

Abstract
BACKGROUND AND PURPOSE
While the slow delayed rectifier K+ current (IKs) is known to be enhanced by the stimulation of β-adrenoceptors in several mammalian species, phosphorylation-dependent regulation of the rapid delayed rectifier K+ current (IKr) is controversial.
EXPERIMENTAL APPROACH
In the present study, therefore, the effect of isoprenaline (ISO), activators and inhibitors of the protein kinase A (PKA) pathway on IKr and IKs was studied in canine ventricular myocytes using the whole cell patch clamp technique.
KEY RESULTS
IKr was significantly increased (by 30–50%) following superfusion with ISO, forskolin or intracellular application of PKA activator cAMP analogues (cAMP, 8-Br-cAMP, 6-Bnz-cAMP). Inhibition of PKA by Rp-8-Br-cAMP had no effect on baseline IKr. The stimulating effect of ISO on IKr was completely inhibited by selective β1-adrenoceptor antagonists (metoprolol and CGP-20712A), by the PKA inhibitor Rp-8-Br-cAMP and by the PKA activator cAMP analogues, but not by the EPAC activator 8-pCPT-2'-O-Me-cAMP. In comparison, IKs was increased threefold by the activation of PKA (by ISO or 8-Br-cAMP), and strongly reduced by the PKA inhibitor Rp-8-Br-cAMP. The ISO-induced enhancement of IKs was decreased by Rp-8-Br-cAMP and completely inhibited by 8-Br-cAMP.
CONCLUSIONS AND IMPLICATIONS
The results indicate that the stimulation of β1-adrenoceptors increases IKr, similar to IKs, via the activation of PKA in canine ventricular cells.
Keywords: β-adrenoceptors, isoprenaline, cAMP, PKA, delayed rectifier K+ current, IKr, IKs, dog myocytes
Introduction
Delayed rectifier K+ currents play a pivotal role in the repolarization of the cardiac action potential. In the ventricular myocardium of most mammalian species, including dog and human, the delayed rectifier K+ current (IKr) is composed of two independent components, a rapid IKr and a slow delayed rectifier K+ current (IKs) (Gintant, 1996; Li et al., 1996). Both components are controlled by catecholamines, and are important targets for antiarrhythmic drug action. While IKs is known to be enhanced by the stimulation of β-adrenoceptors, the phosphorylation-dependent regulation of IKr is controversial (Thomas et al., 2004). Protein kinase A (PKA)-mediated phosphorylation of expressed HERG channels significantly decreased IKr tail currents (Thomas et al., 1999; Wei et al., 2002). Similarly, stimulation of β-adrenoceptors by 10 µM isoprenaline (ISO) decreased IKr in voltage-clamped guinea pig ventricular myocytes (Karle et al., 2002). In contrast to these results, Heath and Terrar (2000) found that IKr was enhanced by ISO in guinea pig ventricular cells, provided the conditions necessary to activate the PKC pathway were met, that is, Ca2+ current was not blocked, cytosolic Ca2+ was not buffered and the cell interior was not dialyzed. In the absence of relevant human data, we decided to study the effects of β-adrenoceptor stimulation on delayed rectifier K+ currents in ventricular cardiomyocytes of the dog, a species that has action potential characteristics and properties of the underlying transmembrane ion currents most resembling those in humans (Szabóet al., 2005; Szentandrássy et al., 2005). It was found that ISO increased IKr significantly in canine ventricular myocytes by the activation of PKA and this effect is mediated by the stimulation of β1-adrenoceptors.
Methods
Single canine ventricular myocytes were obtained from hearts of adult mongrel dogs using the segment perfusion technique (Magyar et al., 2000). The animals (10–20 kg) were anaesthetized with ketamine hydrochloride (Calypsol, Richter Gedeon Rt., Budapest, Hungary) 5 mg·kg−1 i.v. plus xylazine (CP-Xylazine, Sedaxylan, Eurovet Animal Health BV, Bladel, the Netherlands) 0.04 mg·kg−1. After the chest had been opened, the heart was rapidly removed and the left anterior descending coronary artery was perfused using a Langendorff apparatus. Ca2+-free JMM solution (Joklik modification of Eagle's Minimum Essential Medium, Sigma-Aldrich Co., St. Louis, MO, USA), supplemented with taurine (2.5 g·L−1), pyruvic acid (175 mg·L−1), ribose (750 mg·L−1), allopurinol (13.5 mg·L−1) and NaH2PO4 (200 mg·L−1), was used during the initial 5 min of perfusion to remove Ca2+ and blood from the tissue. After the addition of NaHCO3 (1.3 g·L−1), the pH of this perfusate was adjusted to 6.9 by equilibrating the solution with a mixture of 95% O2 and 5% CO2. Cell dispersion was performed for 30 min in the same solution containing, in addition, collagenase (660 mg·L−1, Worthington CLS-II, Worthington Biochemical Co., Lakewood, NJ, USA), bovine serum albumin (2 g·L−1) and CaCl2 (50 µM). During the isolation procedure, the solutions were gassed with carbogen and the temperature was maintained at 37°C. The cells were rod shaped and showed clear striation when the external calcium was restored.
IKr and IKs were recorded at 37°C from Ca2+-tolerant canine ventricular cells superfused with oxygenated Tyrode solution containing (in mM) NaCl 140, KCl 5.4, CaCl2 2.5, MgCl2 1.2, HEPES 5, glucose 10, at pH 7.4. This superfusate was supplemented with 5 µM nifedipine plus 1 µM E-4031 when measuring IKs, or 5 µM nifedipine plus 1 µM HMR-1556 when recording IKr, in order to eliminate L-type Ca2+ currents, IKr, or IKs respectively. Suction pipettes, fabricated from borosilicate glass, had tip resistances of 1.5–2 MΩ after being filled with pipette solution composed of (in mM) K-aspartate 100, KCl 45, MgCl2 1, HEPES 5, EGTA 10, K-ATP 3. The pH of this solution was adjusted to 7.2 with KOH. Membrane currents were recorded with an Axopatch-200B amplifier (Axon Instruments Inc., Foster City, CA, USA) using the whole cell configuration of the patch clamp technique. After a high (1–10 GΩ) resistance seal had been established by gentle suction, the cell membrane beneath the tip of the electrode was disrupted by further suction or by applying 1.5 V electrical pulses for 1 ms. Ion currents were normalized to cell capacitance, determined in each cell using short hyperpolarizing pulses from −10 mV to −20 mV. The series resistance was typically 4–8 MΩ before compensation (usually 50–80%) prior to the measurement. Experiments were discarded when the amplitude of IKr or IKs was unstable within the initial 5 min of the experiment, or the series resistance was high or increased during the measurement. Outputs from the clamp amplifier were digitized at 20 kHz using an A/D converter (Digidata-1200, Axon Instruments) under software control (pClamp 6.0, Axon Instruments).
All values presented are arithmetic means ± SEM. Statistical significance of differences was evaluated by using one-way analysis of variance followed by Student's t-test for paired or unpaired data, as appropriate. Differences were considered significant when the P value was less than 0.05.
The principles of laboratory animal care (NIH publication no. 85-23, revised 1985) and current version of the Hungarian Law on the Protection of Animals were strictly followed throughout the experiments. Drug and molecular target nomenclature conforms to Guide to Receptors and Channels (Alexander et al., 2009).
Results
Effect of ISO on IKr
IKr was activated by 250 ms depolarizing pulses to +10 mV applied at a rate of 0.05 Hz. IKr was characterized as tail current amplitudes determined as the difference between the peak current and the pedestal value observed following repolarization to the holding potential of −40 mV in the presence of 5 µM nifedipine plus 1 µM HMR-1556. Exposure of myocytes to ISO for 3–4 min increased IKr tail amplitude in a readily reversible manner (Figure 1A–C). The effect of ISO developed rapidly; the maximal effect was typically achieved within 2–3 min. These current tails (including the baseline amplitude as well as the ISO-induced component) were fully eliminated by 1 µM E-4031 (the IKr tail amplitude decreased to 0.01 ± 0.002 pA/pF in the presence of E-4031, n= 5); therefore, the current can be considered to be purely IKr mediated by HERG channels (Figure 1D). The ISO-induced enhancement of IKr was caused by a leftward shift in the voltage-dependence of activation of IKr (the midpoint potential was shifted from +5.6 ± 0.9 mV to −4.6 ± 1.2 mV), while no significant change was found in the slope factor (6.7 ± 0.5 vs. 7.4 ± 0.5 mV−1) in the four myocytes studied (Figure 1E).
Figure 1.
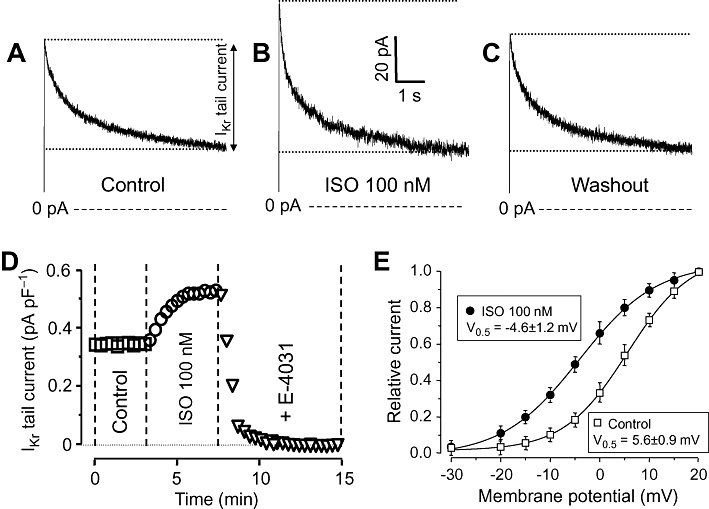
Effects of isoprenaline (ISO) on rapid delayed rectifier K+ (IKr) current. (A–C) IKr current traces showing tail currents as relaxation of current during repolarization to −40 mV under control conditions (A), following exposure to 100 nM ISO (B) and after washing the cells in ISO-free solution (C). (D) Representative experiment demonstrating the effect of 100 nM ISO on IKr and the full suppression of the ISO-induced current by 1 µM E-4041. (E) Voltage-dependence of the activation of IKr in control and in the presence of 100 nM ISO. Tail current amplitudes measured at each test potential was normalized to the respective tail current obtained at +20 mV. Symbols and bars are means ± SEM, n= 4.
The stimulating effect of ISO on IKr was concentration dependent; 1 µM ISO increased IKr by 37 ± 3% (Figure 2A). By fitting these results to the Hill equation, an EC50 value of 13.6 ± 2.5 µM and a Hill coefficient of close to unity were obtained. Because 100 nM ISO resulted in a nearly maximal activation of IKr, this concentration was used in the following experiments. The effect of ISO was fully prevented by pretreatment with either 100 nM metoprolol or 300 nM CGP-20712A – both are known to be selective inhibitors of β1-adrenoceptors at these concentrations (Figure 2B,C).
Figure 2.
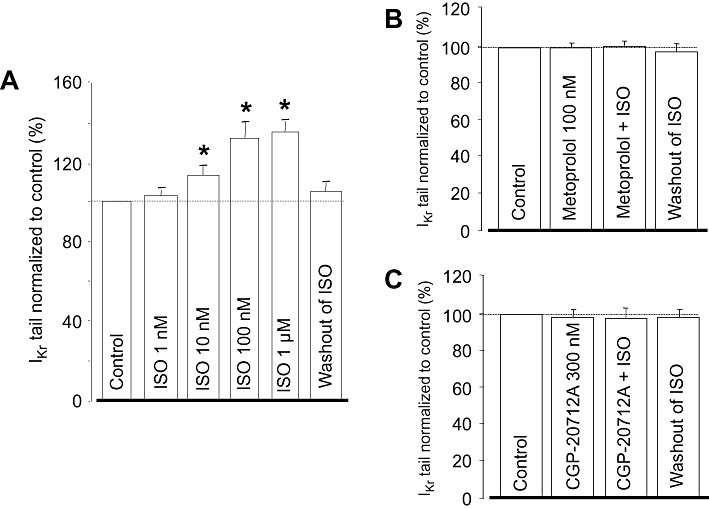
(A) Cumulative concentration-dependent effect of isoprenaline (ISO) on rapid delayed rectifier K+ (IKr) tail current amplitude studied in eight myocytes. (B,C) Effects of 100 nM ISO in the presence of 100 nM metoprolol (n= 6) and 300 nM CGP-20712A (n= 4). IKr amplitudes were normalized to their respective control values. Columns and bars indicate means ± SEM. *Denotes significant (P < 0.05) differences from control (100%) determined using paired t-test.
The signal transduction pathway mediating the ISO-induced stimulation of IKr was investigated using specific PKA activators and inhibitors. The effect of ISO was mimicked (i.e. IKr was increased in a similar extent) by various types of PKA activators, including 3 µM forskolin, 250 µM intracellular cAMP or 8-Br-cAMP (Figure 3). Similar results were obtained with 100 µM 6-bnz-cAMP (selective PKA activator with no effect on EPAC), while the same concentration of 8-pCPT-2'-O-Me-cAMP, a cAMP analogue known to activate EPAC without altering the activity of PKA (Holz et al., 2008), failed to enhance IKr. Intracellular application of 100 µM Rp-8-Br-cAMP, a cAMP analogue that is a selective PKA inhibitor, had no effect on baseline IKr.
Figure 3.
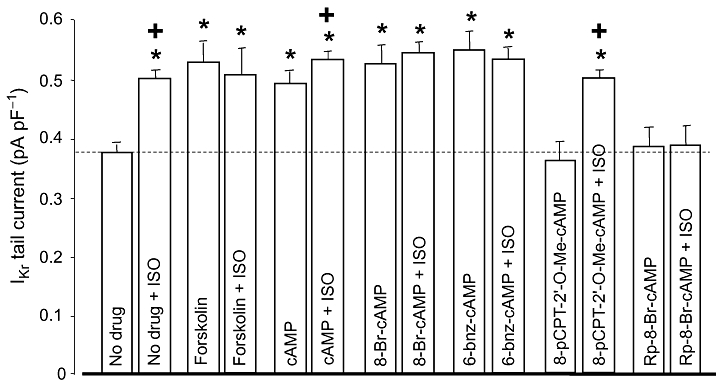
Role of the cAMP/PKA pathway in regulation of rapid delayed rectifier K+ current (IKr). Left-hand column of each pair shows IKr tail current amplitudes measured in control (no drug, n= 13), following superfusion with forskolin (3 µM, n= 6), and after internal application of cAMP (250 µM, n= 9), 8-Br-cAMP (250 µM, n= 8), 6-bnz-cAMP (100 µM, n= 8), 8-pCPT-2'-O-Me-cAMP (100 µM, n= 6) and Rp-8-Br-cAMP (100 µM, n= 6). Right-hand column of each pair indicates IKr tails obtained following superfusion with 100 nM ISO. Data are means ± SEM. *Indicates significant (P < 0.05) differences from the control (no drug) IKr amplitude, determined using unpaired t-test. +Denotes ISO-induced differences from the respective pre-ISO values, determined using paired t-test. ISO, isoprenaline; PKA, protein kinase.
The effect of ISO on IKr was fully prevented by pretreatment with some PKA activators (forskolin, 8-Br-cAMP and 6-bnz-cAMP), and by the PKA inhibitor (Rp-8-Br-cAMP). It was only partially eliminated in the presence of cAMP, while the EPAC activator 8-pCPT-2'-O-Me-cAMP had no effect (Figure 3). These results indicate that the ISO-induced enhancement of IKr is critically dependent on the activation of PKA.
Effect of ISO on IKs
IKs was activated by 3 s long depolarizing pulses to +30 mV delivered at a rate of 0.1 Hz from the holding potential of −40 mV. Tail currents, obtained after repolarization in the presence of 5 µM nifedipine plus 1 µM E-4031, were used to characterize IKs. Exposure of myocytes to 10 and 100 nM ISO increased IKs tail amplitude to 194 ± 28 and 293 ± 41%, of the control, respectively, in a largely reversible manner (Figure 4). The current was fully eliminated by 1 µM HMR-1556, indicating that it was purely IKs.
Figure 4.
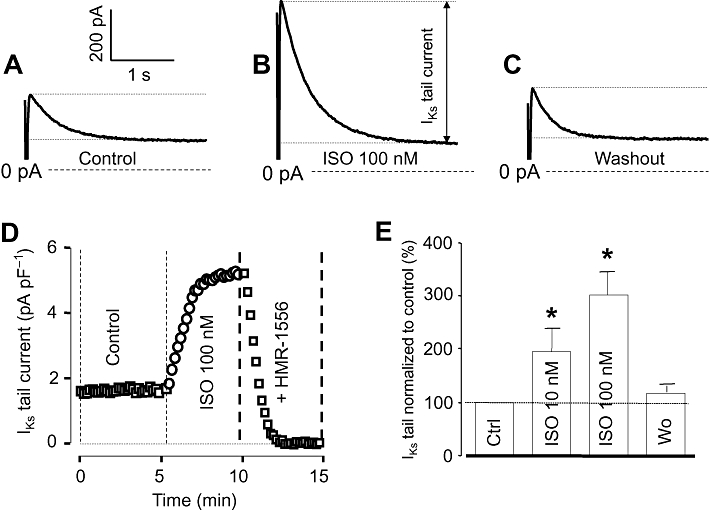
Effects of isoprenaline (ISO) on slow delayed rectifier K+ (IKs) current. (A–C) IKs tail current traces obtained at repolarization to −40 mV under control conditions (A), following exposure to 100 nM ISO (B) and after washing the cells in ISO-free solution (C). (D) Representative experiment demonstrating the effect of 100 nM ISO on IKs and the full suppression of the ISO-induced current by 1 µM HMR-1556. (E) Average data obtained in 11 myocytes showing the cumulative effect of 10 nM and 100 nM ISO on IKs tail current amplitude. Columns and bars are means ± SEM values. *Indicates significant (P < 0.05) differences from control determined using paired t-test.
Baseline IKs was significantly reduced by selective inhibition of PKA using 100 µM intracellular Rp-8-Br-cAMP (1.63 ± 0.22 pA/pF in control, n= 13 vs. 0.47 ± 0.06 pA/pF in the presence of Rp-8-Br-cAMP, n= 5). On the other hand, full activation of PKA by loading the pipette with 250 µM of the non-hydrolyzable cAMP derivative 8-Br-cAMP, increased IKs to 4.53 ± 0.63 pA/pF (n= 5), which was three times higher than its control value (Figure 5). The effect of ISO on IKs was reduced in the presence of Rp-8-Br-cAMP, and ISO failed to enhance IKs any more when PKA was fully activated by 8-Br-cAMP.
Figure 5.
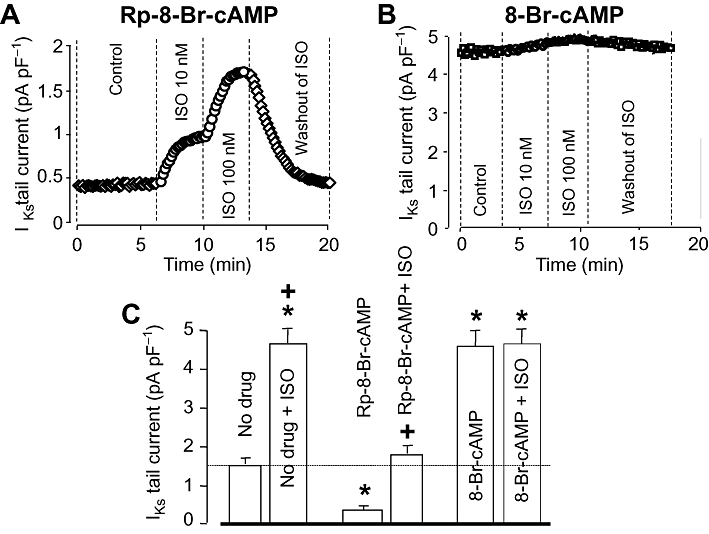
Role of the cAMP/PKA pathway in the regulation of slow delayed rectifier K+ (IKs) current. (A, B) Representative experiments demonstrating the effects of ISO in the presence of Rp-8-Br-cAMP (A) and 8-Br-cAMP (B). In (C), left-hand column of each pair shows IKs tail current amplitude measured in control (no drug, n= 11), and after internal application of Rp-8-Br-cAMP (100 µM, n= 5) and 8-Br-cAMP (250 µM, n= 5). Right-hand column of each pair indicates IKs tails obtained following superfusion with 100 nM ISO. Data are means ± SEM. *Indicates significant (P < 0.05) differences from the control (no drug)IKs amplitude determined using unpaired t-test. +Denotes ISO-induced differences from the respective pre-ISO values determined using paired t-test. ISO, isoprenaline; PKA, protein kinase.
Discussion and conclusions
In the present study, the effects of the β-adrenoceptor agonist ISO on the two components of the delayed rectifier K+ current, IKr and IKs, were studied and compared in canine myocytes. This is the first time an enhancement of IKr by ISO has been demonstrated in canine ventricular cells, which may be an important mechanism of defense against the lengthening of action potentials in the case of β-adrenoceptor stimulation. This ISO-induced enhancement of IKr seems to be mediated by the activation of PKA, because the effect of ISO was eliminated after either inhibition or full activation of PKA. It must be noted, however, that cAMP – in contrast to 8-Br-cAMP and 6-bnz-cAMP – failed to fully prevent the action of ISO. This may be explained by the proper compartmentalization of the PKA-channel complex, suggesting that the submembrane phosphodiesterase barrier may limit the accessibility of PKA from the intracellular side (Jurevicius and Fischmeister, 1996; Fischmeister et al., 2006). Thus cAMP – but not 8-Br-cAMP – might partially be degraded locally by phosphodiesterase.
In strong support of the PKA-dependent enhancement of IKr is the finding that when the current was observed in the presence of cAMP analogues, it was markedly elevated resulting in permanent activation of the enzyme. However, in contrast to our results, IKr was shown to be reduced following activation of PKA in oocytes expressing HERG channels (Thomas et al., 1999; Wei et al., 2002). The reason for this discrepancy is not clear; it may be due to the lack of other important members of the underlying signal transduction pathway in the oocytes, but it may reflect interspecies' differences as well.
Similar to our results, Heath and Terrar (2000) found that IKr was enhanced by 10 µM ISO in guinea pig ventricular cells if the conditions required to activate the conventional PKC isoenzymes were met, that is, Ca2+ current was not blocked, cytosolic Ca2+ was not buffered and the cell interior was not dialyzed. They concluded that this stimulating effect was mediated via the activation of the PKC pathway, involving crosstalk between PKA and PKC. However, the activation of the conventional PKC isoforms with thymelatoxin was shown to decrease IKr in oocytes (Thomas et al., 2003). Furthermore, our experimental conditions did not favour the activation of conventional PKC isoenzymes, as Ca2+ current was blocked by 5 µM nifedipine, the cytosolic Ca2+ was strongly buffered by 10 mM EGTA and the cell interior was dialyzed.
Similar to IKr, IKs was also equally enhanced by exposure to ISO and intracellular application of 8-Br-cAMP; however, marked differences were observed between IKr and IKs in response to ISO after inhibition of PKA. Rp-8-Br-cAMP strongly compromised baseline IKs, but failed to modify baseline IKr at all. This may indicate a more marked contribution of the cAMP/PKA pathway to the basal activity of IKs compared with that of IKr. On the other hand, pretreatment with Rp-8-Br-cAMP fully prevented the effect of ISO on IKr, but only decreased it on IKs. Thus, it appears that a moderate suppression of the cAMP/PKA pathway is sufficient to blunt the effect of ISO on IKr, which may be the consequence of a less effective stimulus transduction targeting the HERG channel. However, it is also possible that different PKA isoenzymes with different sensitivities to inhibitors are involved in mediating the effects of β-adrenoceptor stimulation to IKr and IKs. Hence, further studies are required to elucidate the differences between the fine-tuning of β-adrenoceptor stimulation of IKr and IKs.
In summary, IKr, similar to IKs, is enhanced by ISO in canine ventricular myocytes via the activation of the cAMP/PKA system. Due to the particular importance of β-adrenoceptor stimulation in controlling cardiac repolarization and the susceptibility to arrhythmias, the detailed mechanism of regulation may be a promising subject of further studies.
Acknowledgments
Financial support for the studies was provided by grants from the Hungarian Research Fund (OTKA-K68457, OTKA-K73160, CNK-77855), the Hungarian Ministry of Health (ETT-060/2006), and the Medical and Health Science Center of University of Debrecen (MEC-14/2008). The authors thank Mrs Vighné Katalin Horváth for her excellent technical assistance.
Glossary
Abbreviations
- EPAC
exchange protein directly activated by cAMP
- IKr
rapid delayed rectifier K+ current
- IKs
slow delayed rectifier K+ current
- ISO
isoprenaline
- PKA
protein kinase A
- PKC
protein kinase C
Conflict of interest
None
Supplemental material
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. 2009;158(Suppl 1):S1–S180. doi: 10.1111/j.1476-5381.2011.01649_1.x. 4th. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischmeister R, Castro LRV, Abi-Geres A, Rochais F, Jurevicius J, Leroy J, et al. Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ Res. 2006;99:816–828. doi: 10.1161/01.RES.0000246118.98832.04. [DOI] [PubMed] [Google Scholar]
- Gintant G. Two components of delayed rectifier current in canine atrium and ventricle. Circ Res. 1996;78:26–37. doi: 10.1161/01.res.78.1.26. [DOI] [PubMed] [Google Scholar]
- Heath BM, Terrar DA. Protein kinase C enhances the rapidly activating delayed rectifier potassium current, IKr, through a reduction in C-type inactivation in guinea-pig ventricular myocytes. J Physiol. 2000;522:391–402. doi: 10.1111/j.1469-7793.2000.t01-2-00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz GG, Chepurny OG, Schwede F. Epac-selective cAMP analogs: new tools with which to evaluate the signal transduction properties of cAMP-regulated guanine nucleotide exchange factors. Cell Signal. 2008;20:10–20. doi: 10.1016/j.cellsig.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurevicius J, Fischmeister R. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by β-adrenergic agonists. Proc Natl Acad Sci USA. 1996;93:295–299. doi: 10.1073/pnas.93.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karle CA, Zitron E, Zhang W, Kathöfer S, Schoels W, Kiehn J. Rapid component IKr of the guinea-pig cardiac delayed rectifier K+ current is inhibited by beta1-adrenoreceptor activation, via cAMP/protein kinase A-dependent pathways. Cardiovasc Res. 2002;53:355–362. doi: 10.1016/s0008-6363(01)00509-0. [DOI] [PubMed] [Google Scholar]
- Li G-R, Feng J, Yue L, Carrier M, Nattel S. Evidence for two components of delayed rectifier K+ current in human ventricular myocytes. Circ Res. 1996;78:689–696. doi: 10.1161/01.res.78.4.689. [DOI] [PubMed] [Google Scholar]
- Magyar J, Bányász T, Szigligeti P, Körtvély Á, Jednákovits A, Nánási PP. Electrophysiological effects of bimoclomol in canine ventricular myocytes. Naunyn Schmiedebergs Arch Pharmacol. 2000;361:303–310. doi: 10.1007/s002109900164. [DOI] [PubMed] [Google Scholar]
- Szabó G, Szentandrássy N, Bíró T, Tóth IB, Czifra G, Magyar J, et al. Asymmetrical distribution of ion channels in canine and human left ventricular wall: epicardium versus midmyocardium. Pflügers Arch. 2005;450:307–316. doi: 10.1007/s00424-005-1445-z. [DOI] [PubMed] [Google Scholar]
- Szentandrássy N, Bányász T, Bíró T, Szabó G, Tóth B, Magyar J, et al. Apico-basal inhomogeneity in distribution of ion channels in canine and human ventricular myocardium. Cardiovasc Res. 2005;65:851–860. doi: 10.1016/j.cardiores.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Thomas D, Zhang W, Karle CA, Kathöfer S, Schöls W, Kübler W, et al. Delation of protein kinase A phosphorylation sites in the HERG potassium channel inhibits activation shift by protein kinase A. J Biol Chem. 1999;274:27457–27462. doi: 10.1074/jbc.274.39.27457. [DOI] [PubMed] [Google Scholar]
- Thomas D, Zhang W, Wu K, Wimmer AB, Gut B, Wendt-Nordal G, et al. Regulation of HERG potassium channel activation by protein kinase C independent of direct phosphorylation of the channel protein. Cardiovasc Res. 2003;59:14–26. doi: 10.1016/s0008-6363(03)00386-9. [DOI] [PubMed] [Google Scholar]
- Thomas D, Kiehn J, Katus HA, Karle CA. Adrenergic regulation of the rapid component of the cardiac delayed rectifier potassium current, IKr, and the underlying hERG ion channel. Basic Res Cardiol. 2004;99:279–287. doi: 10.1007/s00395-004-0474-7. [DOI] [PubMed] [Google Scholar]
- Wei Z, Thomas D, Karle CA, Kathöfer S, Schenkel J, Kreye VA, et al. Proein kinase A-mediated phosphorylation of HERG potassium channels in a human cell line. Chin Med J. 2002;115:668–676. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.