

Abstract
BACKGROUND AND PURPOSE
β-Adrenoceptor (β-AR)-mediated inotropic effects are attenuated and Gi proteins are up-regulated in heart failure (HF). Muscarinic receptors constitutively inhibit cAMP formation in normal rat cardiomyocytes. We determined whether constitutive activity of muscarinic receptors to inhibit adenylyl cyclase (AC) increases in HF and if so, whether it modifies the reduced β-AR- or emergent 5-HT4-mediated cAMP-dependent inotropic effects.
EXPERIMENTAL APPROACH
Contractility and AC activity were measured and related to each other in rat ventricle with post-infarction HF and sham-operated (Sham) controls with or without blockade of muscarinic receptors by atropine and inactivation of Gi protein by pertussis toxin (PTX).
KEY RESULTS
Isoprenaline-mediated inotropic effects were attenuated and basal, isoprenaline- and forskolin-stimulated AC activity was reduced in HF compared with Sham. Atropine or PTX pretreatment increased forskolin-stimulated AC activity in HF hearts. β-AR-stimulated AC and maximal inotropic response were unaffected by atropine in Sham and HF. In HF, the potency of serotonin (5-HT) to evoke an inotropic response was increased in the presence of atropine with no change in the maximal inotropic response. Interestingly, PTX pretreatment reduced the potency of 5-HT to evoke inotropic responses while increasing the maximal inotropic response.
CONCLUSIONS AND IMPLICATIONS
Although muscarinic constitutive inhibition of AC is increased in HF, it does not contribute to the reduced β-AR-mediated inotropic effects in rat ventricle in HF. The data support the hypothesis that there are differences in the functional compartmentation of 5-HT4 and β-AR AC signalling in myocardium during HF.
Keywords: compartmentation, serotonin (5-HT), contractility, Gi protein, β-adrenoceptors
Introduction
A fundamental hallmark of the failing heart is the reduced contractile response to activation of β-adrenoceptors (β-ARs) resulting from increased sympathetic drive with accompanying desensitization of the β-ARs (Tilley and Rockman, 2006). The maximal contractile response to Ca2+ is similar in failing and non-failing human (Ginsburg et al., 1983; Fowler et al., 1986; Harding et al., 1990) or rat myocardium (Anand et al., 1997), indicating that decreased β-AR responsiveness does not result merely from the dysfunction of the myocardial contractile machinery. Reduced β-AR responsiveness is associated with a selective reduction of β1-AR density resulting in an increased β2/β1-AR ratio, increased expression and activity of β-AR kinases and an increase in cardiac Gi protein levels (El-Armouche et al., 2003).
The level of mRNA and protein expression of Gi is up-regulated in various models of heart failure (HF) (El-Armouche et al., 2003). Allegedly, increased Gi protein levels are part of an adaptive response to ischaemia, protecting cardiomyocytes from apoptosis (DeGeorge Jr et al., 2008). In addition, numerous reports propose that increased Gi levels also contribute to the reduction of β-AR-mediated contractile responsiveness observed in the failing myocardium (Janssen et al., 2002; El-Armouche et al., 2003; Rau et al., 2003). In direct support of this hypothesis, inactivation of Gi with pertussis toxin (PTX) partially restored attenuated β-AR-mediated contractility in various models of HF, including myocardial infarction in rats (Kompa et al., 1999), a spontaneously hypertensive rat model (Xiao et al., 2003) and in myocytes from failing human heart (Brown and Harding, 1992).
Of the adrenoceptor subtypes, β2-ARs display constitutive activity that is considerably higher than β1-ARs (Engelhardt et al., 2001). In failing rat myocytes, enhanced Gi signalling selectively inhibits the inotropic effects mediated by β2-ARs but not those mediated by β1-ARs (Xiao et al., 2003), probably because the β2-AR but not β1-AR couples to both Gs and Gi (Daaka et al., 1997). In support of this, PTX inactivation of Gi restored the β2-AR-mediated contractility (Xiao et al., 2003). These studies indicate that up-regulation of Gi activity with a concomitant potentiation of β2-AR-Gi constitutive signalling, at least in part, contributes to the reduced total β-AR-mediated contractile responses in the failing heart.
Analogous to the β2-AR system, constitutive activity of muscarinic receptors exerts a mild, continuous inhibition of adenylyl cyclase (AC) activity in normal rat ventricular cardiomyocyte membranes (Ricny et al., 2002), probably though M2 receptors (Jakubik et al., 1995). It is well established that the muscarinic system antagonizes the inotropic responses mediated by β-ARs (Dhein et al., 2001). This indirect negative inotropic response is mediated by the M2 receptor, the predominant muscarinic receptor subtype in the mammalian cardiac ventricle (Dhein et al., 2001), through activation of PTX sensitive Gi proteins and inhibition of AC (Brodde et al., 2001). Furthermore, β-AR-mediated AC activation is tonically inhibited by agonist-free (‘empty’) muscarinic receptors in rabbit myocardium (Akaishi et al., 1997). Previously, we reported that muscarinic M2 receptor expression is increased in the rat failing left ventricle compared with that of sham-operated rats (Hussain et al., 2009). In this study, we investigated whether increased Gi expression coupled with an increase in muscarinic M2 receptors in failing heart also enhances muscarinic constitutive inhibition of AC. In addition, we determined the functional effects of muscarinic constitutive inhibition of AC upon (i) the reduced β-AR-mediated inotropic effect; and (ii) the emergent cAMP-mediated inotropic effect of 5-HT (5-hydroxytryptamine, serotonin) through 5-HT4 receptors in the rat failing ventricle (Qvigstad et al., 2005a).
Methods
The experiments and animal care were conducted in accordance with ‘Regulations on Animal Experimentation’ under The Norwegian Animal Welfare Act and were approved by the Norwegian Animal Research Authority. Two animals per cage were housed in a temperature-regulated room with a 12 h/12 h light/dark cycle and given access to food and water ad libitum.
Induction of myocardial infarction and congestive HF
Male Wistar rats (Møllegaard Breeding and Research Centre, Skensved, Denmark) weighing about ∼300–350 g, were intubated and ventilated with 68% N2O, 29% O2 and 2–3% isoflurane (Abbot Scandinavia, Solna, Sweden). As described earlier (Sjaastad et al., 2000) an extensive myocardial infarction was induced by proximal ligation of the left coronary artery. Mortality rate was ∼10% the first day and ∼65% of surviving rats developed HF by 6 weeks post-infarction. Six weeks after infarction the rats were anaesthetized again, ventilated with isoflurane and left ventricular pressures were measured. The criterion for inclusion in the HF group was a left ventricular end-diastolic pressure ≥15 mmHg. Sham-operated animals (Sham) underwent the same surgical procedures, except the ligation of the coronary artery. In a subset of rats, PTX was administered at a dose of 60 µg·kg−1 i.p. 3 days prior to isolation of the muscles. Data from animals treated with PTX were included only if carbachol inhibition of the β-AR-mediated inotropic response was completely abolished.
Isolated papillary muscles and ventricular strips
Posterior left ventricular papillary muscles and left ventricle strips (diameter ∼1.0 mm) were prepared, mounted in 31°C organ baths containing physiological salt solution with 1.8 mM Ca2+, equilibrated and field-stimulated at 1 Hz (Skomedal et al., 1982; 1997;). Contraction–relaxation cycles were recorded and analysed as previously described (Sjaastad et al., 2003; Qvigstad et al., 2005b). Maximal development of force (dF/dt)max, time to peak force (TPF), time to 80% relaxation (TR80) and relaxation time (RT; RT = TR80-TPF) were measured. Inotropic responses were expressed as increases in (dF/dt)max and lusitropic responses were expressed as changes in RT. The descriptive parameters at the end of the equilibration period were used as basal (control) values. Prazosin (1 µM), a blocker of α1-adrenoceptors was added 90 min prior to agonist stimulation in the different experiments. The 5-HT4 selective antagonist GR113808 (1 µM) was given subsequent to 5-HT and completely reversed the 5-HT-evoked inotropic response, confirming the effect was 5-HT4 receptor-mediated (data not shown) as previously reported (Qvigstad et al., 2005a). The agonists in the study were added to the organ bath cumulatively (concentration–response relationships) or as a bolus. Concentration–response curves were constructed by estimating centiles (EC10 to EC100) and calculating the corresponding means, and the horizontal positioning is expressed as −logEC50 values (Sjaastad et al., 2003).
Membrane preparation and AC activity assay
Membranes were prepared as previously described (Krobert et al., 2001). AC activity was measured and analysed by determining conversion of [α-32P]-ATP to [32P]-cAMP in membranes as previously described (Krobert et al., 2001). Increases in AC activity induced by isoprenaline or forskolin (experiments performed in triplicates) are reported as % increase over basal or control (stimulated in the absence of atropine).
Experimental design to evaluate possible influences of constitutive muscarinic activity
To evaluate constitutive muscarinic receptor activity, the influence of the non-selective muscarinic inverse agonist atropine was determined on basal activation of AC and that stimulated by isoprenaline, 5-HT or forskolin and on functional effects of isoprenaline and 5-HT. The influence of prior PTX inactivation of Gi upon atropine-mediated changes in AC activity and contractility were evaluated to substantiate that the effects of atropine were mediated through constitutive muscarinic receptor activation of Gi.
Statistics
Data are expressed as mean ± SEM. P < 0.05 was considered to represent statistically significant differences (Student's t-test and anova). When appropriate, Bonferroni corrections were made to correct for multiple comparisons.
Drugs and solutions
5-HT hydrochloride, (-)isoprenaline hydrochloride, timolol maleate, prazosin hydrochloride, atropine sulphate, lidocaine (2-diethylamino-N-[2,6-dimethylphenyl]-acetamide) hydrochloride, GR113808 (1-[2-[(methylsulphonyl)-amino]ethyl]-4-piperidinyl]methyl 1-methyl-1H-indole-3-carboxylate) and L-ascorbic acid were purchased from Sigma-Aldrich (St. Louise, MO, USA). Isoflurane (1-chloro-2,2,2-trifluoroethyl difluoromethyl ether: Forene) was from Abbot Scandinavia (Solna, Sweden). PTX was from Merck chemicals (Nottingham, UK). Drug and molecular target nomenclature conforms with the British Journal of Pharmacology Guide to Receptors and Channels (Alexander et al., 2008).
Results
Animal characteristics
All rats in the HF group had large antero-lateral infarctions and signs of congestion, including tachypnea, pleural effusion and increased lung weight. Animals in the HF group had significantly increased left ventricular end-diastolic pressure, heart weight/body weight ratio, lung weight and an increased time to reach peak force in the contraction–relaxation cycle (Sjaastad et al., 2003), all indicators of congestive HF. Animal characteristics and haemodynamic data at 6 weeks after infarction are given in Table 1.
Table 1.
Animal and papillary muscle/ventricular strip characteristics
| Sham (n= 31) | HF (n= 81) | |
|---|---|---|
| Body weight, g | 391 ± 6 | 378 ± 3 |
| Heart weight, g | 1.48 ± 0.07 | 2.60 ± 0.05* |
| Heart/body weight, g·kg−1 | 3.79 ± 0.17 | 6.89 ± 0.14* |
| LVEDP, mmHg | 3.4 ± 0.3 | 23.3 ± 1.2* |
| LVSP, mmHg | 121 ± 6 | 97 ± 2* |
| Lung weight, g | 1.54 ± 0.04 | 3.89 ± 0.12* |
| Basal Fmax, mN/mm2 | 5.5 ± 0.3 | 5.3 ± 0.2 |
Student's t-test,
P < 0.05 versus Sham.
Fmax, maximal developed force; HF, heart failure; LVEDP, left ventricular end-diastolic pressure; LVSP, left ventricular systolic pressure; Sham, sham-operated.
The maximal inotropic response to β-AR stimulation is reduced and the potency of isoprenaline is increased in HF
Basal contractile force did not significantly differ between Sham (5.6 ± 0.3 mN·mm−2) and HF (5.0 ± 0.2 mN·mm−2) papillary muscles. A maximally stimulating concentration of the β-AR agonist isoprenaline (10 µM) elicited a large sustained inotropic response [(dF/dt)max 145 ± 11% above basal, n= 14] in the ventricle of Sham (Figure 1). The maximal response to isoprenaline (reached between 0.1 and 1 µM) was significantly reduced (38 ± 4% above basal, n= 21, P < 0.05) in HF rat ventricle, approximating ∼26% of the response in Sham (Figure 1). Interestingly, the potency of isoprenaline was increased in the HF group (−LogEC50 8.55 ± 0.07 M) compared with Sham (−LogEC50 7.44 ± 0.06 M, Figure 1). The potency of forskolin was also significantly increased in the HF group (−LogEC50 6.44 ± 0.16 M, n= 7) compared with Sham (−LogEC50 5.61 ± 0.11 M, n= 8, P < 0.05). Maximal forskolin-stimulated inotropic effects were also significantly reduced in HF compared with Sham (62 ± 14 and the 127 ± 18% above basal, respectively, P < 0.05). Although maximal forskolin-stimulated inotropic response was modestly higher than β-AR-mediated in the HF group, maximal forskolin-evoked inotropic responses were similar to those evoked by isoprenaline in the Sham.
Figure 1.
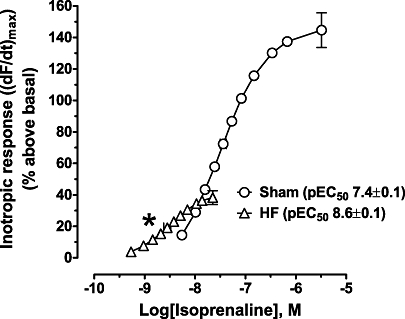
The inotropic response to isoprenaline is reduced, but the potency increased in heart failure (HF). Concentration–response curves for isoprenaline expressed as increase of maximum dF/dt [(dF/dt)max] as % above basal in sham-operated (Sham) (n= 14) or HF (n= 21) papillary muscles. Basal Fmax (mN/mm2) was 5.1 ± 0.3 and 4.8 ± 0.5 for Sham and HF respectively. Data are mean ± SEM. *P < 0.05 versus Sham. CSA, cross-sectional area.
AC activity induced by stimulation of β-ARs and forskolin is reduced in HF
Basal AC activity in HF ventricle was reduced by ∼16% compared with Sham (26 ± 1 and 31 ± 2 pmol·mg protein−1·min−1, n= 25 and n= 13 for HF and Sham, respectively, P < 0.05). The absolute isoprenaline-stimulated AC activity above basal was decreased by ∼24% in HF ventricle compared with Sham (Figure 2A). The absolute forskolin-stimulated AC activity above basal was decreased by ∼32% in HF ventricle compared with Sham (Figure 2B). Isoprenaline- and forskolin-stimulated AC activities, expressed as a percentage change over basal (to account for the changes in basal AC activity), were also significantly reduced (isoprenaline: 148 ± 6 vs. 124 ± 7%; 1 µM forskolin: 565 ± 19 vs. 427 ± 12%; 10 µM forskolin: 1056 ± 44 vs. 836 ± 20% for Sham and HF, respectively, all P < 0.05).
Figure 2.
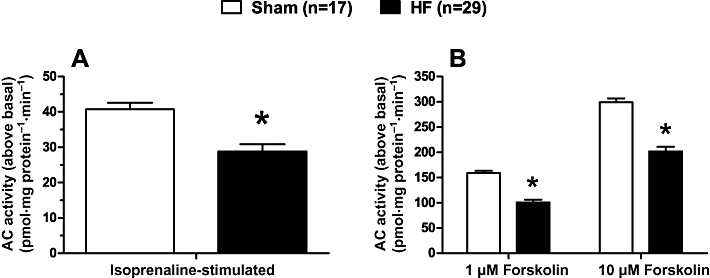
The stimulation of adenylyl cyclase (AC) activity by isoprenaline and forskolin is reduced in heart failure (HF). (A) Isoprenaline (10 µM) – or (B) forskolin-stimulated AC activity in membranes of left ventricle prepared from sham-operated (Sham) or HF rats. Data are mean ± SEM. *P < 0.05 versus Sham.
Constitutive inhibition of AC by muscarinic receptors is increased in HF
Forskolin-stimulated AC activity was significantly increased in HF ventricle compared with Sham after inactivation of muscarinic receptors by the non-selective muscarinic inverse agonist atropine (Figure 3A), indicative of an increase in muscarinic constitutive activity in HF (Ricny et al., 2002). The magnitude of the absolute increase in AC activation (∼4 pmol·mg protein−1·min−1) approximated a ∼20% increase relative to basal AC activity (Figure 3B). However, β-AR-stimulated AC activity was not increased by atropine inactivation of muscarinic receptors (Figure 3A). Muscarinic constitutive AC inhibition was abolished in HF ventricle by prior inactivation of Gi with PTX (Figure 3A,B). PTX inactivation of Gi alone significantly increased forskolin-stimulated AC activity (∼20%) in HF ventricle (Figure 4). These data support the hypothesis that atropine's effect is through removal of the constitutive activation of Gi, because the relative increase in AC activity elicited by either atropine or PTX treatment was similar (∼20%) (Figures 3B and 4).
Figure 3.
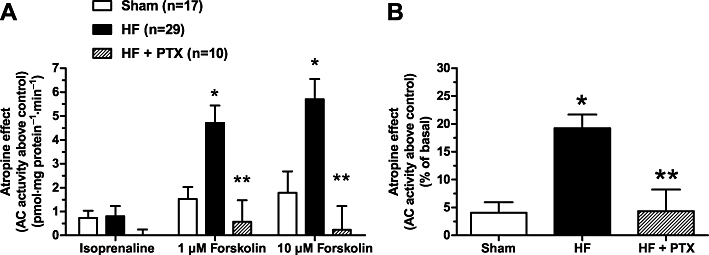
Atropine increases forskolin-stimulated adenylyl cyclase (AC) activity more in HF than sham-operated (Sham), and the effect of atropine is eliminated by pertussis toxin (PTX). The figure shows the effect of blocking muscarinic receptor constitutive activity with atropine (1 µM) on isoprenaline (10 µM)- or forskolin (1 and 10 µM)-activated AC activity in membranes of left ventricle from Sham, heart failure (HF) or HF rats pretreated with PTX. (A) Data shown are the mean ± SEM of the absolute difference in AC activity induced by isoprenaline or forskolin in the presence of atropine minus that in the absence of atropine (control). (B) The combined mean of the difference scores of 1 and 10 µM forskolin-stimulated AC activity in membranes of left ventricle (same data from A) reported as a percentage of basal AC activity for each respective group. *P < 0.05 versus Sham only. **P < 0.05 versus HF group.
Figure 4.
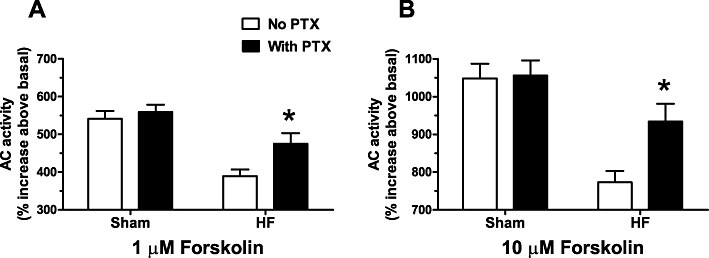
Pertussis toxin (PTX) pretreatment counteracts the reduction in forskolin-stimulated adenylyl cyclase (AC) activity in heart failure (HF). The figure shows the effect of pretreatment with PTX on 1 µM (A) and 10 µM (B) forskolin-stimulated AC activity in membranes of left ventricle from sham-operated (Sham) or HF rats. Data are mean ± SEM and are reported as % increase over basal. *P < 0.05 versus without PTX.
Muscarinic receptor constitutive activity modulates only 5-HT4-mediated contractility
Neither the potency (pEC50 of 7.5 ± 0.1 and 8.5 ± 0.1 for Sham and HF respectively) nor the efficacy (Figure 5A) of isoprenaline to induce an inotropic effect was changed in Sham or HF ventricle after inactivation of muscarinic receptor constitutive activity by the non-selective muscarinic inverse agonist atropine. Likewise, neither the potency nor efficacy of forskolin to induce an inotropic effect was increased by atropine (data not shown). However, in the presence of atropine, the potency of the 5-HT4-stimulated inotropic effect was significantly increased by a 0.25 ± 0.08 log unit shift (Figure 5B; pEC50= 7.33 ± 0.05 without atropine and pEC50= 7.56 ± 0.05 with atropine; P < 0.05) without an increase in the maximal inotropic effect (Figure 5B). Prior inactivation of Gi with PTX blocked the ability of atropine to increase 5-HT potency in HF (data not shown). PTX inactivation of Gi significantly reduced the potency of the 5-HT4-stimulated inotropic effect by 0.5 ± 0.2 log unit (Figure 5B; pEC50= 6.85 ± 0.2 with PTX pretreatment and pEC50= 7.35 ± 0.05 without PTX) while increasing the maximal 5-HT inotropic effect by ∼107% (∼36% and 17% above basal, respectively, Figure 5B). Activation of AC by 5-HT was not detectable in HF ventricular membranes, but was detectable (∼6% increase over basal) in HF ventricles pretreated with PTX (basal AC 17.6 ± 1.8 pmol·mg protein−1·min−1, 5-HT-stimulated 18.6 ± 1.8 pmol·mg protein−1·min−1, n= 8, P < 0.05), consistent with the increased 5-HT4-mediated inotropic effect.
Figure 5.
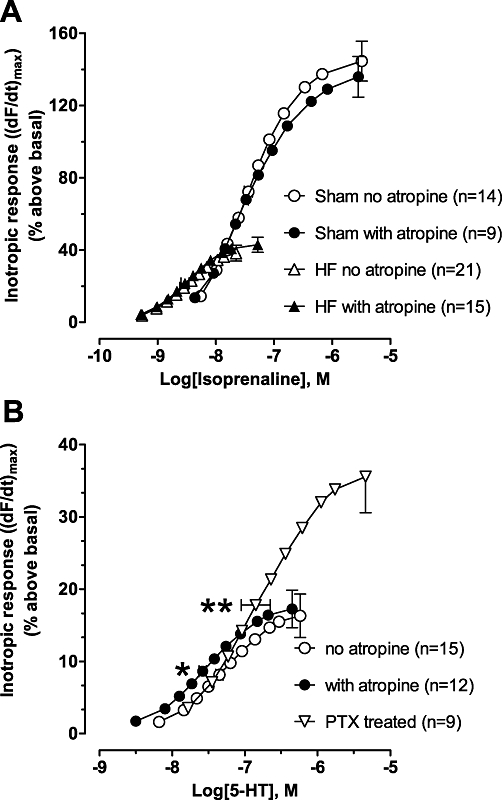
Atropine sensitizes heart failure (HF) ventricles to 5-hydroxytryptamine (5-HT)- but not isoprenaline-evoked inotropic effects. (A) Concentration–response curves of the inotropic response to isoprenaline in HF or sham-operated (Sham) papillary muscles in the absence or presence of 1 µM atropine. Inotropic response is expressed as increase of maximum dF/dt [(dF/dt)max] as % above basal. Basal Fmax (mN/mm2) was 5.1 ± 0.3 and 5.9 ± 0.4 for Sham and 4.8 ± 0.5 and 5.0 ± 0.3 for HF in the absence and presence of atropine, respectively. Data are mean ± SEM. (B) Effects of atropine (1 µM) and pertussis toxin (PTX) on the concentration-response curve of the inotropic response to 5-HT in HF papillary muscles. The 5-HT concentration-response curve for the PTX-treated group is the combined data of muscles with and without atropine from nine rats, because atropine did not significantly alter potency in PTX-treated rats. Basal Fmax (mN/mm2) was 5.4 ± 0.4 and 5.2 ± 0.5 in the absence and presence of atropine, respectively, and 4.9 ± 0.3 in the PTX-treated group. Data are mean ± SEM. *P < 0.05 versus no atropine. **P < 0.05 versus non-PTX treated.
Discussion and conclusions
Constitutive muscarinic receptor activity is enhanced in failing ventricle
The present study indicates that constitutive activity of muscarinic receptors to inhibit AC is increased in the left ventricle of rats with HF compared with Sham. Our data do not support the hypothesis that an increase of constitutive muscarinic receptor inhibition of AC contributes to the reduced β-AR-mediated inotropic effect in failing ventricle. However, muscarinic receptor constitutive inhibition of AC appears to reduce the magnitude and potency of cAMP-mediated inotropic effects evoked by stimulation of 5-HT4 receptors in failing ventricle. Our results are consistent with previous reports that muscarinic receptors constitutively inhibit AC activity in normal rat ventricle (Ricny et al., 2002); and to our knowledge, these data are the first to demonstrate a functional significance of constitutive muscarinic receptor activity altering contractility in the failing heart.
Muscarinic constitutive inhibition of AC was significantly enhanced in HF ventricle (∼20% increase relative to basal AC; Figure 3A,B), by a magnitude that corresponds well with the increase in M2 receptor expression (∼26%) previously reported by us (Hussain et al., 2009). The absolute magnitude of muscarinic constitutive inhibition of forskolin-stimulated AC remained constant in both Sham and HF, despite a doubling in forskolin-stimulated AC activity when the concentration increased from 1–10 µM (see Figures 2 and 3). Therefore, muscarinic constitutive inhibition of AC is independent of forskolin concentration, indicating muscarinic constitutive inhibitory effects are limited to a localized subset of the AC that is accessible to forskolin.
Constitutive muscarinic receptor activity is dependent on Gi
Inactivation of Gi with PTX completely blocked the ability of atropine to increase forskolin-stimulated AC activity, indicating atropine's effect is probably due to binding at the Gi-coupled muscarinic M2 receptor (Brodde et al., 2001). Inactivation of Gi alone also increased forskolin-stimulated AC activity only in HF, not Sham (Figure 4). This finding is in accordance with previous reports indicating Gi activity is increased in the failing heart (El-Armouche et al., 2003). In fact, atropine and PTX pretreatment both increased forskolin-stimulated AC activity by relatively the same magnitude (∼20% increase when compared with basal and ∼20% increase above basal, respectively, Figures 3B and 4). The data do not allow us to determine if the increased effect of atropine in failing heart results from the generalized increase in Gi activity only or from other mechanisms regulating AC activity. An increase in muscarinic receptor levels in failing rat ventricle (Hussain et al., 2009) may also be a contributing factor. Interestingly, neither the potency nor efficacy of isoprenaline-evoked inotropic effects was enhanced by atropine (Figure 5A). This finding is consistent with the absence of atropine effects upon isoprenaline activation of AC (Figure 3A). However, β-AR-mediated activation of AC was potentiated by PTX inactivation of constitutive Gi activity in rabbit ventricular myocardium (Akaishi et al., 1997), indicating the functional role of constitutive Gi activity varies among species.
Inactivation of Gi enhances the 5-HT-evoked inotropic effect and normalizes the relationship between inotropic and lusitropic concentration–response curves
Inactivation of Gi with PTX did not increase 5-HT potency (as expected if removing tonic Gi inhibition similar to atropine) but rather reduced the potency of 5-HT to elicit inotropic effects (rightward shift in EC50, Figure 5B). Typically, a reduction of drug potency is indicative of a desensitized receptor system and would normally be coupled with a concomitant reduction in receptor–effector coupling (efficacy). However and perhaps the most intriguing finding of this study, the reduced 5-HT potency was coupled with the unexpected and counter-intuitive finding that 5-HT-evoked maximal inotropic effects were significantly amplified after Gi inactivation (Figure 5B).
We propose that this paradoxical effect of Gi inactivation upon the 5-HT4-evoked inotropic effect possibly results from a restoration in the correlation between the inotropic and lusitropic concentration–response curves by PTX. The potency of 5-HT to mediate the inotropic effect is higher compared with the lusitropic effect in non-PTX-treated rats (Figure 6A). At higher 5-HT concentrations the divergence is greater, indicating 5-HT elicits a lusitropic effect well after the inotropic effect has reached maximum (∼4.0 µM and 0.4 µM, respectively; Figure 6A). Similar results have been reported for β-AR-evoked inotropic effects in failing rat ventricle (Qvigstad et al., 2005b). In contrast, the maximal inotropic and lusitropic effects were achieved over the same concentration range for β-AR stimulation in Sham (Qvigstad et al., 2005b). In HF ventricle, possibly the lusitropic effect is increased relative to the inotropic component, impeding development of the maximal inotropic response.
Figure 6.
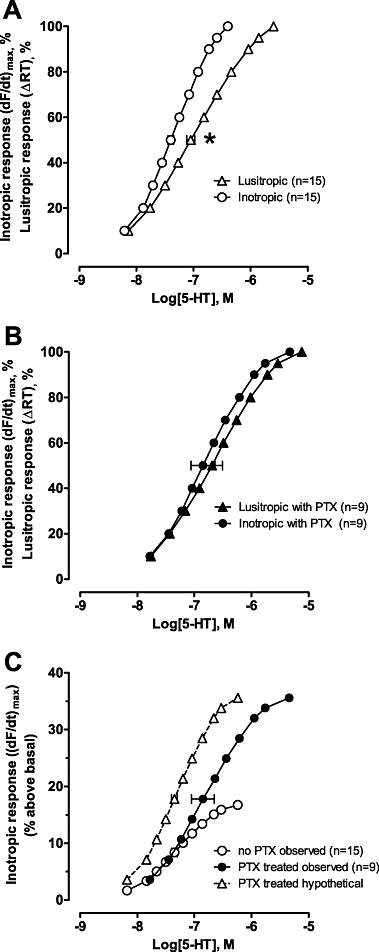
Concentration–response curves for functional effects of 5-hydroxytryptamine (5-HT) in heart failure (HF) papillary muscles from (A) rats without or (B) pretreated with pertussis toxin (PTX). Inotropic [increase of (dF/dt)max] and lusitropic responses [reduction of relaxation time (ΔRT)] are expressed as % of maximum within each experiment. Data are mean ± SEM. *P < 0.05 inotropic versus lusitropic EC50 in non-PTX treated rats. (C) Inotropic [increase of (dF/dt)max] concentration-response curves for 5-HT in HF papillary muscles from rats pretreated with or without PTX plotted as the actual group mean (% above basal). The maximal 5-HT-evoked inotropic effect plotted was the mean from muscles with and without atropine (16.7 and 35.6% above basal without and with PTX pretreatment respectively). The hypothetical curve (open triangles) in (C) illustrates the expected concentration-response curve for 5-HT if PTX treatment did not change potency (obtained by multiplying the response at each concentration with the same factor to adjust for increased efficacy). Thus, the unexpected reduction in potency of the observed data is clearly visualized, revealing additional effects of PTX.
In PTX-treated rats, the maximal inotropic and lusitropic effects were achieved over a similar concentration range of 5-HT stimulation (Figure 6B). Therefore, inactivation of Gi normalizes the relationship between inotropic and lusitropic concentration–response curves similar to that observed with β-AR-mediated effects in Sham or normal rat ventricle. The current data do not distinguish if PTX inactivation of Gi directly enhances the 5-HT inotropic effect or modifies the interaction of the lusitropic effect upon the inotropic. However, because PTX removal of Gi inhibition did not increase the maximal 5-HT lusitropic effect (no change in relaxation time, data not shown) or the concentration–response range (see Figure 6A,B), it seems more likely that Gi inactivation directly modifies the maximal inotropic response. In support of this, PTX inactivation of Gi increased 5-HT-evoked AC activity in the current study, increased 5-HT4(b)-mediated cAMP formation in HEK293 cells (Pindon et al., 2002) and L-type Ca2+ current in rat cardiomyocytes (Pindon et al., 2002), all changes supportive of an increased inotropic effect.
The 5-HT4 receptor subtype mediating inotropic effects in failing ventricle may couple dually with Gs and Gi
The 5-HT4(b) receptor subtype, that couples dually to Gs and Gi (Pindon et al., 2002), may account for the sensitivity of the 5-HT inotropic response to Gi inhibition and the divergence in lusitropic and inotropic effects as discussed above. As seen in Figure 6C, the 5-HT concentration–response curves in PTX-pretreated hearts were virtually identical to those in untreated hearts at lower 5-HT concentrations, but the maximal effect was achieved at higher 5-HT concentrations. This effect of PTX is unlikely to be due to an alteration in Gs-protein activation, because PTX inactivation of Gi altered GTPγS binding and AC activation of only the dually coupled 5-HT4(b) and not the Gs-coupled 5-HT4(a) subtype (Pindon et al., 2002). Our data fits best with a model whereby dual coupling is dependent on agonist concentration, similar to that reported for the dopamine D2 receptor (Chang et al., 1997). Thus, the possible dual coupling of 5-HT4(b) receptors to Gi at higher 5-HT concentrations may constrain the maximal 5-HT4-evoked inotropic effect by impeding Gs activation. Removal of the Gi inhibition by PTX would allow further development of the inotropic response at the higher 5-HT concentrations (extending the concentration–response range), giving the false appearance of a reduction in 5-HT potency (EC50 rightward shift, Figures 5B and 6C). In comparison, if we assume dual coupling to Gs and Gi is independent of agonist concentration, after PTX inactivation of Gi, the 5-HT inotropic effect would be expected to be larger (increased Gs activation of AC) over the entire concentration range (EC50 remains the same) to reach the maximal inotropic response (Figure 6C, hypothetical curve). In fact, the EC50 of 5-HT to evoke inotropic effects in PTX-treated heart begins to approach the EC50 of 5-HT to activate AC at 5-HT4(b) receptors expressed in HEK293 cells (pEC50 of 6.91 and ∼6.7–7.2, respectively; Bruheim et al., 2003), indicating PTX restores the relationship of the potency to activate AC with the functional response. Regardless of mechanism, in rat ventricles, clearly Gi plays a key role in the compartmentation of 5-HT4-evoked AC signalling to access the contractile apparatus.
Muscarinic, β-AR and 5-HT receptor systems are localized to different functional compartments
Given the relatively large absolute muscarinic constitutive inhibition revealed by forskolin-stimulated AC (∼16% of the isoprenaline-stimulated increase of AC), it is intriguing that atropine did not significantly increase isoprenaline-stimulated AC activity in HF (Figure 3A). The differential sensitivity of β-AR and 5-HT receptor systems to muscarinic constitutive activity may, in part, be due to differences in compartmentation. Head et al. (2005) have shown that β-ARs and muscarinic M2 receptors with their associated signalling components (Gs and Gi respectively) vary in their subcellular distribution in adult rat cardiomyocytes. Whereas the β1-AR was primarily localized to low-density gradient fractions with higher levels of caveolin, M2 receptors were only found in high-density fractions with lower caveolin levels (Steinberg, 2004; Head et al., 2005). Interestingly, the cholinergic agonist carbachol promotes the translocation of M2 receptors into the low-density fraction (Feron et al., 1997) and may account for M2-mediated accentuated antagonism upon β1-AR stimulation (Iancu et al., 2007; 2008;). Importantly, atropine did not induce translocation of M2 and also prevented translocation by carbachol (Feron et al., 1997). These findings are highly relevant, because they provide a structural basis for the possibility that β1-ARs and M2 receptors reside in different subcellular microdomains, possibly explaining why atropine did not modify β-AR-mediated inotropic responses in the current study. It is reasonable to propose that in normal rat ventricle, 5-HT4 receptors are localized in subcellular microdomains that are functionally defined by an uncoupling of the production of cAMP from the phosphorylation of contractile proteins [analogous to prostaglandin receptors (Warrier et al., 2007)] that mediate inotropic support [5-HT does not evoke inotropic effects in normal rat ventricle (Qvigstad et al., 2005a)]. Perhaps, 5-HT4 receptors are localized to the same subcellular microdomain as muscarinic M2 receptors, and thereby are susceptible to the enhanced muscarinic constitutive inhibition of AC in HF ventricle.
In summary, these data demonstrate that muscarinic constitutive activation of AC is increased in HF rat ventricle. Removal of constitutive muscarinic inhibition of AC by atropine and PTX inactivation of Gi revealed differences in functional compartmentation of 5-HT4 and β-AR AC signalling in HF myocardium. Additional biochemical studies coupled with confocal microscopic evidence are required to substantiate and further clarify the role of muscarinic receptors and Gi to regulate compartmentation of 5-HT4- and β-AR-mediated inotropic and lusitropic effects in the failing ventricle.
Acknowledgments
This work was supported by the Norwegian Council on Cardiovascular Diseases, the Research Council of Norway, the Anders Jahre Foundation for the promotion of science, the Family Blix foundation, the Helgesen Foundation and grants from the University of Oslo. The experiments were performed in accordance with all regulations concerning biomedical research in Norway.
Conflict of interest
The authors state no conflict of interest.
Supplemental material
References
- Akaishi Y, Hattori Y, Kanno M, Sakuma I, Kitabatake A. Agonist-independent tonic inhibitory influence of Gi on adenylate cyclase activity in rabbit ventricular myocardium and its removal by pertussis toxin: a role of empty receptor-mediated Gi activation. J Mol Cell Cardiol. 1997;29:765–775. doi: 10.1006/jmcc.1996.0315. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl)(2):S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand IS, Liu D, Chugh SS, Prahash AJ, Gupta S, John R, et al. Isolated myocyte contractile function is normal in postinfarct remodeled rat heart with systolic dysfunction. Circulation. 1997;96:3974–3984. doi: 10.1161/01.cir.96.11.3974. [DOI] [PubMed] [Google Scholar]
- Brodde OE, Bruck H, Leineweber K, Seyfarth T. Presence, distribution and physiological function of adrenergic and muscarinic receptor subtypes in the human heart. Basic Res Cardiol. 2001;96:528–538. doi: 10.1007/s003950170003. [DOI] [PubMed] [Google Scholar]
- Brown LA, Harding SE. The effect of pertussis toxin on β-adrenoceptor responses in isolated cardiac myocytes from noradrenaline-treated guinea-pigs and patients with cardiac failure. Br J Pharmacol. 1992;106:115–122. doi: 10.1111/j.1476-5381.1992.tb14302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruheim S, Krobert KA, Andressen KW, Levy FO. Unaltered agonist potency upon inducible 5-HT7(a) but not 5-HT4(b) receptor expression indicates agonist-independent association of 5-HT7(a) receptor and Gs. Receptors Channels. 2003;9:107–116. [PubMed] [Google Scholar]
- Chang A, Shin SH, Pang SC. Dopamine D2 receptor mediates both inhibitory and stimulatory actions on prolactin release. Endocrine. 1997;7:177–182. doi: 10.1007/BF02778139. [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- DeGeorge BR, Jr, Gao E, Boucher M, Vinge LE, Martini JS, Raake PW, et al. Targeted inhibition of cardiomyocyte Gi signaling enhances susceptibility to apoptotic cell death in response to ischemic stress. Circulation. 2008;117:1378–1387. doi: 10.1161/CIRCULATIONAHA.107.752618. [DOI] [PubMed] [Google Scholar]
- Dhein S, van Koppen CJ, Brodde OE. Muscarinic receptors in the mammalian heart. Pharmacol Res. 2001;44:161–182. doi: 10.1006/phrs.2001.0835. [DOI] [PubMed] [Google Scholar]
- El-Armouche A, Zolk O, Rau T, Eschenhagen T. Inhibitory G-proteins and their role in desensitization of the adenylyl cyclase pathway in heart failure. Cardiovasc Res. 2003;60:478–487. doi: 10.1016/j.cardiores.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Engelhardt S, Grimmer Y, Fan GH, Lohse MJ. Constitutive activity of the human β1-adrenergic receptor in β1-receptor transgenic mice. Mol Pharmacol. 2001;60:712–717. [PubMed] [Google Scholar]
- Feron O, Smith TW, Michel T, Kelly RA. Dynamic targeting of the agonist-stimulated M2 muscarinic acetylcholine receptor to caveolae in cardiac myocytes. J Biol Chem. 1997;272:17744–17748. doi: 10.1074/jbc.272.28.17744. [DOI] [PubMed] [Google Scholar]
- Fowler MB, Laser JA, Hopkins GL, Minobe W, Bristow MR. Assessment of the β-adrenergic receptor pathway in the intact failing human heart: progressive receptor down-regulation and subsensitivity to agonist response. Circulation. 1986;74:1290–1302. doi: 10.1161/01.cir.74.6.1290. [DOI] [PubMed] [Google Scholar]
- Ginsburg R, Esserman LJ, Bristow MR. Myocardial performance and extracellular ionized calcium in a severely failing human heart. Ann Intern Med. 1983;98:603–606. doi: 10.7326/0003-4819-98-5-603. [DOI] [PubMed] [Google Scholar]
- Harding SE, Jones SM, O'Gara P, Vescovo G, Poole-Wilson PA. Reduced β-agonist sensitivity in single atrial cells from failing human hearts. Am J Physiol. 1990;259:H1009–H1014. doi: 10.1152/ajpheart.1990.259.4.H1009. [DOI] [PubMed] [Google Scholar]
- Head BP, Patel HH, Roth DM, Lai NC, Niesman IR, Farquhar MG, et al. G-protein-coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin-3-associated microdomains in adult cardiac myocytes. J Biol Chem. 2005;280:31036–31044. doi: 10.1074/jbc.M502540200. [DOI] [PubMed] [Google Scholar]
- Hussain RI, Qvigstad E, Birkeland JA, Eikemo H, Glende A, Sjaastad I, et al. Activation of muscarinic receptors elicits inotropic responses in ventricular muscle from rats with heart failure through myosin light chain phosphorylation. Br J Pharmacol. 2009;156:575–586. doi: 10.1111/j.1476-5381.2009.00016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iancu RV, Jones SW, Harvey RD. Compartmentation of cAMP signaling in cardiac myocytes: a computational study. Biophys J. 2007;92:3317–3331. doi: 10.1529/biophysj.106.095356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iancu RV, Ramamurthy G, Warrier S, Nikolaev VO, Lohse MJ, Jones SW, et al. Cytoplasmic cAMP concentrations in intact cardiac myocytes. Am J Physiol Cell Physiol. 2008;295:C414–C422. doi: 10.1152/ajpcell.00038.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubik J, Bacakova L, el-Fakahany EE, Tucek S. Constitutive activity of the M1-M4 subtypes of muscarinic receptors in transfected CHO cells and of muscarinic receptors in the heart cells revealed by negative antagonists. FEBS Lett. 1995;377:275–279. doi: 10.1016/0014-5793(95)01360-1. [DOI] [PubMed] [Google Scholar]
- Janssen PM, Schillinger W, Donahue JK, Zeitz O, Emami S, Lehnart SE, et al. Intracellular β-blockade: overexpression of Gα(i2) depresses the β-adrenergic response in intact myocardium. Cardiovasc Res. 2002;55:300–308. doi: 10.1016/s0008-6363(02)00406-6. [DOI] [PubMed] [Google Scholar]
- Kompa AR, Gu XH, Evans BA, Summers RJ. Desensitization of cardiac β-adrenoceptor signaling with heart failure produced by myocardial infarction in the rat. Evidence for the role of Gi but not Gs or phosphorylating proteins. J Mol Cell Cardiol. 1999;31:1185–1201. doi: 10.1006/jmcc.1999.0951. [DOI] [PubMed] [Google Scholar]
- Krobert KA, Bach T, Syversveen T, Kvingedal AM, Levy FO. The cloned human 5-HT7 receptor splice variants: a comparative characterization of their pharmacology, function and distribution. Naunyn-Schiedeberg's Arch Pharmacol. 2001;363:620–632. doi: 10.1007/s002100000369. [DOI] [PubMed] [Google Scholar]
- Pindon A, van Hecke G, van Gompel P, Lesage AS, Leysen JE, Jurzak M. Differences in signal transduction of two 5-HT4 receptor splice variants: compound specificity and dual coupling with Gαs- and Gαi/o-proteins. Mol Pharmacol. 2002;61:85–96. doi: 10.1124/mol.61.1.85. [DOI] [PubMed] [Google Scholar]
- Qvigstad E, Brattelid T, Sjaastad I, Andressen KW, Krobert KA, Birkeland JA, et al. Appearance of a ventricular 5-HT4 receptor-mediated inotropic response to serotonin in heart failure. Cardiovasc Res. 2005a;65:869–878. doi: 10.1016/j.cardiores.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Qvigstad E, Sjaastad I, Bøkenes J, Schiander I, Solberg L, Sejersted OM, et al. Carvedilol blockade of α1- and β-adrenoceptor induced inotropic responses in rats with congestive heart failure. Eur J Pharmacol. 2005b;516:51–59. doi: 10.1016/j.ejphar.2005.04.027. [DOI] [PubMed] [Google Scholar]
- Rau T, Nose M, Remmers U, Weil J, Weissmuller A, Davia K, et al. Overexpression of wild-type Gai-2 suppresses β-adrenergic signaling in cardiac myocytes. FASEB J. 2003;17:523–525. doi: 10.1096/fj.02-0660fje. [DOI] [PubMed] [Google Scholar]
- Ricny J, Gualtieri F, Tucek S. Constitutive inhibitory action of muscarinic receptors on adenylyl cyclase in cardiac membranes and its stereospecific suppression by hyoscyamine. Physiol Res. 2002;51:131–137. [PubMed] [Google Scholar]
- Sjaastad I, Sejersted OM, Ilebekk A, Bjørnerheim R. Echocardiographic criteria for detection of postinfarction congestive heart failure in rats. J Appl Physiol. 2000;89:1445–1454. doi: 10.1152/jappl.2000.89.4.1445. [DOI] [PubMed] [Google Scholar]
- Sjaastad I, Schiander I, Sjetnan A, Qvigstad E, Bøkenes J, Sandnes D, et al. Increased contribution of α1- vs. β-adrenoceptor-mediated inotropic response in rats with congestive heart failure. Acta Physiol Scand. 2003;177:449–458. doi: 10.1046/j.1365-201X.2003.01063.x. [DOI] [PubMed] [Google Scholar]
- Skomedal T, Osnes JB, Øye I. Differences between α-adrenergic and β-adrenergic inotropic effects in rat heart papillary muscles. Acta Pharmacol Toxicol (Copenh) 1982;50:1–12. doi: 10.1111/j.1600-0773.1982.tb00932.x. [DOI] [PubMed] [Google Scholar]
- Skomedal T, Borthne K, Aass H, Geiran O, Osnes JB. Comparison between α1-adrenoceptor-mediated and β-adrenoceptor-mediated inotropic components elicited by norepinephrine in failing human ventricular muscle. J Pharmacol Exp Ther. 1997;280:721–729. [PubMed] [Google Scholar]
- Steinberg SF. β2-Adrenergic receptor signaling complexes in cardiomyocyte caveolae/lipid rafts. J Mol Cell Cardiol. 2004;37:407–415. doi: 10.1016/j.yjmcc.2004.04.018. [DOI] [PubMed] [Google Scholar]
- Tilley DG, Rockman HA. Role of β-adrenergic receptor signaling and desensitization in heart failure: new concepts and prospects for treatment. Expert Rev Cardiovasc Ther. 2006;4:417–432. doi: 10.1586/14779072.4.3.417. [DOI] [PubMed] [Google Scholar]
- Warrier S, Ramamurthy G, Eckert RL, Nikolaev VO, Lohse MJ, Harvey RD. cAMP microdomains and L-type Ca2+ channel regulation in guinea-pig ventricular myocytes. J Physiol. 2007;580:765–776. doi: 10.1113/jphysiol.2006.124891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao RP, Zhang SJ, Chakir K, Avdonin P, Zhu W, Bond RA, et al. Enhanced Gi signaling selectively negates β2-adrenergic receptor (AR) – but not β1-AR-mediated positive inotropic effect in myocytes from failing rat hearts. Circulation. 2003;108:1633–1639. doi: 10.1161/01.CIR.0000087595.17277.73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.