP1 antagonist (Giles et al., 1989; Sturino et al., 2007).
Single-point estimate.
pA2= 9.72 on guinea-pig ileum (Figueiredo et al., 1990).
Unsurmountable block; see text.
Selective α1D antagonist (Saussy et al., 1994).
pA2= 7.48 on guinea-pig vas deferens using sulprostone as agonist (Clarke et al., 2004).
MW, molecular weight; PGE2, prostaglandin E2; TDR4, onset half-time for an antagonist corresponding to a dose-ratio of 4.
Protocol B
Responses on aorta were corrected to the maximum response to PE (30 µM) obtained 40 min after S2 washout. Between-preparations dose-ratios were obtained from S2 log concentration–response curves; a corresponding pA2 value was derived from either the Gaddum–Schild equation (single-point estimate) or linear regression of a conventional Schild plot, log(dose-ratio – 1) versus log[antagonist]. For Schild's analysis, agonist E100 values under antagonist treatments derived from sigmoidal fitting were analysed by a test for linear trend allied to 1-factor anova (GraphPad Prism). For EP3 agonist/EP3 antagonist data, time-matched mean responses for vehicle and antagonist treatments (blue symbols in Figure 2) were compared by planned orthogonal contrasts (planned contrasts) allied to repeated-measures 2-factor anova (SuperANOVA software, Abacus Concepts, Berkeley, CA, USA), a robust method of comparing multiple means (Glass and Hopkins, 1996). Errors associated with a mean value are SEM unless stated otherwise; the significance level was set at P= 0.05.
Chemicals and stock solutions
A 10 mM stock solution of each compound was prepared in ethanol unless stated otherwise. Sources (alphabetical) of prostanoid ligands: Allergan, USA: cicaprost, 3,7-dithia PGE1, L-902688 (1-decarboxy-11-deoxy-16,16-difluoro-16-phenyl-ω-tetranor-1-(5-tetrazolo) PGE1). Bristol-Myers Squibb, USA: BMS-180291 ([1S-(exo,exo)]-2-[[3-[4-[(pentylamino)carbonyl]-2-oxazolyl]-7-oxabicyclo[2.2.1]hept-2-yl]methyl]-benzenepropanoic acid, Ifetroban). Cayman Chemical, USA: BW-245C (((4S)-(3-[(3R,S)-3-cyclohexyl-3-hydroxypropyl]-2,5-dioxo)-4-imidazolidine-heptanoic acid), carbacyclin, (+)-cloprostenol, 16,16-dimethyl PGE2, 16,16-dimethyl PGF2α, I-BOP ([1S-(1α,2β(5Z),3α(1E,3S),4α]-7-[3-(3-hydroxy-4-(4′-iodophenoxy)-1-butenyl)-7-oxabicyclo[2.2.1]heptan-2-yl]-5-heptenoic acid, 0.2 mM, ethanol), I-SAP (7-[(1R,2S,3S,5R)-6,6-dimethyl-3-(4-iodobenzenesulfonylamino) bicyclo[3.1.1]-hept-2-yl]-5(Z)-heptenoic acid, 1 mM, ethanol), latanoprost-free acid, MK-0524 ((R)-2-(4-(chlorobenzyl)-7-fluoro-5-(methylsulphonyl)-1,2,3,4-tetrahydrocyclopenta[b]indolyl-3-yl) acetic acid), PGE2, PGF2α, 17-phenyl-ω-trinor PGE2, 17-phenyl-ω-trinor PGF2α, RO-1138452 (4,5-dihydro-N-[[4-(1-methylethoxy)phenyl)]methyl]-phenyl]-1H-imidazol-2-amine, CAY-10441), SQ-29548 ([1S-[1α,2α(Z),3α,4α]]-7-[3-[[2-[(phenylamino)carbonyl]hydrazino]methyl]-7-oxabicyclo[2.2.1]hept-2-yl]-5-heptenoic acid), sulprostone, U-46619 (15S-hydroxy-11α,9α-epoxymethano-prosta-5Z,13E-dienoic acid). Edinburgh University, UK: EP-045 ((+/−)-9α,11α-ethano-13-(N-phenylcarbamoyl)-hydrazono-ω-heptanor-prosta-5Z-enoic acid), EP-092 ((+/−)-9α,11α-ethano-13-methyl-13-(N-phenylthiocarbamoyl)-hydrazono-ω-heptanor-prosta-5Z-enoic acid). Evotec, UK: ONO-DI-004 (17S-17,20-dimethyl-2,5-ethano-6-oxo PGE1), ONO-AE-248 (11,15-bis(O-methyl) PGE2). GlaxoSmithKline, UK: BW-A868C (3-[(2-cyclohexyl-2-hydroxyethyl)amino]-2,5-dioxo-1-(phenylmethyl)-4-imidazolidine-heptanoic acid, 20 mM. ethanol), L-798106 ([(2E)-N-[(5-bromo-2-methoxyphenyl)-sulphonyl]-3-[2-(2-naphthylmethyl)phenyl]acrylamide sodium salt, 10 mM, DMSO). Target Molecules, UK: (DG)-3ap (1-(3-methoxybenzyl)-3a-methyl-[3,3a,4,5,6-hexahydroindol-2-one-7-acrylic acid, 3,4-difluoro-benzenesulphonamide, 10 mM, DMSO), L-826266 ([(2E)-N-[(5-bromo-2-methoxyphenyl)-sulphonyl]-3-[5-chloro-2-(2-naphthyl methyl) phenyl]acrylamide, 10 mM, DMSO). Tocris Bioscience, UK: ICI-192605 ([2S,4S,5R]-6-(2-o-chlorophenyl-4-o-hydroxyphenyl-1,3-dioxan-5-yl)-hex-4Z-enoic acid).
Other compounds (typically 10 mM stock): Fluka Chemical, Switzerland: phenylephrine HCl (water). Sigma-Aldrich, USA: astemizole (DMSO), atropine sulphate (3 mM, water), BMY-7378 (8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4.5]decane-7,9-dione dihydrochloride; water) (+)-chlorpheniramine maleate (3 mM in water), histamine HCl (water), indomethacin (20 mM, ethanol), terfenadine (1 mM, DMSO). Tocris-Cookson, UK: diphenhydramine HCl (water), doxepin HCl (10 mM, DMSO).
Dilutions were prepared with 0.9% NaCl solution (saline), except for L-798106, L-826266 (DMSO) and terfenadine (DMSO/saline 30:70). (DG)-3ap, EP-045, EP-092, ICI-192605, I-SAP and U-46619 were solubilized with a trace of NaHCO3 on first dilution; similarly for astemizole with 0.002 M HCl. L-798106 and L-826266 were added (in 10 µL) to the tissue bath with a swirling motion over 10 s to prevent local precipitation of solute. The aqueous solubility of L-826266, the most lipophilic compound used, was examined at 600 nm (10 mm pathlength) in a conventional visible spectrophotometer. No light-scattering signal was obtained following dilution of a 3 mM DMSO solution of L-826266 to 3 µM with distilled water (pH 5.4) at 20°C; a threshold signal was obtained for 10 mM/10 µM dilution and larger signals (and obvious opalescence) were found for 10 mM/20–50 µM dilutions. L-826266 (10 µM) in Krebs–Henseleit solution pre-aerated with 95% O2/5% CO2 (pH 7.35) at 37°C in a sealed cuvette showed no light-scattering signal over a period of 3 h.
Nomenclature
Nomenclature for prostanoid and other receptors conforms to the British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2009).
Results
Profiles of EP3 antagonists in human recombinant prostanoid receptor-Ca2+ flux (FLIPR) assays
pEC50 values for standard agonists in the panel of human recombinant receptor FLIPR assays ranged between 7.90 and 9.54, except for carbacyclin (pEC50= 7.18) in the IP (prostacyclin receptor) assay (Table 1). However, this assay had acceptable agonist sensitivity as shown by the higher potency (8.39) of another prostacyclin analogue cicaprost. L-826266 at 10 µM (Schild protocol, 4.5 min treatment) produced a large right-shift of the PGE2 curve in the EP3 assay (Figure 3); there was also modest block of EP4 and TP receptors, but minimal block of DP1, EP1, EP2, FP and IP receptors (Table 1). The antagonist selectivity of (DG)-3ap was similar, except that its EP1 affinity was higher and its TP affinity was minimal. Antagonism appeared surmountable in all assays exhibiting a measurable right-shift (mean dose-ratio > 4.0, pA2 > 5.5). The reduction of Ca2+ flux seen with high concentrations of PGE2 in the EP4 assay may be due to agonist desensitization. For completeness, the pKi value for L-798106 in a similar human rc-EP3 receptor-FLIPR assay (Jugus et al., 2009) has been included in Table 1.
Table 1.
Affinities of EP3 antagonists in human recombinant prostanoid receptor/Ca2+ flux (FLIPR) assays
| Human prostanoid receptor | ||||||||
|---|---|---|---|---|---|---|---|---|
| DP1 | EP1 | EP2 | EP3 | EP4 | FP | IP | TP | |
| Standard agonist | BW-245C (7.90 ± 0.07) | PGE2(9.16 ± 0.10) | PGE2(8.27 ± 0.03) | PGE2(8.90 ± 0.13) | PGE2(9.29 ± 0.04) | 17-Phenyl PGF2α(8.05 ± 0.08) | Carbacyclin (7.18 ± 0.30) | U-46619 (9.54 ± 0.21) |
| Antagonist | pA2 | |||||||
| (DG)-3ap | <5.0 | 6.02 | <5.0 | 8.30 | 5.67 | <5.0 | 5.06 | <5.0 |
| L-826266 | 5.34 | 5.14 | <5.0 | 7.97 | 5.74 | <5.0 | <5.0 | 6.39 |
| pKi | ||||||||
| L-798106 | – | – | – | 7.77 | – | – | – | – |
Prostanoid receptor and chimeric G-protein were transfected into HEK-293 EBNA cells to facilitate Ca2+ signalling (FLIPR assay). Standard agonists: pEC50 values in parentheses (mean ± SEM, n= 6). pA2 values are single point estimates (10 µM antagonist) under Schild protocol (n= 3). Lower section: FLIPR assay involving inhibition of E80 response to PGE2 in U2OS carrier cell; pKi calculated using the unmodified Cheng–Prusoff equation (Jugus et al., 2009).
Affinities for the EP3 receptor are shown in bold type.
FLIPR, fluorimetric imaging plate reader.
Figure 3.
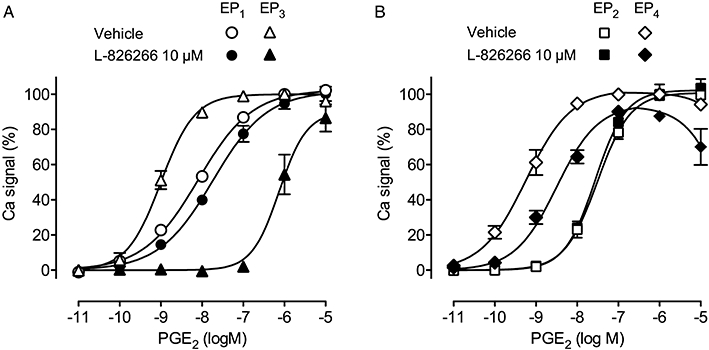
Antagonism of prostaglandin E2 (PGE2)-induced Ca2+ flux by L-826266 (10 µM) in human recombinant (A) EP1/EP3 and (B) EP2/EP4 receptor systems. Each receptor was co-transfected with chimeric G-protein into HEK-293 EBNA cells; Ca2+ flux was measured by Fluo-4 fluorescence (fluorimetric imaging plate reader assay). Responses were normalized to the peak signal for 1 µM PGE2. Vertical bars show SEM (n= 3).
Potencies and onset rates of EP3 agonists on guinea-pig aorta
Potencies (pEC50) and onset rates (T50) for a set of EP3 agonists were measured on guinea-pig aorta. EP3 agonist activity was measured under priming with PE (0.3–1.0 µM) in the presence of the TP antagonist BMS-180291 (300 nM; expected dose-ratio = 2860) as described by Jones and Woodward (2010) (cf. Figure 4A). 17-Phenyl PGE2 was chosen as the EP3 standard agonist owing to its moderately high potency (pEC50= 8.41) and its fairly fast onset and offset. The data are presented in Table 2, with a corresponding correlation plot in Figure 8A. Higher potencies (pEC50 > 8.0) were associated with progressively slower onset rates. Indeed, it was necessary to use a single + maximum dose protocol (Figure 2A) to obtain pEC50 values for the slowest-acting agonists, sulprostone, 16,16-dimethyl PGE2 (DM PGE2) and 11-deoxy-16,16-dimethyl PGE2 (DX-DM PGE2).
Figure 4.
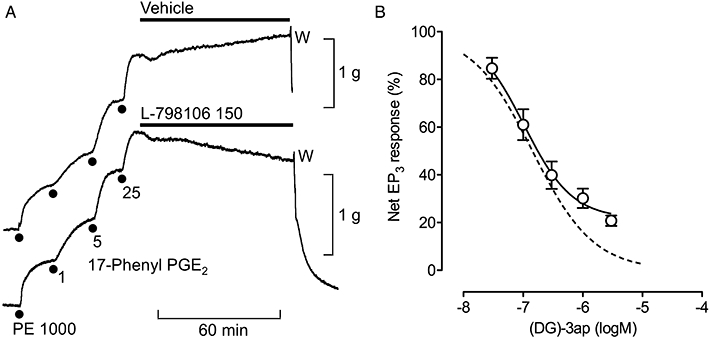
Inhibitory profiles of EP3 antagonists on guinea-pig aorta. Using protocol A, contraction was established with 25 nM 17-phenyl PGE2 under phenylephrine (PE) priming in the presence of the TP antagonist BMS-180291 (300 nM). (A) Slowly developing antagonism by L-798106; the transient initial fall in tension in both records is a typical effect of the vehicle (14 mM DMSO). Concentrations in nM; W, wash. (B) Non-cumulative log concentration–inhibition curve for (DG)-3ap. The broken line is a predicted curve for complete inhibition of the net EP3 response corresponding to a pA2 of 7.92 for (DG)-3ap (protocol B) and nH of 0.85 for 17-phenyl PGE2. Vertical bars show SEM (n= 4–5).
Table 2.
Potencies and onset rates of prostanoid receptor agonists on isolated smooth muscle preparations with corresponding physicochemical data
| Agonist | pEC50 | T50 (min) | pKa | AlogP98 | MW |
|---|---|---|---|---|---|
| Guinea-pig aorta | |||||
| EP3 receptor | |||||
| DM PGE2 | 9.74 ± 0.05a | 14.0 | 4.84 | 3.79 | 380 |
| Sulprostone | 9.55 ± 0.06a | 15.1 | 4.41b | 1.74 | 466 |
| DX-DM PGE2 | 9.21 ± 0.06a | 11.5 | 4.84 | 4.83 | 364 |
| PGE2 | 8.79 ± 0.09c | – | 4.84 | 3.20 | 352 |
| 17-Phenyl PGE2 | 8.41 ± 0.04 | 5.2 | 4.84 | 3.32 | 386 |
| Carbacyclin | 7.84 ± 0.09d | 5.2 | 4.86 | 4.15 | 350 |
| 3,7-Dithia PGE1 | 7.39 ± 0.05 | 3.1 | 3.89 | 2.25 | 386 |
| (+)-Cloprostenol | 7.28 ± 0.09 | 5.3 | 4.84 | 3.04 | 423 |
| PGF2α | 7.05 ± 0.11 | 2.5 | 4.84 | 2.98 | 354 |
| ONO-AE-248 | 6.78 ± 0.10 | 3.7 | 4.84 | 4.02 | 380 |
| Latanoprost-FA | 5.22 ± 0.07 | 2.7 | 4.84 | 3.41 | 390 |
| ONO-DI-004 | <5.4 | 3.4 | 5.24 | 3.48 | 436 |
| TP receptor | |||||
| I-BOP | 9.71 ± 0.09a | 52 | 4.84 | 4.30 | 512 |
| DX-DM PGE2 | 8.19 ± 0.10 | 12.7 | 4.84 | 4.83 | 364 |
| U-46619 | 7.89 ± 0.05 | 9.1 | 4.84 | 4.14 | 350 |
| DM PGF2α | 6.70 ± 0.10 | 9.5 | 4.84 | 3.58 | 382 |
| PGF2α | 6.11 ± 0.06 | 7.7 | 4.84 | 2.98 | 354 |
| Latanoprost-FA | 5.03 ± 0.03 | 6.3 | 4.84 | 3.41 | 390 |
| Guinea-pig trachea | |||||
| TP receptore | |||||
| EP-171 | 10.24 ± 0.14a | 164 | 4.84 | 3.93 | 404 |
| EP-031 | 9.06 ± 0.18a | 33 | 4.84 | 5.17 | 402 |
| DX-CP PGF2α | 8.75 ± 0.16 | 16.2 | 4.84 | 4.33 | 409 |
| U-46619 | 8.23 ± 0.15 | 7.4 | 4.84 | 4.14 | 350 |
| ICI-79939 | 7.69 ± 0.12 | 8.8 | 4.84 | 2.58 | 408 |
| 12,15-ent EP-171f | 7.55 ± 0.16 | 6.5 | 4.84 | 3.93 | 404 |
| DM PGF2α | 7.32 ± 0.14 | 9.1 | 4.84 | 3.58 | 382 |
| PGF2α | 6.20 ± 0.13 | 7.8 | 4.84 | 2.98 | 354 |
| Guinea-pig vas deferens | |||||
| EP3 receptor | |||||
| DM PGE2 | 9.32 ± 0.06 | 1.05 | 4.84 | 3.79 | 380 |
| Sulprostone | 9.30 ± 0.07 | 0.83 | 4.41b | 1.74 | 466 |
| DX-DM PGE2 | 8.50 ± 0.05 | 0.80 | 4.84 | 4.83 | 364 |
| PGE2 | 8.25 ± 0.08 | ∼0.38 | 4.84 | 3.20 | 352 |
| 17-Phenyl PGE2 | 7.56 ± 0.06 | <0.4 | 4.84 | 3.32 | 386 |
| ONO-AE-248 | 6.39 ± 0.08 | <0.4 | 4.84 | 4.02 | 380 |
| 3.7-Dithia PGE1 | 6.20 | <0.4 | 3.89 | 2.25 | 386 |
| ONO-DI-004 | <5.4 | – | 5.24 | 3.48 | 436 |
Agonists are listed according to potency (mean ± SEM) for each tissue/receptor system; n= 4–5; larger for standard agonists (underlined). Antagonists routinely present: aorta/EP3, 300 nM BMS-180291; aorta/TP, 1 µM (DG)-3ap; trachea/TP, 30 µM SC-19220.
Acidity constants (pKa) and n-octanol/water partition coefficients for the unionized species (AlogP98) were calculated using Pipeline Pilot (version 7.5.2) software. Experimental partition coefficient of the unionized ligand between n-octanol and water for PGE2= 2.90 by potentiometric titration (Avdeef et al., 1995). Values of AlogP98 > 5.0 and MW > 450 are shown in bold.
Single + maximum dose protocol.
C1-methylsulphonamide (see Figure 1); all other prostanoids are C1-carboxylic acids.
EP2 agonism suppresses maximum in some preparations.
IP antagonist RO-1138452 (1 µM) present.
Re-analysis of published data (Jones et al., 1989).
Isomer of EP-171 with inversion of configuration at C12 and C15 (Wilson et al., 1988).
CP, 16-p-chlorophenoxy; DM, 16,16-dimethyl; DX, 11-deoxy; FA, free acid. MW, molecular weight; PGE2, prostaglandin E2; T50, onset half-time for an agonist to achieve 50% maximal response.
Figure 8.
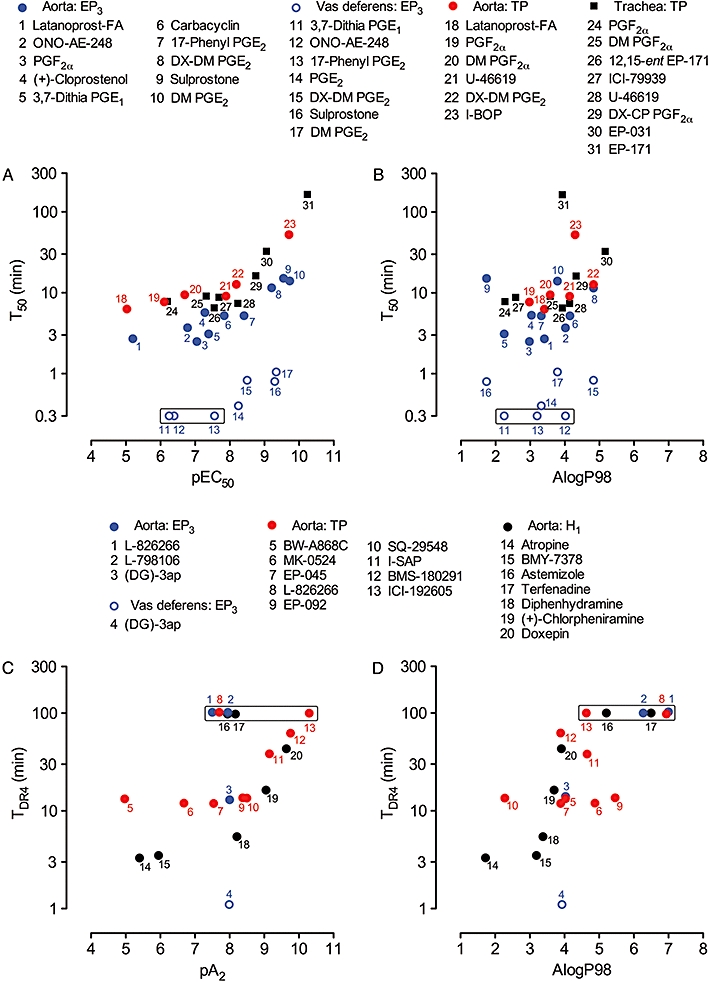
Correlations between onset rate and potency/affinity and between onset rate and lipophilicity on guinea-pig isolated smooth muscle preparations. Upper panels: plots of T50 versus (A) pEC50 and (B) AlogP98 for EP3 and TP agonists. Data points in each box indicate that responses were too rapid for accurate estimation of T50 (arbitrary location on 0.3 min level). Lower panels: plots of TDR4 versus (C) pA2 and (D) AlogP98 for EP3, TP and H1 antagonists. Data points in each box indicate that antagonism was too slow for TDR4 to be obtained (arbitrary location on 100 min level). L-798106 and L-826266 had slow onsets on the vas deferens EP3 system: see text. PGE2, prostaglandin E2; T50, onset half-time for an agonist to achieve 50% maximal response; TDR4, onset half-time for an antagonist corresponding to a dose-ratio of 4.
Only the prostacyclin analogue carbacyclin (1–500 nM) showed evidence of IP receptor-mediated relaxation; hence its activity was estimated in the presence of the IP receptor antagonist RO-1138452 (300 nM; expected dose-ratio = 75; Jones et al., 2006). Interference from EP1 receptors appeared unlikely in view of the weak agonism (20 ± 6% of E100 at 4 µM, n= 4) of the selective EP1 agonist ONO-DI-004 (Okada et al., 2000; Cao et al., 2002). The selective EP4 agonist L-902688 (Young et al., 2004; Foudi et al., 2008) induced only slight contraction at 1 µM (7 ± 2% of E100, n= 4); there was no evidence for a typical EP4 relaxant action. Hence any EP4 affinity of the antagonists studied (cf. human data in Table 1) is unlikely to be of any consequence.
Potencies and onset rates of TP agonists on guinea-pig aorta and trachea
Thromboxane agonists induced strong contraction of the aorta in the presence of the EP3 antagonist (DG)-3ap (1 µM; expected dose-ratio = 84). The standard agonist was U-46619 (pEC50= 7.89). Data are presented in Table 2, which also contains TP agonist data for guinea-pig trachea derived from re-analysis of published data (Jones et al., 1989). The standard agonist was U-46619 (pEC50= 8.23) and the sensitive EP1 contractile system present was blocked by SC-19220 (30 µM, expected dose-ratio = 30). The potency-rate trends (Figure 8A) are similar to that found for the EP3 system.
Single + maximum dose protocols were again required for the highest potency agonists: I-BOP (Sessa et al., 1990) on aorta and EP-171 and EP-031 on trachea. These prostanoids, together with DX-CP PGF2α and ICI-79939, contain a 16-phenoxy group (cf. sulprostone in Figure 1) with a para-halogen substituent. EP-171 was about 500 times more potent and had a much slower onset of action than its diastereoisomer 12,15-ent EP-171.
Potencies and onset rates of EP3 agonists on guinea-pig vas deferens
EP3 agonists rapidly inhibited twitch contractions of the guinea-pig vas deferens preparation induced by short EFS trains. PGE2 (standard agonist, pEC50= 8.25) had a T50 of ∼23 s (Table 2). The higher-potency agonists sulprostone, DM PGE2 and DX-DM PGE2, had slower onsets. Onset half-times for the least potent agonists, 17-phenyl PGE2, ONO-AE-248 and 3,7-dithia PGE1, were too fast to measure accurately (no effect on 5 s EFS twitch, >50% inhibition of 25 s twitch); T50 values are quoted as <0.4 min in Table 2 and data points are located along the 0.3 min level in Figure 8A. ONO-DI-004 at 0.1–4 µM had no effect on the twitch strength.
Affinities and onset rates of EP3 antagonists on guinea-pig aorta
To enable valid comparisons of the different receptor systems, antagonist affinity was estimated as a pA2 value and onset rate as TDR4 (Table 3); correlations are shown in Figure 8C. Descriptions in the text relate to use of inhibition-curve protocols (protocol A, Figure 2B) unless stated otherwise.
Under PE priming (DG)-3ap (30–3000 nM) inhibited established contraction of guinea-pig aorta induced by 17-phenyl PGE2; steady-state was achieved within 30–40 min. However, inhibition of the net EP3 response was incomplete and deviated from the predicted inhibition curve (see Methods) over the higher (DG)-3ap range (Figure 4B). Because pA2 showed a significant negative correlation with log[(DG)-3ap] (slope =−0.44, r2= 0.63, P < 0.001), the pA2 range for all individual estimations is given in Table 3. It is noteworthy that, under priming with 25 nM 17-phenyl PGE2, net contractions to PE, histamine or U-46619 were only reduced by 85–90% by concentrations of phentolamine (3 µM), diphenhydramine (3 µM) and BMS-180291 (300 nM) sufficient to abolish α1, H1 and TP agonist activity respectively. For purposes of correlation, a TDR4 of 14 min was derived for (DG)-3ap using the 30 and 100 nM data only.
The onset of EP3 antagonism by L-798106 (50–1000 nM, Figures 4A and 5A) was much slower than that for a similar concentration of (DG)-3ap; L-826266 (100–1000 nM) was even slower, with a particularly pronounced lag-phase (Figure 5A). Because TDR4 values could not be estimated, data points in Figure 8C are located along the 100 min level. The three EP3 antagonists had similar onset rates when the selective EP3 agonist ONO-AE-248 (1.5 µM) was substituted for 17-phenyl PGE2 in protocol A (data not shown).
Figure 5.
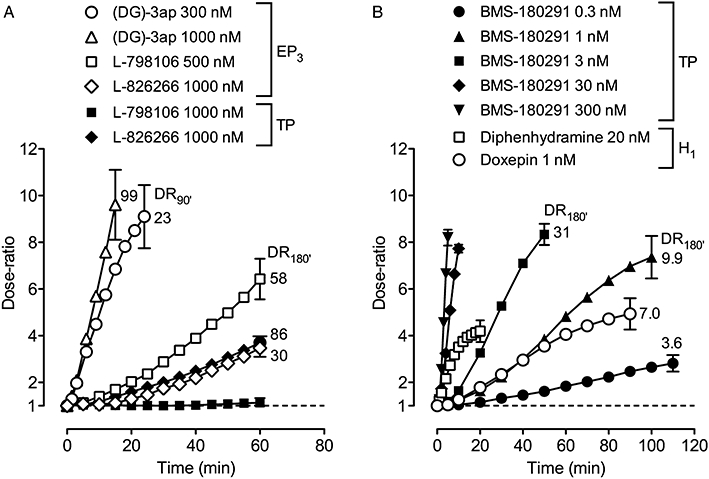
Onset rates for antagonists on guinea-pig aorta under protocol A presented as dose-ratio – time plots: (A) EP3 antagonists versus 17-phenyl PGE2 and U-46619; (B) TP antagonist BMS-180291 versus U-46619 and H1 antagonists diphenhydramine and doxepin versus histamine. For clarity, SEM (n= 4–5) for each final time-point only is shown. The value adjacent to a curve is the mean dose-ratio achieved after continuation of antagonist treatment for a total of 90 min (DR90′) or 180 min (DR180′) under protocol B. BMS-180291 (300 nM) was present throughout the EP3 measurements in (A).
The pA2 values of the three EP3 antagonists were determined using protocol B (Figure 2B); antagonist contact was extended to 90 min for (DG)-3ap and 180 min for L-798106 and L-826266. In each case, planned contrasts allied to repeated-measures 2-factor anova (Figure 2B) revealed good matching of preparations in S1 and no effect of the EP3 antagonist on the PE priming response in S2 (P > 0.05). Analysis of 17-phenyl PGE2 log concentration–response curves (Figure 6A and B) showed a slight suppression of the agonist maximum (fitted values) with increasing antagonist concentration for each antagonist (post-test for linear trend, P < 0.05). Schild plots are shown in Figure 6C. The regression slopes were not significantly different from unity (P > 0.05); pA2 values are presented in Table 3. Although a full investigation of offset kinetics was not performed, EP3 antagonism by (DG)-3ap (1 µM) was found to be slowly reversible (original right-shift of 17-phenyl PGE2 curve = 1.96 log units, subsequent left-shift for 120 min wash = 0.89 log units), whereas 0.5 µM L-798106 and 1 µM L-826266 showed no reversal under this protocol.
Figure 6.
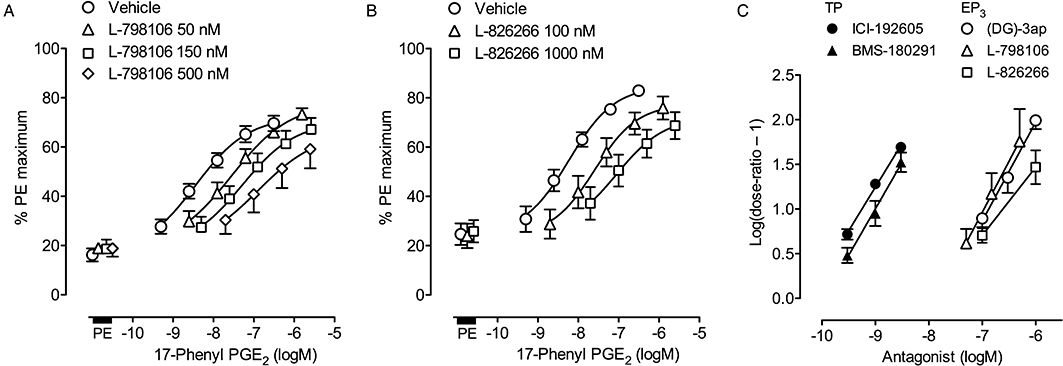
Estimation of pA2 values for prostanoid antagonists on guinea-pig aorta using a Schild protocol (protocol B, Figure 2B). (A) and (B) EP3 assay: log concentration–response curves for 17-phenyl PGE2 in the presence of L-798106 and L-826266 (180 min treatment); PE, phenylephrine priming. BMS-180291 (300 nM) was present for all tests. (C) Schild plots: TP agonist was U-46619; EP3 agonist was 17-phenyl PGE2. Vertical bars show SEM (all n= 4).
Affinities and onset rates of TP and H1 antagonists on guinea-pig aorta
Low concentrations of BMS-180291 (0.3–3.0 nM, Figure 5B) and I-SAP (1–10 nM) very slowly inhibited established contraction to 30 nM U-46619, while ICI-192605 (0.3–3 nM) had not achieved steady-state after 90 min treatment (location on 100 min level in Figure 8C). Higher concentrations of each antagonist caused progressively faster inhibition of U-46619-induced contraction as shown for BMS-180291 in Figure 5B. Under protocol B (180 min treatment), ICI-192605 and BMS-180291 caused parallel displacements of the U-46619 log concentration–response curve. The corresponding Schild plots (Figure 5C) had slopes of 0.98 (95% CI 0.82–1.14) and 0.96 (95% CI 0.76–1.16) and pA2 values were estimated as 10.24 ± 0.03 and 9.98 ± 0.07 respectively.
The established response to 30 nM U-46619 was not affected by 60 min exposure to 1000 nM L-798106 (Figure 5A). In contrast, L-826266 at 1000 nM caused a very slow inhibition similar to that found for the EP3 system in the aorta. Under protocol B (180 min), the parallel right-shift of the U-46619 curve (dose-ratio = 85.5 ± 11.8) afforded a pA2 of 7.92 ± 0.06, n= 4); cumulative PE responses obtained between S1 and S2 were not affected by the L-826266 treatment (dose-ratio = 0.94 ± 0.05, n= 4).
Within the set of H1 antagonists studied, diphenhydramine (10–50 nM) rapidly inhibited histamine-induced contraction, while doxepin (0.5–2.0 nM) was much slower, requiring ∼90 min to approach steady-state (Figures 5B and 8C). Doxepin at 100 nM abolished the histamine response within 12–15 min. Parallel displacement of the histamine curve was found with 1 nM doxepin: pA2= 9.78 ± 0.05 (n= 4, protocol B, 180 min exposure). Inhibition by both terfenadine and astemizole (10–50 nM) was slow and had not reached steady-state after 90 min contact. The pA2 values presented in Table 3 are based on right-shifts at E50 for pretreatment with 25 nM for 180 min. Higher concentrations of both antagonists reduced the maximum response to histamine (e.g. to 46 ± 6% and 74 ± 4% of E100, respectively, at 100 nM, n= 4). Unsurmountable H1 antagonism was previously reported for astemizole and terfenadine on guinea-pig aorta and trachea respectively (Dodel and Borchard, 1992; Christophe et al., 2003).
EP3 antagonists on guinea-pig vas deferens
Inhibition of the EFS-twitch induced by 15 nM PGE2 was rapidly reversed by 30–1000 nM (DG)-3ap (Figure 7); a dose-ratio of 11.5 was achieved after 50 s exposure to 1000 nM (DG)-3ap. TDR4 was estimated to be about 1 min. L-798106 (1000 nM) and L-826266 (1000 nM) produced much slower reversal of PGE2-induced inhibition; reversals at lower concentrations were even slower and more variable and TDR4 estimation was abandoned. Under protocol B (30 min pretreatment) (DG)-3ap (30–1000 nM) caused parallel right-shifts of the PGE2 log concentration–response curve. Schild analysis gave a slope of 1.11 (95% C1 = 0.87–1.35) and pA2 of 7.84 ± 0.05. Pretreatment with 1000 nM L-798106 and 1000 nM L-826266 for 60 min also caused parallel right-shifts of the PGE2 curve, yielding pA2 values of 7.53 and 7.25 respectively. Following repeated washing of preparations exposed to 30 and 1000 nM (DG)-3ap, the 17-phenyl PGE2 curve returned to its original location (dose-ratio < 2) within 30 and 150 min respectively. In contrast, L-798106 and L-826266 exhibited slower reversal of antagonism: right-shift for 1000 nM treatment/subsequent left-shift for 150 min wash = 1.53/0.81 and 1.28/0.45 log units respectively.
Figure 7.
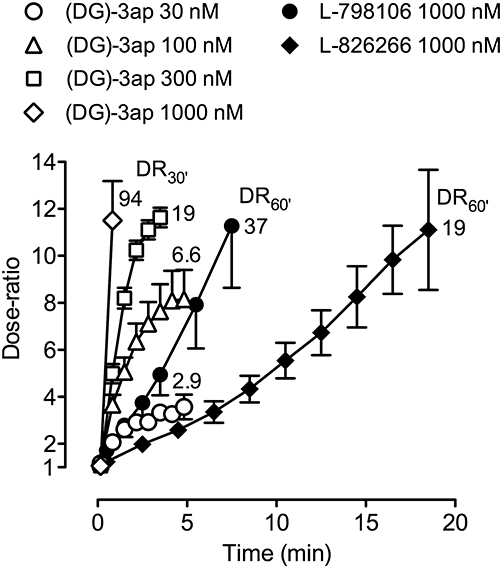
Onset rates of EP3 antagonists on guinea-pig vas deferens presented as dose-ratio – time plots. Under protocol A, inhibition of electrical field stimulation twitch contraction induced by 15 nM prostaglandin E2 was reversed by antagonist application. Vertical bars show SEM (all n= 4). The first two data points on each (DG)-3ap curve correspond to 10 and 50 s after antagonist addition. The value adjacent to each curve is the mean dose-ratio achieved after continuation of antagonist treatment for a total of 30 min (DR30′) or 60 min (DR60′) under protocol B.
Prediction of physiochemical parameters
Acidity constants (pKa) and n-octanol-water partition coefficients (AlogP98) of the ligands studied were predicted (Tables 2 and 3). The prostanoid agonists and antagonists are weak acids with pKa values between 3.89 and 5.44 (≥99.0% ionized at pH 7.4). As amine bases, the H1 antagonists (pKa 8.52–9.47) are ≥93% ionized at pH 7.4, except for astemizole (pKa 6.71, 17% ionized). Figure 8B and D shows correlation plots for agonist T50 versus AlogP98 and antagonist TDR4 versus AlogP98 respectively.
Discussion
Pharmacological profiles of the EP3 receptor antagonists
Before considering tissue disposition as a cause of the large differences in the onset (and offset) kinetics of the EP3 antagonists, it is important to exclude an extraordinary interaction with the EP3 receptor. The α, β-unsaturated amide group common to the three antagonists (Figure 1) could potentially form a stable covalent link with a nucleophilic centre in the EP3 receptor (Michael addition reaction, see Perlmutter, 1992). However, a reversible competitive interaction appears more likely given the reversibility of antagonism seen for all three antagonists in the vas deferens assay and for (DG)-3ap in the guinea-pig aorta assay. This proposition is supported by the surmountable antagonism observed in the human FLIPR and guinea-pig vas deferens and aorta assays. The slight suppression of the agonist maximum by each EP3 antagonist on the aorta may have been due to activation of an opposing EP2 system by the high concentrations of 17-phenyl PGE2 used.
In relation to specificity, (DG)-3ap and L-826266 showed moderately high selectivity for EP3 versus EP1, EP2 and EP4 subtypes in functional assays involving human rc-prostanoid receptor (Table 1). Similarly, L-798106 was reported to be highly EP3-selective based on ligand binding to human rc-prostanoid receptors: pKi = 4.41, <4.3, 9.22 and 6.05 for EP1, EP2, EP3 and EP4 respectively (Belley et al., 2006). However, it appeared to have lower affinity for human EP3 receptors (pKi = 7.77) in a FLIPR assay similar to the one used by us (Jugus et al., 2009). On guinea-pig aorta, L-826266 showed similar affinities for EP3 and TP receptors (pA2= 7.71 and 7.92 respectively). However, α1-adrenoceptor-mediated responses were not inhibited (similarly for (DG)-3ap and L-798106), which argues against any non-specific suppression of contractility. (DG)-3ap did not completely block the net EP3 response on the aorta (protocol A, Figure 4B); this finding affords no hindrance (apart from difficulty in estimating a pA2 value) as a fraction of the established synergism may be sustained by the α1-adrenoceptor stimulus alone.
Explaining slow onset by high-affinity binding under the limited diffusion model
Rang (1966) developed the limited diffusion model to explain the inability of acceptor association/dissociation rate constants alone to account for the slow onsets/offsets of particular ligands in solid tissue systems. Under this model (Figure 9), the build-up of ligand concentration in the centre of the tissue is retarded by binding of the ligand to its cell-surface acceptors (e.g. ion channels, receptors) as it diffuses through the e.c.f.; on washout, this cell-surface reservoir delays outward diffusion of the ligand leading to a correspondingly slow offset. The model was refined (Colquhoun et al., 1972; Colquhoun and Ritchie, 1972) to explain the slow paralysis of non-myelinated nerve fibres by the Na+ channel blocker tetrodotoxin (binding pKd = 8.5 for rabbit vagus nerve). Retardation of diffusion, which may be as large as 1000-fold, will increase as ligand affinity and acceptor density increase and as the extracellular channels become narrower.
The correlation plots for prostanoid agonists in Figure 8A show two components, a flat component where onset rates change little with increasing potency (pEC50 < 8.0) and a steeper component where slowing of onset is associated with increase in potency (see Figure 9 for a schematic plot). Jones et al. (1989) used ‘receptor binding under the limited diffusion model’ to explain the steep component for TP agonists on guinea-pig trachea (data re-analysed here) and other isolated preparations. Low onset rates were associated with high binding affinities for the (human platelet) TP receptor. The same reasoning could also apply to agonist and antagonist onset rate data obtained in this study:
Guinea-pig aorta/TP agonists. I-BOP, the most potent and slowest onset agonist, has a binding pKd of 8.66 for the human platelet TP receptor compared with 6.92 for the less potent and faster U-46619 (Morinelli et al., 1989).
Guinea-pig aorta/EP3 agonists. Onset half-time positively correlates with both potency and binding affinity for mouse rc-EP3 receptors: sulprostone (pKi = 9.22), 17-phenyl PGE2 (8.43), ONO-AE-248 (7.82–8.12) (Kiriyama et al., 1997; Suzawa et al., 2000).
Guinea-pig aorta/TP and H1 antagonists. The TP antagonist ICI-192605 had the slowest onset and highest affinity (pA2= 10.24). Similarly slow onset profiles have been encountered previously during development of TP antagonists related to EP-092 (Wilson et al., 1988). For example, steady-state block of U-46619 on guinea-pig trachea was not achieved by 5 h exposure to sub-nanomolar concentrations of EP-225 (rac-9α,11α-ethano-10a-homo-13-methyl-13-(N-(4-methoxy)-phenylthiocarbamoyl)-hydrazono-3-oxa-ω-heptanor-prostanoic acid); its pA2 value was estimated as ≥10.5 (R.L. Jones and N.H. Wilson, unpubl. obs.). The steep trend for the higher-affinity H1 antagonists is a significant finding because these ligands are (amine) bases as opposed to (carboxylic) acids. Each of the high-affinity TP and H1 antagonists (pA2 > 9.0, Table 3) rapidly inhibited established contraction at concentrations 100–1000×Kd, which is explicable by the magnitude of the k1[ligand] term for the ligand-receptor interaction (Figure 9, upper middle box) and also by overpowering of the limited diffusion mechanism.
The flat components of the correlation plots may indicate that ligand residence time on the receptor is too short to retard inward diffusion significantly (another factor is rate-limiting – see later).
Overall, onsets rates for EP3 agonists and antagonists on guinea-pig vas deferens were much faster than on guinea-pig aorta (Figure 8A). For agonists with pEC50 < 8.0, this discrepancy may reflect the kinetic properties of the transduction processes involved: Gi-coupling to Ca2+ influx channels in sympathetic varicosities in vas deferens (Ito and Tajima, 1979) may be faster than Gi-driven suppression of cAMP levels/G12/13 activation of Rho-kinase in smooth muscle cells of aorta (Shum et al., 1993). Similarly, the much faster onset of EP3 antagonism by (DG)-3ap in vas deferens relative to aorta (Figure 8C) could be due to more rapid decay of the transduction processes. The faster onset rates of the higher-potency EP3 agonists (e.g. DM PGE2) on the vas deferens may also reflect a reduced influence of the limited diffusion mechanism due to a lower EP3 receptor density and/or easier access of ligands to EP3 receptors located on sympathetic varicosities. If this is the case, then the agonist – EP3 receptor interaction alone could become the rate-limiting process. Assuming sensible values of 10−9 M and 107 M−1·s−1 for Kd and k1 of DM PGE2, respectively, konset for 0.3 nM ligand will be 0.013 s−1 (upper middle box, Figure 9); the corresponding onset half-time is 53 s, close to the experimental value of 63 s. An analogous calculation was used to account for the slower shape-change response of EP-171 (0.1 nM) relative to U-46619 (5–10 nM) in a human platelet suspension, where access to the platelet surface is almost instantaneous (Jones et al., 1989).
Mechanisms whereby lipophilicity could affect onset/offset kinetics
Crucially, four antagonists with modest affinities on guinea-pig aorta are located well above the general trends for onset in Figure 8C: L-798106 on the EP3 system, L-826266 on both EP3 and TP systems and terfenadine and astemizole on the H1 system. We propose that their high lipophilicity, operating within the limited diffusion model, underlies their slow kinetics. The model (Figure 9) accommodates partition into lipid domains within the cell membrane as a means of retarding diffusion of ligand through the e.c.f. (Colquhoun et al., 1972; Colquhoun and Ritchie, 1972). The membrane-water partition coefficient of the ligand and the relative volumes of the aqueous and lipid phases and would be controlling factors. Similar to receptor binding under the model, it is assumed that significant residence in the membrane would only occur when lipophilicity exceeds a critical value, say AlogP98 of 5.0 with respect to Figure 8D. In this context, Austin et al. (2003) showed a positive correlation between lipophilocity (distribution coefficient for all molecular species at pH 7.4) and duration of β2 relaxation on guinea-pig trachea for a large series of benzothiazolinones with dual β2/dopamine D2 agonism (n= 103); a similar trend, but on a smaller scale, was found for D2 relaxation. There was no apparent correlation between β2 agonist potency and duration of action. However, of the 69 compounds evaluated, the durations of 15 with pIC50 > 8.0 exceeded the time limit of the assay (180 min), and so correlative power was reduced in this higher potency range.
A lipophilic ligand may also access the binding site of receptor via lateral diffusion in the cell membrane core. This plasmalemmal diffusion microkinetic model (Figure 9, lower right box) provides one explanation for the slow onset/very persistent relaxation of respiratory smooth muscle preparations induced by salmeterol and several other β2-adrenoceptor agonists (Anderson, 1993; see reviews by Anderson et al., 1994; Waldeck, 1996; Coleman, 2009). This model could explain the slow kinetics of the lipophilic ligands under investigation here. It is of interest that the initial partition into the membrane core could retard centripetal movement of ligand through the e.c.f. in the same way that the limited diffusion (lipid partition) model operates.
Returning to prostanoid agonists, Figure 8B shows no obvious trend for T50 versus AlogP98. This may be due to only one agonist (EP-031) having AlogP98 greater then 5.0. Notwithstanding, we have found (unpubl. obs.) that a EP2 agonist (cinnamic acid derivative 9 in Belley et al., 2005) with high lipophilicity (AlogP98 = 6.64) relaxes guinea-pig aorta much more slowly than PGE2 (AlogP98 = 3.20).
Comments on the correlation analysis
In relation to access of ligand to the e.c.f. (Figure 9, upper left box), there is a general consensus that membrane permeation rates are best correlated to distribution coefficient for all molecular species at pH 7.4 as opposed to partition coefficient of the unionized ligand, usually between n-octanol and water (logP). In the current situation, the trends for prostanoid ligands shown in Figure 8B and D would be similar given the rather small variation in pKa (Tables 2 and 3). Also, it is usually assumed that the unionized molecule is by far the major permeating species. While this may indeed hold for moderately lipophilic ligands (see Saparov et al., 2006 for salicylic acid data), the difference in permeation rates for highly lipophilic compounds may be much less. Roda et al. (1990) reported a difference of only one log unit between the experimental logP values for the ionized and unionized forms of chenodeoxycholic, concluding that the hydrophobicity of the molecule predominates over the effect due to ionization. Given these considerations, we were reluctant at this time to attempt more refined analyses of the correlation data.
The correlation approach would clearly benefit from examination of larger numbers of close congeners for each receptor system. The acyl-sulphonamide series of EP3 antagonists appears to be suitable for attempting lipophilicity changes on a fairly large scale, given the ease of altering the acyl and/or sulphonyl moieties (Figure 1).
Relevance of the current studies
Probing receptor function with a highly lipophilic, slow-onset EP3 antagonist may confound data interpretation; erroneous pA2 values may be generated and specificity may be compromised by the forced use of higher antagonist concentrations. In this context, L-826266 has been used at 30 µM to implicate EP3 receptors in the relaxation of guinea-pig trachea induced by bradykinin (Schlemper et al., 2005). This is a surprising proposal given that guinea-pig trachea is relaxed by EP2 receptor activation (Coleman and Kennedy, 1985). It may be prudent to use an antagonist with moderate lipophilicity, say logP of 3.0–4.0, to ensure that steady-state block occurs within a reasonably short time.
High lipophilicity (typically >logP 5.0) is characteristic of compounds that have low solubility and poor absorption and are rapidly metabolized (Kirkpatrick, 2003). Lipinski's analysis of the properties of orally available marketed drugs (Lipinski et al., 1997) led to the ‘Rule of 5’ being used to guide lead optimization and candidate selection in drug development. Lipinski also noticed that poor absorption and permeation are more likely when two or more of the critical limits are exceeded, for example, logP > 5.0/molecular weight (MW) > 500. This scenario has been discussed for the anti-arrhythmic amiodarone (AlogP98 = 7.24, MW = 645) by Roden (1999) and a comprehensive review of the literature has recently appeared (Waring, 2010). L-798106 and L-826266 exceed both limits, as do terfenadine and astemizole when the MW limit is reduced to 450 (Table 3). In this context, it is somewhat surprising that the acyl-sulphonamide EP3 antagonist DG-041 (Zegar et al., 2007; Heptinstall et al., 2008, Figure 1), which readily exceeds both limits (AlogP98 = 7.67, MW 592), has progressed to early stage clinical trials for preventing thrombosis associated with deterioration of atherosclerotic plaques (see Fabre and Gurney, 2010). EP3 agonists, including PGE2, enhance human platelet aggregation (Matthews and Jones, 1993; Heptinstall et al., 2008). However, our current findings are more concerned with the tissue disposition of a ligand than its initial absorption into the plasma compartment. As such, lipid partition under the limited diffusion model may not markedly affect the interaction of a lipophilic antagonist with the EP3 receptors of freely-circulating platelets because ready access to the plasma membrane is expected. However, the model may have greater relevance to the more complex structure of a long-established plaque where EP3 receptor systems are likely to be operative in compact tissue masses comprised of platelets (and immune cells) and vascular smooth muscle cells (Qian et al., 1994) located adjacently.
Acknowledgments
We appreciate generous gifts of L-798106 from GlaxoSmithKline, UK, BMS-180291 from Bristol-Myers Squibb, USA, and EP-045 and EP-092 from Dr NH Wilson, University of Edinburgh. We thank Prof Ian McGrath for advice on α-adrenoceptors.
Glossary
Abbreviations
- e.c.f.
extracellular fluid
- E50/E100
50% maximal/maximal agonist response
- EFS
electrical field stimulation
- FLIPR
fluorimetric imaging plate reader
- logP
partition coefficient of the unionized ligand, usually between n-octanol and water
- PGE2
prostaglandin E2
- rc-EP3
recombinant EP3 receptor (as expressed in a carrier cell)
- T50
onset half-time for an agonist to achieve 50% maximal response
- TDR4
onset half-time for an antagonist corresponding to a dose-ratio of 4
Conflicts of interest
None.
Supplemental material
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (4th edn) 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GP. Formoterol: pharmacology, molecular basis of agonism, and mechanism of long duration of a highly potent and selective β2-adrenoceptor agonist bronchodilator. Life Sci. 1993;52:2145–2160. doi: 10.1016/0024-3205(93)90729-m. [DOI] [PubMed] [Google Scholar]
- Anderson GP, Lindén A, Rabe KF. Why are long-acting beta-adrenoceptor agonists long-acting? Eur Respir J. 1994;7:569–578. doi: 10.1183/09031936.94.07030569. [DOI] [PubMed] [Google Scholar]
- Austin RP, Barton P, Bonnert RV, Brown RC, Cage PA, Cheshire DR, et al. QSAR and the rational design of long-acting dual D2-receptor/β2-adrenoceptor agonists. J Med Chem. 2003;46:3210–3220. doi: 10.1021/jm020886c. [DOI] [PubMed] [Google Scholar]
- Avdeef A, Box KJ, Takács-Kovák K. pH-metric logP. 6. Effects of sodium, potassium and N-CH3-D-glucamine on the octanol-water partitioning of prostaglandins E1 and E2. J Pharm Sci. 1995;84:523–529. doi: 10.1002/jps.2600840502. [DOI] [PubMed] [Google Scholar]
- Belley M, Gallant M, Roy B, Houde K, Lachance N, Labelle M, et al. Structure-activity relationship studies on ortho-substituted cinnamic acids, a new class of selective EP3 antagonists. Bioorg Med Chem Lett. 2005;15:527–530. doi: 10.1016/j.bmcl.2004.11.051. [DOI] [PubMed] [Google Scholar]
- Belley M, Chan CC, Gareau Y, Gallant M, Juteau H, Houde K, et al. Comparison between two classes of selective EP3 antagonists and their biological activities. Bioorg Med Chem Lett. 2006;16:5639–5642. doi: 10.1016/j.bmcl.2006.08.025. [DOI] [PubMed] [Google Scholar]
- Cao J, Shayibuzhati M, Tajima T, Kitazawa T, Taneike T. In vitro pharmacological characterization of the prostanoid receptor population in the non-pregnant porcine myometrium. Eur J Pharmacol. 2002;442:115–123. doi: 10.1016/s0014-2999(02)01489-9. [DOI] [PubMed] [Google Scholar]
- Christophe B, Carlier B, Gillard M, Chatelain P, Peck M, Massingham R. Histamine H1 receptor antagonism by cetirizine in isolated guinea pig tissues: influence of receptor reserve and dissociation kinetics. Eur J Pharmacol. 2003;470:87–94. doi: 10.1016/s0014-2999(03)01781-3. [DOI] [PubMed] [Google Scholar]
- Clarke DL, Giembycz MA, Patel HJ, Belvisi MG. E-ring 8-isoprostanes inhibit ACh release from parasympathetic nerves innervating guinea-pig trachea through agonism of prostanoid receptors of the EP3-subtype. Br J Pharmacol. 2004;141:600–609. doi: 10.1038/sj.bjp.0705648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RA. On the mechanism of the persistent action of salmeterol: what is the current position? Br J Pharmacol. 2009;158:180–182. doi: 10.1111/j.1476-5381.2009.00370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RA, Kennedy I. Characterisation of the prostanoid receptors mediating contraction of guinea-pig isolated trachea. Prostaglandins. 1985;29:363–375. doi: 10.1016/0090-6980(85)90096-6. [DOI] [PubMed] [Google Scholar]
- Coleman RA, Kennedy I, Sheldrick RL. New evidence with selective agonists and antagonists for the subclassification of PGE2-sensitive (EP) receptors. Adv Prostaglandin Thromboxane Leukot Res. 1987;17A:467–470. [PubMed] [Google Scholar]
- Colquhoun D, Ritchie JM. The kinetics of the interaction between tetrodotoxin and mammalian nonmyelinated nerve fibers. Mol Pharmacol. 1972;285:285–292. [PubMed] [Google Scholar]
- Colquhoun D, Henderson R, Ritchie JM. The binding of labelled tetrodotoxin to non-myelinated nerve fibres. J Physiol. 1972;227:95–126. doi: 10.1113/jphysiol.1972.sp010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig DA. The Cheng-Prusoff relationship: something lost in the translation. Trends Pharmacol Sci. 1993;14:89–91. doi: 10.1016/0165-6147(93)90070-z. [DOI] [PubMed] [Google Scholar]
- Daray FM, Minvielle AI, Puppo S, Rothlin RP. Pharmacological characterization of prostanoid receptors mediating vasoconstriction in human umbilical vein. Br J Pharmacol. 2003;139:1409–1426. doi: 10.1038/sj.bjp.0705375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodel R, Borchard U. The affinity of antihistamines to peripheral histamine H1-receptors. Inflam Res. 1992;36(Suppl. 2):C410–C413. [Google Scholar]
- Fabre JE, Gurney ME. Limitations of current therapies to prevent thrombosis: a need for novel strategies. Mol Biosyst. 2010;6:305–315. doi: 10.1039/b914375k. [DOI] [PubMed] [Google Scholar]
- Figueiredo A, Ribeiro CA, Gonçalo M, Almeida L, Poiares-Baptista A, Teixeira F. Mechanism of action of doxepin in the treatment of chronic urticaria. Fundam Clin Pharmacol. 1990;4:147–158. doi: 10.1111/j.1472-8206.1990.tb00482.x. [DOI] [PubMed] [Google Scholar]
- Foudi N, Kotelevets L, Louedec L, Leséche G, Henin D, Chastre E, et al. Vasorelaxation induced by prostaglandin E2 in human pulmonary vein: role of the EP4 receptor subtype. Br J Pharmacol. 2008;154:1631–1639. doi: 10.1038/bjp.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallant M, Carrière MC, Chateauneuf A, Denis D, Gareau Y, Godbout C, et al. Structure-activity relationship of biaryl acylsulfonamide analogues on the human EP3 prostanoid receptor. Bioorg Med Chem Lett. 2002;12:2583–2586. doi: 10.1016/s0960-894x(02)00518-8. [DOI] [PubMed] [Google Scholar]
- Giles H, Leff P, Bolofo ML, Kelly MG, Robertson AD. The classification of prostaglandin DP-receptors in platelets and vasculature using BW A868C, a novel, selective and potent competitive antagonist. Br J Pharmcol. 1989;96:291–300. doi: 10.1111/j.1476-5381.1989.tb11816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass GV, Hopkins KD. Statistical Methods in Education and Psychology. Boston: Allyn and Bacon; 1996. Multiple comparisons and trend analysis; pp. 444–481. [Google Scholar]
- Heptinstall S, Espinosa DI, Manolopoulos P, Glenn JR, White AE, Johnson A, et al. DG-041 inhibits the EP3 prostanoid receptor – a new target for inhibition of platelet function in atherosclerotic disease. Platelets. 2008;19:605–613. doi: 10.1080/09537100802351073. [DOI] [PubMed] [Google Scholar]
- Ito Y, Tajima K. Electrophysiological analysis of the actions of prostaglandin on neuromuscular transmission in the guinea-pig vas deferens. J Physiol. 1979;29:521–537. doi: 10.1113/jphysiol.1979.sp013054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RL, Woodward DF. Interaction of prostanoid EP3 and TP receptors in guinea-pig isolated aorta: contractile self-synergism of 11-deoxy-16,16-dimethyl PGE2. Br J Pharmacol. 2011;162:521–531. doi: 10.1111/j.1476-5381.2010.01039.x. accompanying manuscript. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RL, Wilson NH, Lawrence RA. EP 171: a high affinity thromboxane A2-mimetic, the actions of which are slowly reversed by receptor blockade. Br J Pharmacol. 1989;96:875–887. doi: 10.1111/j.1476-5381.1989.tb11898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RL, Wise H, Clark R, Whiting RL, Bley KR. Investigation of the prostacyclin (IP) receptor antagonist RO1138452 on isolated blood vessel and platelet preparations. Br J Pharmacol. 2006;149:110–120. doi: 10.1038/sj.bjp.0706841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RL, Giembycz MA, Woodward DF. Prostanoid receptor antagonists: development strategies and therapeutic applications. Br J Pharmacol. 2009;158:104–145. doi: 10.1111/j.1476-5381.2009.00317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jugus MJ, Jaworski JP, Patra PB, Jin J, Morrow DM, Laping NJ, et al. Dual modulation of urinary bladder activity and urine flow by prostanoid EP3 receptors in the conscious rat. Br J Pharmacol. 2009;158:372–381. doi: 10.1111/j.1476-5381.2009.00275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S. Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br J Pharmacol. 1997;122:217–224. doi: 10.1038/sj.bjp.0701367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick P. Pressures in the pipeline. Nat Rev Drug Discov. 2003;2:337. doi: 10.1038/nrd1015. [DOI] [PubMed] [Google Scholar]
- Lawrence RA, Jones RL, Wilson NH. Characterization of receptors involved in the direct and indirect actions of prostaglandins E and I on the guinea-pig ileum. Br J Pharmacol. 1992;105:271–278. doi: 10.1111/j.1476-5381.1992.tb14245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazareno S, Birdsall NJ. Estimation of competitive antagonist affinity from functional inhibition curves using the Gaddum, Schild and Cheng-Prusoff equations. Br J Pharmacol. 1993;109:1110–1119. doi: 10.1111/j.1476-5381.1993.tb13737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leff P, Dougall IG. Further concerns over Cheng-Prusoff analysis. Trends Pharmacol Sci. 1993;14:110–112. doi: 10.1016/0165-6147(93)90080-4. [DOI] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Mannhold R, Poda GI, Ostermann C, Tetko IV. Calculation of molecular lipophilicity: state-of-the-art and comparison of log P methods on more than 96,000 compounds. J Pharm Sci. 2008;98:861–893. doi: 10.1002/jps.21494. [DOI] [PubMed] [Google Scholar]
- Matias I, Chen J, De Petrocellis L, Bisogno T, Ligresti A, Fezza F, et al. Prostaglandin ethanolamides (prostamides): in vitro pharmacology and metabolism. J Pharmacol Exp Ther. 2004;309:745–757. doi: 10.1124/jpet.103.061705. [DOI] [PubMed] [Google Scholar]
- Matthews JS, Jones RL. Potentiation of aggregation and inhibition of adenylate cyclase in human platelets by prostaglandin E analogues. Br J Pharmacol. 1993;108:363–369. doi: 10.1111/j.1476-5381.1993.tb12810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinelli TA, Oatis JE, Jr, Okwu AK, Mais DE, Mayeux PR, Masuda A, et al. Characterization of an 125I-labelled thromboxane A2/prostaglandin H2 receptor agonist. J Pharmacol Exp Ther. 1989;251:557–562. [PubMed] [Google Scholar]
- O'Connell M, Zeller W, Burgeson J, Mishra RK, Ramirez J, Kiselyov AS, et al. Peri-substituted hexahydro-indolones as novel, potent and selective human EP3 receptor antagonists. Bioorg Med Chem Lett. 2009;19:778–782. doi: 10.1016/j.bmcl.2008.12.027. [DOI] [PubMed] [Google Scholar]
- Okada Y, Hara A, Ma H, Xiao CY, Takahata O, Kohgo Y, et al. Characterization of prostanoid receptors mediating contraction of the gastric fundus and ileum: studies using mice deficient in prostanoid receptors. Br J Pharmacol. 2000;131:745–755. doi: 10.1038/sj.bjp.0703627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter P. Conjugated Addition Reactions in Organic Synthesis. Oxford: Pergamon; 1992. p. 114. [Google Scholar]
- Pratt WB. The entry, distribution and elimination of drugs. In: Pratt WB, Taylor P, editors. Principles of Drug Action. New York: Churchill-Livingstone; 1990. pp. 201–296. 3rd. [Google Scholar]
- Qian YM, Jones RL, Chan KM, Stock AI, Ho JK. Potent contractile actions of prostanoid EP3-receptor agonists on human isolated pulmonary artery. Br J Pharmacol. 1994;113:369–374. doi: 10.1111/j.1476-5381.1994.tb16997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rang HP. The kinetics of action of acetylcholine antagonists in smooth muscle. Proc Royal Soc Lond B. 1966;164:488–510. doi: 10.1098/rspb.1966.0045. [DOI] [PubMed] [Google Scholar]
- Roda A, Minutello A, Angellotti MA, Fini A. Bile acid structure-activity relationship: evaluation of bile acid lipophilicity using 1-octanol/water partition coefficient and reverse phase HPLC. J Lipid Res. 1990;31:1433–1443. [PubMed] [Google Scholar]
- Roden DM. Mechanisms underlying variability in response to drug therapy: implications for amiodarone use. Am J Cardiol. 1999;84:29R–36R. doi: 10.1016/s0002-9149(99)00699-2. [DOI] [PubMed] [Google Scholar]
- Saparov SM, Antonenko YN, Pohl P. A new model of weak acid permeation through membranes revisited: does Overton still rule? Biophys J. 2006;90:L86–L88. doi: 10.1529/biophysj.106.084343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saussy DL, Goetz AS, King HK, True TA. BMY 7378 is a selective antagonist of α1D receptors. Can J Physiol Pharmacol. 1994;72:A323. doi: 10.1016/0014-2999(94)00751-r. [DOI] [PubMed] [Google Scholar]
- Schlemper V, Medeiros R, Ferreira J, Campos MM, Calixto JB. Mechanisms underlying the relaxation response induced by bradykinin in the epithelium-intact guinea-pig trachea in vitro. Br J Pharmacol. 2005;145:740–750. doi: 10.1038/sj.bjp.0706222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senchyna M, Crankshaw DJ. Characterization of the prostanoid TP receptor population in human nonpregnant myometrium. J Pharmacol Exp Ther. 1996;279:262–270. [PubMed] [Google Scholar]
- Sessa WC, Halushka PV, Okwu A, Nasjletti A. Characterization of the vascular thromboxane A2/prostaglandin endoperoxide receptor in rabbit aorta. Regulation by dexamethasone. Circ Res. 1990;67:1562–1569. doi: 10.1161/01.res.67.6.1562. [DOI] [PubMed] [Google Scholar]
- Shum WW, Le GY, Jones RL, Gurney AM, Sasaki Y. Involvement of Rho-kinase in contraction of guinea-pig aorta induced by prostanoid EP3 receptor agonists. Br J Pharmacol. 1993;139:1449–1461. doi: 10.1038/sj.bjp.0705393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth EM, Grosser T, Wang M, Yu Y, FitzGerald GA. Prostanoids in health and disease. J Lipid Res. 2009;50:S423–S428. doi: 10.1194/jlr.R800094-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturino CF, O'Neill G, Lachance N, Boyd M, Berthelette C, Labelle M, et al. Discovery of a potent and selective prostaglandin D2 receptor antagonist, [(3R)-4-(4-chloro-benzyl)-7-fluoro-5-(methylsulfonyl)-1,2,3,4-tetrahydrocyclopenta[b] indol-3-yl]-acetic acid (MK-0524) J Med Chem. 2007;50:794–806. doi: 10.1021/jm0603668. [DOI] [PubMed] [Google Scholar]
- Suzawa T, Miyaura C, Inada M, Maruyama T, Sugimoto Y, Ushikubi F, et al. The role of prostaglandin E receptor subtypes (EP1, EP2, EP3, and EP4) in bone resorption: an analysis using specific agonists for the respective EPs. Endocrinology. 2000;141:1554–1559. doi: 10.1210/endo.141.4.7405. [DOI] [PubMed] [Google Scholar]
- Waldeck B. Some pharmacodynamic aspects on long-acting β-adrenoceptor agonists. Gen Pharmacol. 1996;27:575–580. doi: 10.1016/0306-3623(95)02052-7. [DOI] [PubMed] [Google Scholar]
- Waring MJ. Lipophilicity in drug discovery. Expert Opin Drug Discovery. 2010;5:235–248. doi: 10.1517/17460441003605098. [DOI] [PubMed] [Google Scholar]
- Wilson NH, Jones RL, Marr CG, Muir G. Synthesis of oxabicyclo[2.2.1]heptane prostanoids having thromboxane-like activity at subnanomolar concentrations. Eur J Med Chem. 1988;23:359–364. [Google Scholar]
- Woodward DF, Krauss AH, Chen J, Liang Y, Li C, Protzman CE, et al. Pharmacological characterization of a novel antiglaucoma agent, Bimatoprost (AGN 192024) J Pharmacol Exp Ther. 2003;305:772–785. doi: 10.1124/jpet.102.047837. [DOI] [PubMed] [Google Scholar]
- Woodward DF, Krauss AH, Wang JW, Protzman CE, Nieves AL, Liang Y, et al. Identification of an antagonist that selectively blocks the activity of prostamides (prostaglandin-ethanolamides) in the feline iris. Br J Pharmacol. 2007;150:342–352. doi: 10.1038/sj.bjp.0706989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RN, Billot X, Han YX, Slipetz DA, Chauret N, Belley M, et al. Discovery and synthesis of a potent, selective and orally bioavailable EP4 receptor agonist. Heterocycles. 2004;64:437–446. [Google Scholar]
- Zegar S, Tokar C, Enache LA, Rajagopol V, Zeller W, O'Connell M, et al. Development of a scalable proces for DG-041, a potent EP3 receptor antagonist, via tandem Heck reactions. Org Process Res Dev. 2007;11:747–753. [Google Scholar]
- Zhang R, Ogletree ML, Moreland S. Characterization of thromboxane A2/prostaglandin endoperoxide receptors in aorta. Eur J Pharmacol. 1996;317:91–96. doi: 10.1016/s0014-2999(96)00697-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.