

Abstract
BACKGROUND AND PURPOSE
By interacting with trkB receptors, brain-derived neurotrophic factor (BDNF) triggers various signalling pathways responsible for neurone survival, differentiation and modulation of synaptic transmission. Numerous reports have implicated BDNF and trkB in the pathogenesis of various central nervous system affections and in cancer, thus representing trkB as a promising therapeutic target. In this study, we used an antibody-based approach to search for trkB-selective functional reagents.
EXPERIMENTAL APPROACH
Six commercially available polyclonal and monoclonal antibodies were tested on recombinant and native, human and rodent trkB receptors. Functional and pharmacological characterization was performed using a modified version of the KIRA-elisa method and radioligand binding studies. Western blot analyses and neurite outgrowth assays were carried out to determine the specificity and selectivity of antibody effects. The survival properties of one antibody were further assessed on cultured neurones in a serum-deprived paradigm.
KEY RESULTS
The functional trkB-selective antibodies showed distinct pharmacological profiles, ranging from partial agonists to antagonists, acting on trkB receptors through allosteric modulations. The same diversity of effects was observed on the mitogen-activated protein kinase signalling pathway downstream of trkB and on the subsequent neurite outgrowth. One antibody with partial agonist activity demonstrated cell survival properties by activating the Akt pathway. Finally, these antibodies were functionally validated as true trkB-selective ligands because they failed activating trkA or trkC, and contrary to BDNF, none of them bind to p75NTR.
CONCLUSIONS AND IMPLICATIONS
These trkB-selective antibodies represent a novel class of pharmacological tools to explore the pathophysiological roles of trkB and its potential therapeutic relevance for the treatment of various disorders.
Keywords: BDNF, trkB receptor, functional ligand, agonist, antagonist, antibody, brain disease, cancer
Introduction
Tyrosine kinase receptors are a family of single transmembrane glycoproteins that mediate various trophic effects of the neurotrophins, including nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT3) and NT4/5. Each neurotrophin binds preferentially to a specific receptor: NGF binds mainly to trkA, BDNF and NT4/5 to trkB, and NT3 to trkC. In addition, the p75NTR binds all mature neurotrophins with approximately equal affinity and can act as a co-receptor for trk receptors in regulating both affinity and amplitude of response for a preferred neurotrophin (Hempstead et al., 1991; Benedetti et al., 1993). Binding of BDNF to trkB triggers receptor dimerization that leads to conformational changes and activation of intracellular signal transduction pathways, such as mitogen-activated protein kinase (MAPK), phosphatidylinositol-3 kinase and phospholipase C-γ (Huang and Reichardt, 2003). In adult humans, trkB is one of the most widely distributed neurotrophic receptors in the brain, the expression of which is particularly high in areas such as the neocortex, hippocampus, striatum and brainstem (Muragaki et al., 1995). Numerous studies have demonstrated crucial roles for BDNF in a wide range of neuronal processes such as neurogenesis, differentiation, survival and modulation of synaptic plasticity (for review, see Huang and Reichardt, 2001; Poo, 2001; Chao, 2003; Lu, 2003).
Given its neurotrophic effects and its contribution to the regulation of synaptic transmission and plasticity, BDNF has been strongly associated with many pathophysiological states, such as amyotrophic lateral sclerosis (Mojsilovic-Petrovic et al., 2006), Alzheimer's disease (Murer et al., 2001), pain (Pezet and McMahon, 2006), epilepsy (McNamara et al., 2006) and various neuropsychiatric disorders (Nestler et al., 2002; Durany and Thome, 2004; Chen et al., 2006; Shaltiel et al., 2007). Recent reports have also suggested a crucial role for trkB in tumour formation, angiogenesis and metastasis, pointing at the receptor for BDNF as a new promising target for anti-cancer therapies (Douma et al., 2004; Geiger and Peeper, 2005; Desmet and Peeper, 2006). For all these diseases, research has reached the point of designing strategies for therapeutic intervention at the BDNF/trkB system. However, most of the TrkB-targeting compounds that have been reported so far suffer from either lack of specificity and/or efficacy in appropriate models, thus representing an ongoing and formidable challenge. In addition, because p75NTR induces cell death, inhibits morphogenesis and negatively regulates synaptic transmission (Huang and Reichardt, 2001; Chao, 2003; Lu et al., 2005), it is highly desirable, from a therapeutic point of view, to develop agents that are functionally active on trkB receptors with no effect on p75NTR.
A strategy to overcome these limitations lies in the use of trkB-specific antibodies. In the past, antibodies directed against trkA receptors had exhibited agonist activity, mimicking the effect of the natural mediators by cross-linking the receptor and facilitating its autophosphorylation (LeSauteur et al., 1996; Kramer et al., 1997; Cattaneo et al., 1999). More recently, a study reported the ability of trkB-selective monoclonal antibodies to activate the BDNF receptor and trigger subsequent activation of trkB-dependent signalling pathways (Qian et al., 2006). It has also been recently shown that a monoclonal antibody can bind and activate trkC receptors and subsequently trigger survival signals (Guillemard et al., 2009). These previous studies validated the use of trkB antibodies as functional ligands to explore the roles of the BDNF receptor and its potential therapeutic utility. In the present study, we investigated the effects of several monoclonal and polyclonal trkB antibodies from commercial sources for their ability to modulate trkB activity. Active antibodies demonstrate distinct pharmacological properties depending on cell types, species, and presence or absence of p75NTR. These differences lead to specific effects on trkB-related molecular signalling and subsequent cellular processes. The broad range of action demonstrated by these antibodies forms a new class of useful pharmacological tools for the study of the BDNF/trkB system.
Methods
Drugs, compounds and antibodies
K252a, doxycycline and 3,3′,5′,5-tetramethyl-benzidine substrate (TMB) were purchased from Calbiochem (Nottingham, UK), Clontech (Mountain View, CA, USA) and Sigma (St. Louis, MO, USA) respectively. BDNF, NGF and NT-3 were purchased from Peprotech (London, UK). KIRA-elisa and Western blots were performed using the following antibodies: rabbit polyclonal anti-GFP (Clontech), rabbit polyclonal anti-TrkB (Upstate Biotechnologies, Lake Placid, NY, USA), mouse monoclonal anti-phosphotyrosine (4G10; Upstate Biotechnologies), mouse monoclonal anti-GFP (Chemicon, Temecula, CA, USA), mouse monoclonal anti-TrkB (BD Biosciences, Franklin Lakes, NJ, USA), anti-Akt (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-phospho(Ser473)-Akt antibodies (Cell Signaling, Danvers, MA, USA), anti-MAPK (Cell Signaling), anti-phospho(Thr202/Tyr204)-MAPK (Cell Signaling) and a streptavidin-peroxydase system (Amersham Biosciences, Munich, Germany). A full description of the functional anti-trkB antibodies used in this study can be found in Table 1.
Table 1.
Description of six commercially available polyclonal or monoclonal antibodies directed against trkB
| Code | Commercial supplier | Source | Specificity# | Immunogen |
|---|---|---|---|---|
| pAb-UB1 | Upstate Biotechnology (Ref. 07-225) | Rabbit polyclonal (IgG) | Rodent | Entire Extracellular domain |
| pAb-SA3 | Sigma-Aldrich (Ref. T1941) | Goat polyclonal (IgG) | Human | Entire Extracellular domain |
| pAb-BD5 | BD Biosciences (Ref. T1430) | Rabbit polyclonal (IgG) | Rodent | Extracellular Subdomains d4 and d5 |
| mAb-BD5 | BD Biosciences (Ref. T16020) | Mouse monoclonal Clone 47 (IgG1) | Rodent | Extracellular Subdomains d4 and d5 |
| mAb-RDS6 | R&D Systems (Ref. MAB1494) | Rat Monoclonal Clone 225105 (IgG2B) | Rodent | Entire Extracellular domain |
| mAb-AC7 | Abnova Corporation (Ref. H4915-M02) | Mouse monoclonal Clone 3D12 (IgG2A) | Human | Entire Extracellular domain |
Specificity as described by suppliers.
Plasmid constructs and cell lines
Plasmid constructs were generated using standard cloning techniques and were confirmed by sequencing. The human recombinant trkB receptor was fluorescently tagged at its C-terminus with the enhanced cyan fluorescent protein (rhtrkB-ECFP) by cloning the full human trkB sequence (Nakagawara et al., 1994) (kindly provided by G.M. Brodeur) into the pECFP-N1 expression vector (Clontech). For inducible expression, rhtrkB-ECFP was subcloned into the tetracycline-responsive (TetOn) vector pTRE2 (Clontech). Chinese hamster ovary cells (CHO) stably expressing rhTrkB-ECFP (TetOn-rhtrkB cells) were generated by transfection of the pTRE2-rhtrkB-ECFP plasmid into CHO-K1 TetOn cells (Clontech) followed by clonal selection with 0.25 mg·mL−1 hygromycin B (Invitrogen, Carlsbad, CA, USA) and 0.1 mg·mL−1 geneticin (G418; Invitrogen). Resistant clones were selected based on both fluorescence intensity and KIRA-elisa profile after an overnight incubation with 1000 ng·mL−1 doxycycline (Clontech). Cortical neurones were prepared and cultured from E16 mouse embryos as described previously (Lafont et al., 1992). The trkB receptor kinetics in these cell cultures has been previously characterized (Cazorla et al., 2010). The nnr5 PC12-trkA, nnr5 PC12-trkB and nnr5 PC12-trkC cells (kindly provided by M.V. Chao) are NGF non-responding mutant PC12 cells stably transfected with trkA, trkB and trkC cDNA respectively (Green et al., 1986).
Western blot analysis
Cell lysates were prepared in boiling SDS (2%, w/v) as previously described (Cazorla et al., 2010) and subjected to SDS-PAGE. Detection and analysis of MAPK and Akt phosphorylation were performed using anti-total and -phospho (Thr202/Tyr204)-MAPK antibodies (1:1000) or anti-total and (1:1000) -phospho (Ser473)-Akt (1:1000) antibodies respectively. For species' recognition experiments, cortical neurones were cultured for 8 days and TetOn-rhtrkB cells were incubated overnight with or without doxycycline. For selectivity relative to trkA, trkB and trkC, nnr5 PC12 variant cells were used. Cells were solubilized as for KIRA-elisa assays and lysates were centrifuged at 14 000×g for 20 min at 4°C. Cleared supernatant was collected and equal protein amounts were subjected to SDS-PAGE. Proteins were transferred to PVDF membranes and visualized using appropriate HRP-conjugated secondary antibodies. Detection of trkB receptors was performed using 1 µg·mL−1 of the different tested antibodies. Films were quantified by densitometry for MAPK and Akt analysis.
KIRA-elisa analysis
Auto-phosphorylation of the trkB receptor was quantified using a modified version of KIRA-ELISA (Sadick et al., 1997; Cazorla et al., 2010). Briefly, TetOn-rhtrkB cells were seeded on flat-bottom 96-well culture plates (4 × 104 cells per well) and incubated overnight with 1000 ng·mL−1 doxycycline to induce the expression of trkB receptors. Cortical neurones were seeded on polyornithin-coated flat-bottom 96-well culture plates (12 × 104 cells per well) and cultured for 7–8 days at 37°C in 5% CO2. Fluorescence levels in TetOn-rhtrkB were verified before each assay. Both cell types were carefully washed four times with Dulbecco's modified Eagle's medium (DMEM; Invitrogen). Because binding kinetics for antibodies are very slow compared with BDNF, polyclonal antibodies (pAbs) and monoclonal antibodies (mAbs) were allowed to bind trkB receptors for 30–60 min followed by 20 min of incubation with BDNF (concentrations are indicated in the legends) in DMEM containing 0.5% BSA and 25 mM HEPES (pH 7.4) at 37°C. Assay was stopped by removing the medium on ice and membranes were solubilized. The following combination of antibodies was used for phosphorylation assays: trkB capture with polyclonal anti-GFP (1:5000) for TetOn-rhtrkB, polyclonal anti-trkB (1 µg·mL−1) for cultured neurones; tyrosine phosphorylation quantification with biotinylated anti-pan-phosphotyrosine (4G10, 0.5 µg·mL−1) and a streptavidin-peroxydase system (1:4000). The use of a biotin-streptavidin system ensured avoiding any crosstalk between the functional antibodies and the KIRA-elisa secondary antibody. The following combination was used for total trkB quantification: trkB capture with monoclonal anti-GFP (1:3000) for TetOn-rhtrkB, monoclonal anti-trkB (1:1000) for cultured neurons; total trkB quantification with polyclonal anti-GFP (1:5000) for TetOn-rhtrkB, polyclonal anti-trkB (1 µg·mL−1) for cultured neurones followed by appropriate secondary antibodies and a peroxydase system. Control experiments ruled out any interaction between KIRA-elisa secondary antibodies and the functional antibodies. In fact, the addition of the functional antibodies to the cells did not influence the total-trkB signal (data not shown). Absorbance was read at 450 nm after the addition of TMB substrate and acidification with 1 N hydrochloric acid. Values are expressed in percentage of untreated controls, after subtraction of background signal defined as the signal obtained in total absence of any trkB phosphorylation (i.e. non-induced TetOn-rhtrkB cells; neurones treated with 10 µM of the non-selective Trk inhibitor K252a; see Cazorla et al., 2010).
[125I]-BDNF competition binding study
Two micrograms of recombinant human BDNF were dissolved in 50 mM phosphate buffer (pH 7.0). Iodination was allowed by incubation with 0.5 mCi 125I and chloramine T, and reaction was stopped by the addition of sodium disulphite. The labelled BDNF was purified by PD-10 desalting columns (Amersham Biosciences) and fractions containing the [125I]-BDNF were pooled (63.3 µCi·µg−1). Competition binding studies were done mainly as previously described (Cazorla et al., 2010). Briefly, 2.5–3 × 105 cells were carefully washed with ice-cold binding buffer (DMEM containing 0.5% BSA and 25 mM HEPES). Antibodies were allowed to bind TrkB for 2 h before subsequent incubation with [125I]-BDNF for another 2 h. All binding experiments were carried out at 4°C to prevent any trkB internalization. Cells were then washed four times with ice-cold binding buffer and lysed with 1 N NaOH. Radioactivity was counted in a 1260 MultiGammaII counter (LKB Wallac, Saint Quentin-en-Yvelines, France). Non-specific [125I]-BDNF binding was measured in the presence of 10 nM unlabelled BDNF and was subtracted from all values. Data are expressed as percentage of values obtained in the absence of the ligand.
Neurite outgrowth assessment
The nnr5 PC12 cells were prepared as described previously (Ilag et al., 1994). nnr5 PC12-TrkB, -TrkA and -TrkC cells were treated with BDNF (1 nM), NGF (2 nM) and NT-3 (10 nM) respectively. The number of cells bearing neurites longer than 2 cell diameters was microscopically determined in each counting field (two fields per well, three wells per condition). Counting was performed blind every 24 h for 3 days. Data presented in the figures are those obtained for 48 h but were similar at 24 and 72 h.
Neurone survival assay
The neuroprotective effects induced by BDNF and pAb-UB1 were assessed on cortical neurones (50 × 103 cells per well in 96-well plates) cultured for 5 days. Culture medium was removed and replaced with serum-free fresh medium containing either 1 nM BDNF, various concentrations of pAb-UB1 or nothing. pAb-UB1 was added every 24 h for 3 days. Cell survival was determined using the CellTiter 96 AQueous Cell Proliferation Assay from Promega (Madison, WI, USA), according to the manufacturer's protocol. Values are expressed as a percentage of the signal obtained in serum-treated cells.
Statistical analysis
Concentration-response and competition curves were obtained using an iterative least-squares method derived from that of Parker and Waud. Eadie–Hofstee plotting of the data provided estimates for EC50 values of BDNF in the presence or in absence of antibodies. KIRA-ELISA competition studies were analysed using one-way analysis of variance (anova) followed by Dunnett's multiple comparison post hoc test. Data obtained in neurite outgrowth experiments, neurone survival assessments and densitometric analyses of MAPK phosphorylation levels were analysed using one-way anova followed by Dunnett's multiple comparison post hoc test. Concentration-response curves and Eadie–Hofstee plots were analysed using two-way anova.
The drug and molecular target nomenclature used in this paper follows Alexander et al. (2009).
Results
Cross-reactivity of commercially available trkB antibodies with the rodent and human forms
pAbs and mAbs directed against the extracellular domain of either human (pAb-SA3 and mAb-AC7) or rodent (pAb-UB1, pAb-BD5, mAb-BD5 and mAb-RDS6) trkB were obtained from commercial sources (Table 1) and were tested for their specificity. For that purpose, two cellular models were used: (i) a CHO TetOn cell line expressing a recombinant human form of trkB receptor in a doxycycline-inducible manner (TetOn-rhtrkB); and (ii) neurones originated from the cerebral cortex of mouse embryos. For both human and rodent trkB receptors, two bands are usually detected: a 145 kDa band (full and active form) and a 95 kDa band (truncated and inactive form) (Klein et al., 1990; Nakagawara et al., 1994). In TetOn-rhtrkB cells, the active form is detected at 175 kDa as trkB receptors have been tagged with a 30 kDa fluorescent protein while the inactive form is detected at 95 kDa as it lacks the entire catalytic domain as well as the 30 kDa tag. Western blot analyses using anti-trkB antibodies in TetOn-rhtrkB (Figure 1A) and in cultured neurones (Figure 1B) demonstrated human and rodent cross-reactivity for pAb-UB1, pAb-SA3, pAb-BD5 and mAb-BD5. mAb-AC7 detected the human form of trkB in the TetOn-rhtrkB cells but did not cross-reacted with the rodent form expressed in the mouse neuronal cells. In our hands, mAb-RDS6, which was raised against the mouse trkB isoform, did not recognize the rodent form and poorly bound the human form. Although pAb-UB1, pAb-SA3, pAb-BD5 and mAb-BD5 were specific to trkB in neurones, pAb-UB1 and pAb-BD5 detected additional bands that are not related to trkB in TetOn-rhtrkB cells. Surprisingly, the inactive 95 kDa form was detected for some antibodies in the absence of doxycycline. Because in TetOn-rhtrkB cells more of the truncated form is present than the active isoform, the 95 kDa band is more likely to be detected even in the absence of doxycycline. In fact, longer exposure blots showed that the antibodies faintly but clearly detect both the 175 and 95 kDa bands in the absence of doxycycline (data not shown). However, when all the said antibodies (pAb-UB1, pAb-SA3, pAb-BD5, mAb-BD5, mAb-RDS6 and mAb-AC7) were tested in non-transfected TetOn cells, no signal could be clearly detected. These observations suggest a small leakage in the regulation of the doxycycline-dependent expression of trkB. Interestingly, although all the antibodies were used at the same concentration (1 µg·mL−1), marked differences in band intensity could be observed. In TetOn-rhtrkB cells, strongest signals were obtained with pAb-SA3, mAb-AC7 and pAb-BD5 (Figure 1A). In neurones, mAb-BD5 produced the most intense signal, though pAb-UB1, pAb-SA3 and pAb-BD5 were relatively efficient in mouse trkB recognition (Figure 1B). It must be noted that even though pAb-UB1, pAb-SA3 and pAb-BD5 are polyclonal antibodies, no significant variation in trkB recognition between different batches was observed among all Western blots.
Figure 1.
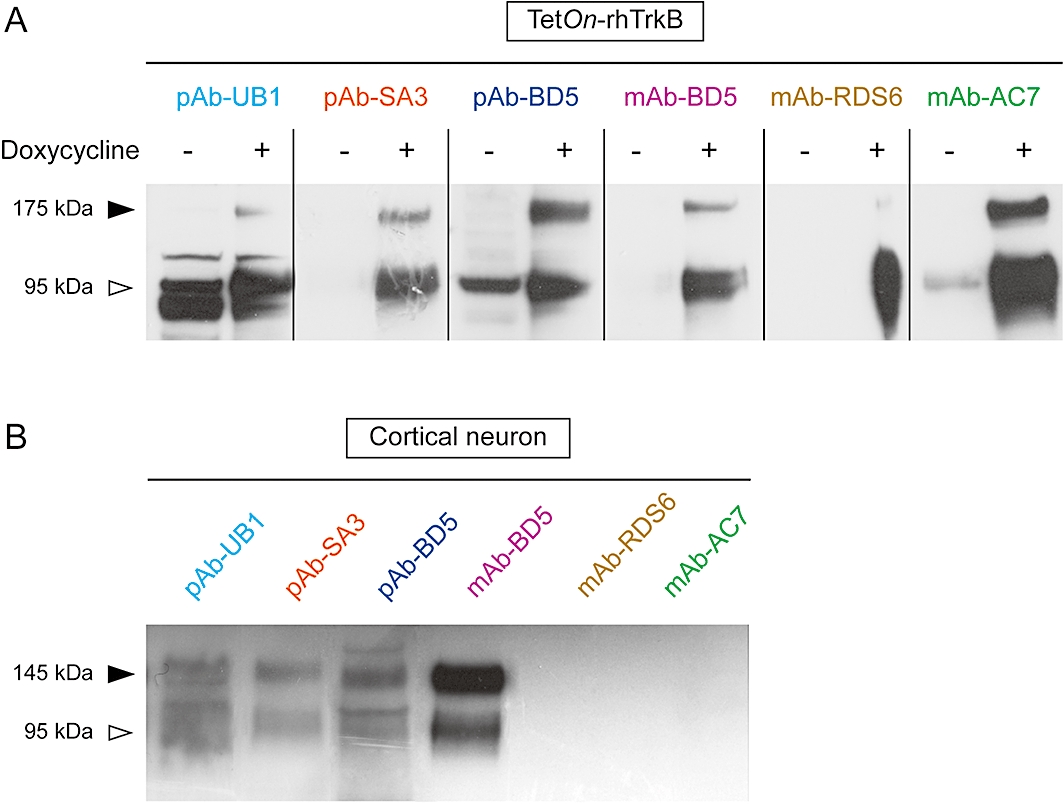
Rodent and human cross-reactive responses for pAbs and mAbs. Antibodies (1 µg·mL−1) were subjected to Western blot analysis to examine their binding properties to (A) human and (B) mouse trkB, in TetOn-rhtrkB treated with or without doxycycline, and neuronal cells respectively. The trkB signal elicits two bands, corresponding to the full length (145 kDa) and truncated (95 kDa) form of the receptor. In TetOn-rhtrkB cells, trkB is C-terminally fused to the fluorescent protein ECFP, resulting in the detection of two bands of approximately 175 kDa (trkB + ECFP) and 95 kDa (truncated trkB). pAbs, polyclonal antibodies; mAbs, monoclonal antibodies; TetOn, tetracycline-responsive; ECFP, enhanced cyan fluorescent protein.
Functional and pharmacological characterization of trkB antibodies
pAbs and mAbs were then assessed for their functional effects on trkB activity in TetOn-rhtrkB cells and cortical neurones. For that purpose, we developed a modified version of KIRA-elisa, a rapid and high-capacity assay that quantifies trkB activation by directly measuring its level of phosphorylation. In addition, KIRA-elisa assay demonstrates higher data accuracy than those obtained with other methods (Cazorla et al., 2010).
BDNF competition studies were performed using increasing concentrations of antibodies in the absence or presence of BDNF in recombinant and neuronal cells. Figure 2A shows an example of staining after incubating either living TetOn-rhtrkB cells with increasing concentrations of anti-human trkB pAb-SA3 or living neurones with increasing concentrations of anti-rodent trkB pAb-UB1.
Figure 2.
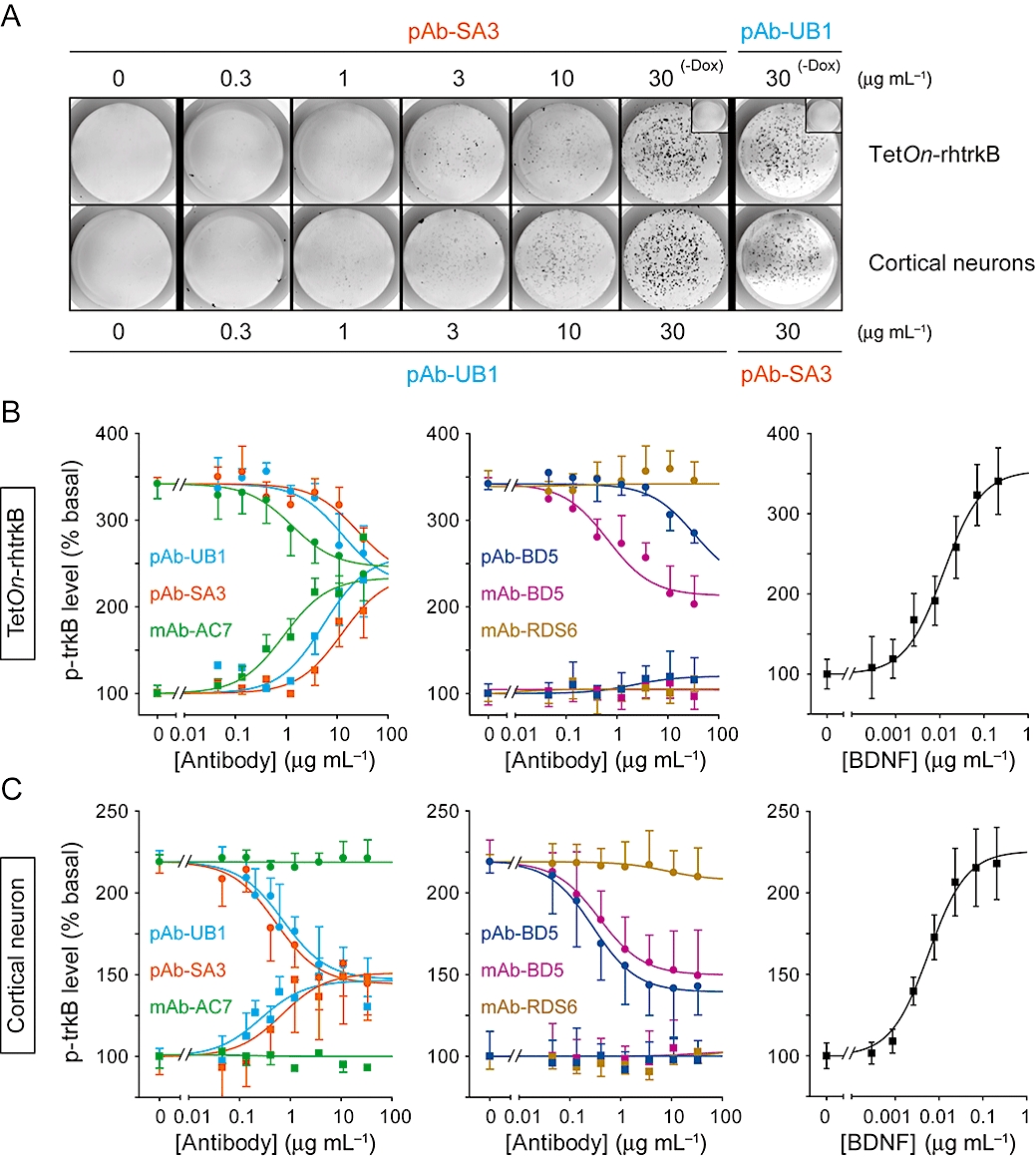
Distinct functional modulations of trkB activity by pAbs and mAbs in both recombinant and native systems. The phosphorylation levels of trkB were quantified by KIRA-elisa assays after treatment with BDNF and antibodies, alone or in combination. (A) Representative fluorescent photomicrographs of TetOn-rhtrkB cells and cortical neurones after treatment with increasing concentrations of pAb-SA3 and pAb-UB1 as for KIRA-elisa assays. Insets show signal obtained with 30 µg·mL−1 of pAb-UB1 or pAb-SA3 in non-induced TetOn-rhtrkB cells (-Dox). (B,C) Increasing concentrations of pAbs and mAbs in the presence (circles) or absence (squares) of BDNF (B, TetOn-rhtrkB, 1 nM; C, neurones, 0.4 nM). BDNF concentration-response curves are shown for comparison. Absorbance read at 450 nm was normalized as percentage of basal trkB activity. Data are mean ± SEM of values obtained in triplicate in three experiments. pAbs, polyclonal antibodies; mAbs, monoclonal antibodies; TetOn, tetracycline-responsive; BDNF, brain-derived neurotrophic factor.
In TetOn-rhtrkB cells, pAb-UB1, pAb-SA3 and mAb-AC7 acted as partial agonists because they partially reproduced the maximal effect of BDNF (342.1 ± 89%) when applied alone, and partially reduced the phosphorylation of trkB in the presence of the neurotrophin (Figure 2B). In contrast, pAb-BD5 and mAb-BD5 did not demonstrate agonist activity but were able to partially decrease the BDNF-induced trkB activation, suggesting a neutral antagonist profile instead. In the absence of BDNF, pAb-UB1, pAb-SA3 and mAb-AC7 (partial agonist antibodies) demonstrated similar intrinsic activities (pAb-UB1, 259.6 ± 35.4%; pAb-SA3, 237.6 ± 42.6%; mAb-AC7, 221.7 ± 14.7%) while pAb-BD5 and mAb-BD5 (neutral antagonist antibodies) remained without effects. Intrinsic potencies (EC50) of pAb-UB1, pAb-SA3 and mAb-AC7 were 2- to 3-log units lower than BDNF potency (0.01 ± 0.006 µg·mL−1) and were significantly different (P < 0.01, one-way anova), with mAb-AC7 being the most potent antibody (pAb-UB1, 5.5 ± 1.5 µg·mL−1; pAb-SA3, 12.4 ± 2.4 µg·mL−1; mAb-AC7, 0.7 ± 0.2 µg·mL−1). In the presence of BDNF, pAb-UB1, pAb-SA3 and pAb-BD5 partially inhibited trkB activity with similar amplitudes of effect (pAb-UB1, −33.3 ± 3.2%; pAb-SA3, −29.1 ± 9.8%; pAb-BD5, −29.2 ± 8.2%) and slightly different potencies (IC50, pAb-UB1, 12.7 ± 3.2 µg·mL−1; pAb-SA3, 25.1 ± 4.8 µg·mL−1; pAb-BD5, 50.1 ± 6.1 µg·mL−1; P < 0.05, one-way anova). mAb-BD5 and mAb-AC7 appeared to be the most efficient antibodies (mAb-AC7, −43.1 ± 9.2%; mAb-BD5, −64.9 ± 11.2%; and IC50, mAb-AC7, 1.3 ± 1.1 µg·mL−1; mAb-BD5, 1.3 ± 1.4 µg·mL−1; P < 0.0001, one-way anova). mAb-RDS6 was found to be ineffective either in the absence or in the presence of BDNF. This latter result was not surprising because, contrary to other antibodies, mAb-RDS6 only faintly recognized the active form of human trkB expressed in TetOn-rhtrkB cells (175 kDa) (Figure 1A).
In mouse cortical neurones, the pattern of activity of all antibodies was similar to that observed in the recombinant system, though with some differences in amplitudes of effect and potencies (Figure 2C). When applied alone, pAb-UB1 and pAb-SA3 still acted as partial agonists but with a reduced intrinsic activity (BDNF, 219.3 ± 22.2%; pAb-UB1, 145.8 ± 6.1%; pAb-SA3, 148.5 ± 18.9%). However, they both were more potent with an EC50 shifted by one-log to the left (pAb-UB1, 0.2 ± 0.3 µg·mL−1; pAb-SA3, 0.7 ± 0.4 µg·mL−1). Because in neurones BDNF EC50 was also shifted to the left by one-log, pAb-UB1 and pAb-SA3 still have a two-log lower potency than BDNF. Again, pAb-BD5 and mAb-BD5 remained without effects when applied alone. In the presence of BDNF, pAb-UB1, pAb-SA3 and pAb-BD5 had increased inhibition and greater potencies (IC50 shifted to the left by one to two-log) than in TetOn-rhtrkB cells. Compared with the recombinant cells in which mAb-BD5 was the most efficient antagonist, all antibodies had very comparable effects in neurones (pAb-UB1, −61.6 ± 10.4%; pAb-SA3, −62.7 ± 3.3%; pAb-BD5, −65.2 ± 17.6%; mAb-BD5, −58.6 ± 18.1%; and IC50, pAb-UB1, 0.7 ± 0.3 µg·mL−1; pAb-SA3, 0.5 ± 0.2 µg·mL−1; pAb-BD5, 0.2 ± 0.1 µg·mL−1; mAb-BD5, 0.3 ± 0.2 µg·mL−1). As previously observed, mAb-RDS6 was ineffective on rodent trkB. Finally, mAb-AC7 was not able to activate rodent TrkB in neurones. Together with the lack of labelling previously observed in Western blots (Figure 1B), these data confirm that mAb-AC7 specifically bind and activate human but not rodent trkB. The pharmacological characteristics (intrinsic activities relative to BDNF, EC50 and IC50) of all antibodies are summarized in Table 2. It should be noted that control experiments performed with respective reconstitution buffer of each antibody (as described by the manufacturer) in the presence or in the absence of BDNF ruled out possible non-specific effects (data not shown).
Table 2.
Summary of pharmacological and functional properties of the six antibodies tested
| Specificity | Selectivity | Pharmacological properties$ | Mode of action | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Code | Human | Rodent | TrkA | TrkB | TrkC | p75 | Recombinant | Native | Function | Binding |
| pAb-UB1 | + | + | − | + | − | − | Partial agonist I.A. 65% EC50 5 µg·mL−1 | Partial agonist I.A. 40% EC50 0.5 µg·mL−1 | Prevents effects of BDNF | Prevents binding of BDNF |
| pAb-SA3 | + | + | − | + | − | − | Partial agonist I.A. 60% EC50 10 µg·mL−1 | Partial agonist I.A. 40% EC50 0.5 µg·mL−1 | Prevents effects of BDNF | Prevents binding of BDNF |
| pAb-BD5 | + | + | −(#) | + | − | − | Partial antagonist Inhib. −30% IC50 50 µg·mL−1 | Partial antagonist Inhib. −65% IC50 0.5 µg·mL−1 | BDNF overcomes inhibition | Does not prevent binding of BDNF |
| mAb-BD5 | + | + | − | + | − | − | Partial antagonist Inhib. −65% IC50 1 µg·mL−1 | Partial antagonist Inhib. −60% IC50 0.5 µg·mL−1 | BDNF overcomes inhibition | n.d. |
| mAb-RDS6 | ∼ | − | − | ∼ | − | − | Inactive | Inactive | Inactive | n.d. |
| mAb-AC7 | + | − | − | + | − | − | Partial agonist I.A. 50% EC50 1 µg·mL−1 | Inactive | n.d. | n.d. |
All antibodies are affinity purified. The concentrations indicated reflect the real concentration in antibody.
pAb-BD5 binds TrkA but does not affect its activity
I.A. intrinsic activity (as % of BDNF); Inhib., inhibition; n.d., not determined; BDNF, brain-derived neurotrophic factor.
Mode of action of the functional trkB antibodies
To investigate the molecular mechanism underlying these effects on trkB receptors, binding experiments using iodinated BDNF and KIRA-elisa assays were performed in the native system (i.e. cortical neurones co-expressing native trkB and p75NTR).
Consistent with results described previously in neuronal cells, mAb-RDS6 and mAb-AC7 did not alter the BDNF concentration-response curve (Figure 3A,B). Neurone-active partial agonists pAb-UB1 and pAb-SA3 partially decreased the BDNF response in a non-competitive way, as demonstrated by the decrease in maximal response (pAb-UB1, −71.7 ± 3.7%; pAb-SA3, −37.7 ± 5.5%) and no significant change in BDNF EC50 (BDNF alone, 117.9 ± 20.5 pM; BDNF + pAb-UB1, 162.6 ± 30.7 pM; BDNF + pAb-SA3, 110.2 ± 55.3 pM). Conversely, antagonist antibodies pAb-BD5 and mAb-BD5 decreased the BDNF response through a competitive mechanism, as shown by the rightward shift of the BDNF curve (BDNF + pAb-BD5, 385.6 ± 12.7 pM, P < 0.01, one-way anova, n= 3; BDNF + mAb-BD5, 1.10 ± 0.17 nM, P < 0.01, one-way anova, n= 3) and no significant change in maximal effect (Figure 3A). All these observations were confirmed by an Eadie–Hofstee plot of the curves (Figure 3B).
Figure 3.
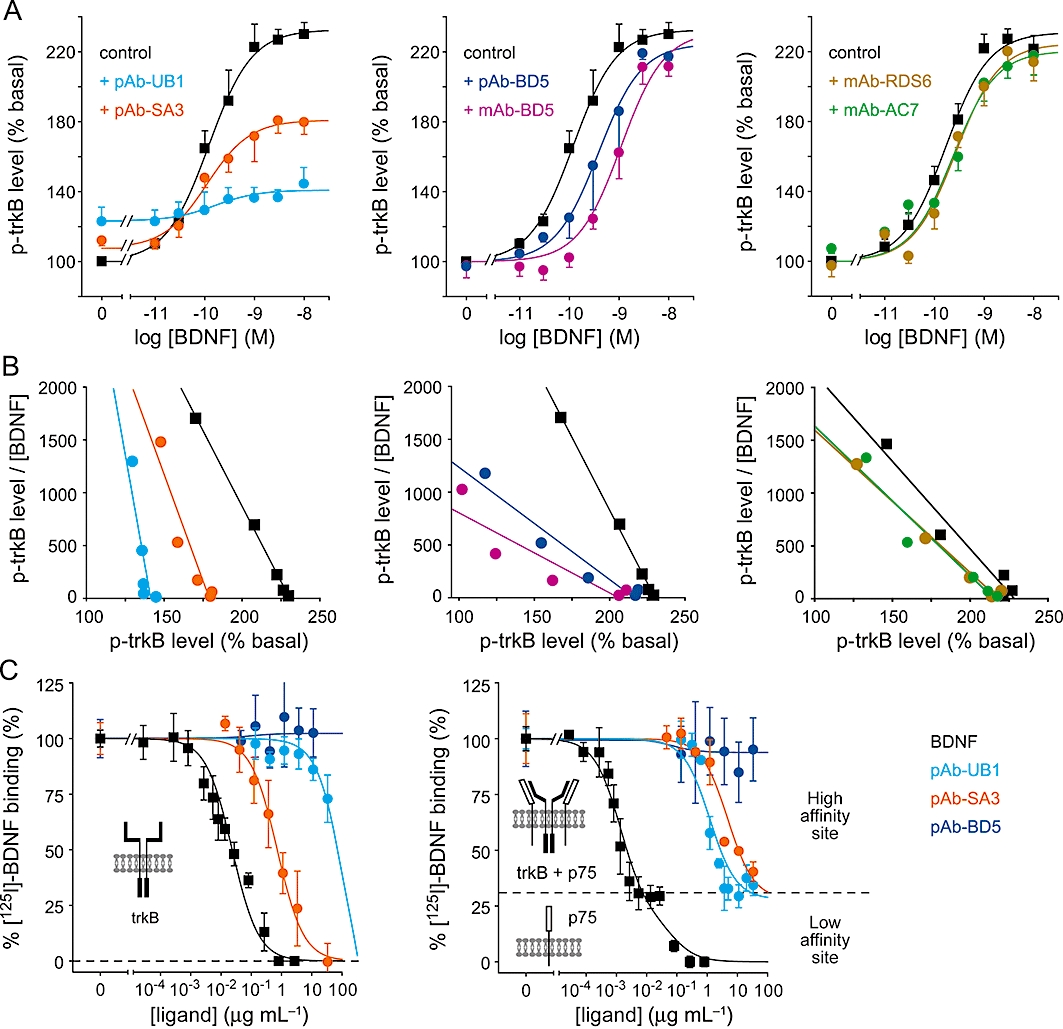
Mechanism of action of pAbs and mAbs. (A) BDNF concentration-response curves in the absence or presence of antibodies (10 µg·mL−1) were obtained in neurones. The addition of pAb-UB1 and pAb-SA3 (left panel) resulted in a non-competitive partial agonism and did not significantly change BDNF EC50. pAb-BD5 and mAb-BD5 (middle panel) elicited a competitive effect, as demonstrated by the significant rightward shift of the BDNF curve. mAb-RDS6 and mAb-AC7 (right panel) were inactive in neurones. (B) Eadie–Hofstee plots of the data obtained in (A) confirmed the non-competitive actions of pAb-UB1 and pAb-SA3 (parallel lines, left panel), the competitive effect elicited by pAb-BD5 and mAb-BD5 (convergent lines, middle panel) and the lack of effect of mAb-RDS6 and mAb-AC7 (right panel). Values are expressed as percentage of basal trkB activity. Data are mean ± SEM of 3–4 experiments performed in triplicate, except for Eadie–Hofstee plots where data are means. (C) Effect of pAbs on the binding of [125I]-BDNF in TetOn-rhtrkB cells (left panel) and in neurones (right panel). Insets show the conformations of the receptors supposed to be responsible for the different BDNF binding sites. Data are mean ± SEM of 2–4 experiments performed in triplicate. pAbs, polyclonal antibodies; mAbs, monoclonal antibodies; BDNF, brain-derived neurotrophic factor; TetOn, tetracycline-responsive.
Binding studies using [125I]-BDNF on both recombinant human and rodent neuronal trkB receptors also revealed distinct binding properties for polyclonal antibodies (Figure 3C). In fact, while pAb-UB1 and pAb-SA3 were able to prevent the binding of [125I]-BDNF to trkB receptors, pAb-BD5 had no effect. Interestingly, pAb-UB1, which was raised against the rodent form, was more efficient in rodent neurones than in TetOn-rhtrkB cells (human, 125.4 ± 23.9 µg·mL−1 and rodent, 1.2 ± 0.3 µg·mL−1, P < 0.001, t-test, n= 2–4). Conversely, pAb-SA3, which was originally designed to recognize the human form, was more efficient in TetOn-rhtrkB cells than in neuronal cultures (human, 0.7 ± 0.2 µg·mL−1 and rodent, 4.8 ± 0.1 µg·mL−1, P < 0.01, t-test, n= 2–4). These results are consistent with the band intensities observed previously in Western blots of human or rodent trkB with pAb-UB1 and pAb-SA3, and may reflect differences in affinity (Figure 1). It must be noted that in neurones that co-express trkB and p75NTR, BDNF completely inhibited the binding of iodinated BDNF on both the trkB + p75NTR complex and p75NTR (high- and low-affinity sites, respectively), consistent with previous reports (Chao and Hempstead, 1995). Contrary to BDNF, pAb-UB1 and pAb-SA3 prevented the binding of [125I]-BDNF on trkB but not on p75NTR, as expected for trkB-selective ligands (Figure 3C). In conclusion, given the nature of antibody–antigen interactions, all these observations strongly suggest an allosteric mechanism of action that influences the activity of the receptor and prevents any effects of BDNF or can be overcome by BDNF, depending on the antibody (e.g. pAb-UB1 vs. pAb-BD5 respectively).
Selectivity of the functional antibodies
The neurotrophin family is composed of NGF that binds trkA, BDNF and NT4/5 that bind trkB, and NT3 that binds trkC. In addition to trk receptors, all these neurotrophins interact with p75NTR. To assess the selectivity of the functional antibodies towards trkB, we took advantage of the nnr5 PC12 cell lines that express endogenous levels of p75NTR (nnr5 PC12), or that were transfected to co-express rodent trkA (nnr5 PC12-trkA) or trkC (nnr5 PC12-trkC) (Green et al., 1986) (Figure 4).
Figure 4.
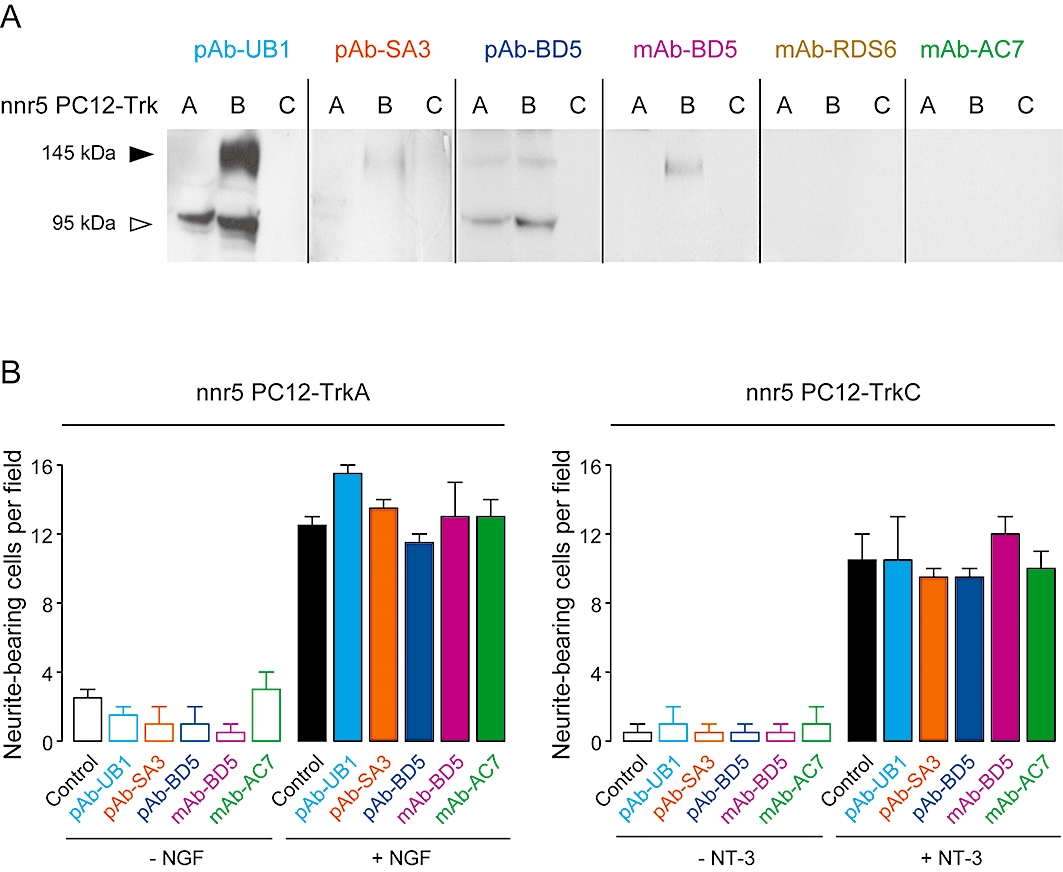
Binding and functional selectivity of pAbs and mAbs for TrkB but not for TrkA or TrkC receptors. (A) Antibodies (10 µg·mL−1) were subjected to Western blot analysis to assess their selectivity with regard to trkA and trkC using nnr5 PC12-trkA and -trkC cells. A representative immunoblot is presented. (B) Quantitative analysis of NGF- (2 nM) and NT3 (10 nM) -induced neurite outgrowth in the presence of 10 µg·mL−1 of antibodies in nnr5 PC12-trkA and -trkC cells respectively. Data are mean ± SEM of results from two experiments. pAbs, polyclonal antibodies; mAbs, monoclonal antibodies; NGF, nerve growth factor.
As in neurones, pAb-UB1, pAb-SA3, pAb-BD5 and mAb-BD5 recognized rat trkB expressed in nnr5 PC12-trkB cells, contrary to mAb-RDS6 and mAb-AC7. Western blotting analysis using these antibodies in nnr5 PC12-trkA and -trkC revealed that pAb-UB1, pAb-SA3 and mAb-BD5 did not recognize the active forms (145 kDa) of trkA and trkC (Figure 4A). Similar to what was observed in non-induced TetOn-rhtrkB cells (Figure 1A), pAb-UB1 and pAb-SA3 also interacted with the truncated form (95 kD) of trkA. A slight signal was detected at 145 kDa in trkA-expressing cells for pAb-BD5, suggesting that this antibody may cross-react to some extent with the active form of trkA. However, functional studies quantifying NGF- and NT3-induced neurite outgrowth in nnr5 PC12-trkA and -trkC, respectively, showed that none of these antibodies was able to affect neurite outgrowth (Figure 4B). These results thus ruled out any non-trkB biological effects for all the antibodies and further demonstrated that their selectivity towards trkB is valid at the functional level even in the case of cross-reactivity.
In addition, Western blots performed in both neuronal and trk-expressing nnr5 PC12 cells revealed no detection of a 75 kDa band (corresponding to p75NTR) for all antibodies (Figures 1B and 4A). The same finding was observed in p75NTR-only-expressing nnr5 PC12 cells (data not shown) and confirms the absence of effect on p75NTR in the [125I]-BDNF binding studies (Figure 3C). These observations are not surprising given that these antibodies have been designed to interact with the extracellular domain of trkB, which differs from p75NTR both at the amino acid sequence and at the structural level.
Molecular and cellular effects of the functional antibodies on trkB-dependent processes
We next proceeded to dissect the molecular and cellular effects of these antibodies. BDNF is known to promote differentiation and survival of trkB-expressing cells (Huang and Reichardt, 2001). The effects of pAbs and mAbs were therefore tested on these TrkB-mediated biological actions in the presence or absence of BDNF using cultures of cortical neurones and nnr5 PC12-trkB cells, both of them co-expressing the rodent form of trkB together with p75NTR.
The effects of pAbs and mAbs on cell differentiation were assessed in the rodent trkB-expressing nnr5 PC12 cells (nnr5 PC12-trkB), in which BDNF has been shown to promote neurite outgrowth (Figure 5). Consistent with previous reports (Ip et al., 1993), an addition of BDNF to nnr5 PC12-trkB cells increased neurite length and number of branch points. The changes in the arbor rearrangement were significantly altered by antibodies (Figure 5A,B), with similar pharmacological profiles to those observed with KIRA-elisa assays. In this cellular model of trkB activation, pAb-UB1 and pAb-SA3 were able to promote neurite outgrowth and, as partial agonists, partially inhibited the effect of BDNF. Both partial antagonists pAb-BD5 and mAb-BD5 significantly impaired the BDNF-induced differentiation of nnr5 PC12-trkB cells and did nothing when incubated alone. Once again, both mAb-RDS6 and mAb-AC7, which did not recognize the rodent form of trkB expressed in nnr5 PC12-trkB, were inactive. Subsequent concentration-response studies confirmed both potency and efficacy of the effect determined by KIRA-elisa for all pAbs and mAbs, as demonstrated by the two representative polyclonal partial agonist and monoclonal partial antagonist antibodies, pAb-UB1 and mAb-BD5 respectively (Figure 5C). trkB-mediated neurite outgrowth involves MAPK signalling activation (Huang and Reichardt, 2001). The effects of antibodies were thus investigated on the MAPK phosphorylation level in nnr5 PC12-trkB cells (Figure 5D). As partial agonists, pAb-UB1 and pAb-SA3 were able to partially activate MAPK in the absence of BDNF and to partially decrease BDNF-induced MAPK phosphorylation, consistent with KIRA-elisa analyses. It must be noted that pAb-UB1 appeared more efficient than pAb-SA3 in this test. On the other hand, pAb-BD5 and mAb-BD5 had their antagonistic profiles confirmed, with partial inhibitory effects relative to the full non-specific trk inhibitor K252a.
Figure 5.
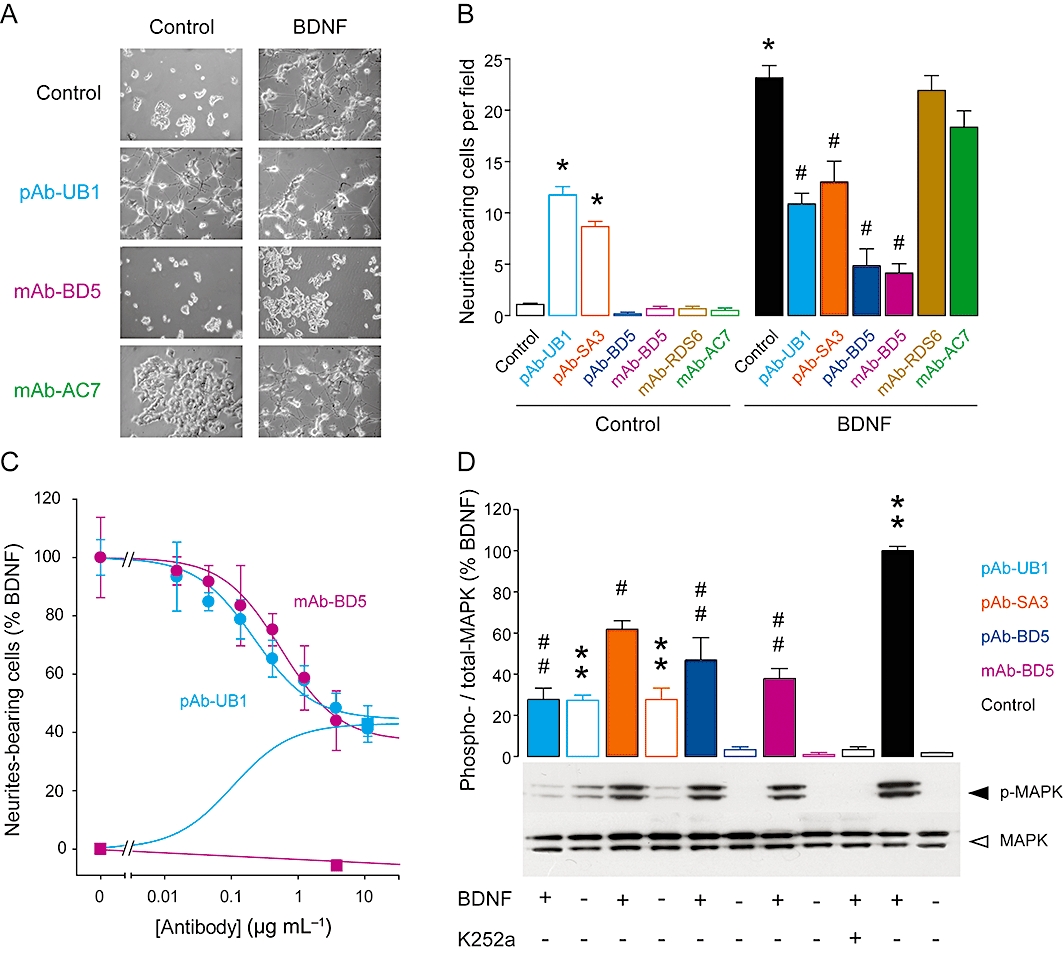
Biological effects of pAbs and mAbs on neurite outgrowth and trkB-dependent MAPK signalling activation. (A) Representative photomicrographs of nnr5 PC12-trkB cells treated for 48 h with 10 µg·mL−1 antibodies (partial agonist effects illustrated by pAb-UB1; antagonist effects illustrated by mAb-BD5; inactive antibody illustrated by mAb-AC7) and 1 nM BDNF, as indicated. (B) Quantitative analysis of BDNF-induced neurite outgrowth in the presence of antibodies. (C) Concentration-response curves of partial agonist pAb-UB1 and antagonist mAb-BD5 obtained in the presence or absence of 1 nM BDNF were performed as in (B). Values are expressed as a percentage of the maximal BDNF response (100%) after subtraction of counting obtained in controls (0%). Data are mean ± SEM of results from two experiments. (D) Representative Western blot analysis of total and phosphorylated MAPK in nnr5 PC12-trkB cells after treatment with BDNF (1 nM), antibodies (10 µg·mL−1) or K252a (100 nM), alone or in combination. Densitometric quantification of MAPK phosphorylation relative to total MAPK is shown and expressed as a percentage of BDNF response after subtraction of background values. Data are mean ± SEM of values obtained in triplicate in two independent experiments; **P < 0.01, compared with basal condition; ##P < 0.01, compared with BDNF; *P < 0.05; #P < 0.05. pAbs, polyclonal antibodies; mAbs, monoclonal antibodies; MAPK, mitogen-activated protein kinase; BDNF, brain-derived neurotrophic factor.
Numerous preclinical data strongly suggest that a trkB agonist, capable of promoting the survival of neurones, is highly desirable for the treatment of neurodegenerative and neurological diseases in which BDNF is down-regulated (Murer et al., 2001; Zuccato et al., 2001; Pezet and McMahon, 2006). In this context, we chose one representative partial agonist, pAb-UB1, to test its ability to promote the survival of cortical neurones. In neuronal cultures, serum deprivation results in the death of differentiated neurones that could be partially rescued by treatment with BDNF (Figure 6). Treatment with pAb-UB1 also protected neurones from serum deprivation-induced injury in a concentration-dependent manner (Figure 6A). The BDNF-induced survival of neurones is linked to the trkB-dependent activation of the Akt signalling pathway (Huang and Reichardt, 2001). Consistent with the survival assays, phospho-Akt Western blot analysis showed an increased signal in neurones treated with both BDNF and pAb-UB1 (Figure 6B).
Figure 6.
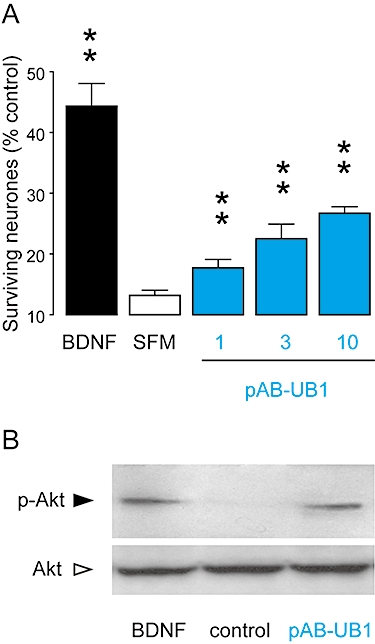
Biological effects of pAbs and mAbs on neurone survival. (A) Increasing concentrations of pAb-UB1 or BDNF (1 nM) were added to cultured neurones for 3 days after serum deprivation. Cell viability data are expressed as percentage of total living cells in the presence of complete culture medium and are mean ± SEM of values obtained in triplicate in three independent experiments; **P < 0.01, compared with serum-deprived condition. (B) Representative immunoblotting analysis of total and phosphorylated Akt in cortical neurones after treatment with BDNF (1 nM) or pAb-UB1 (10 µg·mL−1). pAbs, polyclonal antibodies; mAbs, monoclonal antibodies; BDNF, brain-derived neurotrophic factor; SFM, serum-free medium.
Discussion
Here we describe the ability of selective trkB antibodies from commercial sources to either activate trkB receptors or antagonize BDNF-induced effects in various cell systems (see summary in Table 2). With the exception of mAb-RDS6, which poorly recognizes trkB and is not functional, most of the antibodies selectively bind human and rodent trkB receptors and do not cross-react with trkA and trkC, confirming their specificity as trkB ligands. Additionally, unlike the endogenous ligands for trkB (BDNF and NT4/5), these antibodies do not bind to p75NTR, as demonstrated by the absence of band at 75 kDa in Western blots and by the inability of antibodies to displace BDNF on the low-affinity sites in neurones. These observations are not surprising because these antibodies have been designed against the extracellular part of trkB which does not share any sequence or conformational similarities with p75NTR (Chao and Hempstead, 1995). These observations also have crucial consequences for both therapeutic and fundamental studies. p75NTR is likely to be responsible for most of the ‘negative effects’ of neurotrophins, such as induction of neuronal death events, inhibition of trk-mediated morphogenesis or depression of synaptic transmission (Huang and Reichardt, 2001; Chao, 2003; Lu et al., 2005). It is therefore highly desirable, from a therapeutic point of view, to develop agents that are functionally active on trk receptors with no effect on p75NTR. Furthermore, these antibodies may also represent good pharmacological tools to define the role of trkB with regard to p75NTR in various physiological and pathological situations in which BDNF is involved, such as amyotrophic lateral sclerosis (Mojsilovic-Petrovic et al., 2006), Alzheimer's disease (Murer et al., 2001), pain (Pezet and McMahon, 2006), epilepsy (McNamara et al., 2006), anxiety (Chen et al., 2006), depression (Nestler et al., 2002), schizophrenia (Durany and Thome, 2004), bipolar disorders (Shaltiel et al., 2007) and tumour formation in cancer (Desmet and Peeper, 2006).
Among all the antibodies we tested, some proved to behave as trkB partial agonists (pAb-UB1, pAb-SA3 and mAb-AC7) while others are only antagonists (pAb-BD5 and mAb-BD5) (Table 2). Interestingly, all active antibodies demonstrate partial effects compared with the full activity induced by the endogenous trkB agonist BDNF. In fact, the interaction of mAbs and pAbs with trkB would result in the alteration of the active conformation of trkB by twisting the receptor into a shape that is only partially active. Subsequent binding of BDNF to such trkB receptors would therefore lead to partial activation. As a consequence, the twisted-shape conformation induced by partial agonist antibodies is active by itself to a certain extent either by favouring the recruitment of another trkB monomer or by allowing the docking of intracellular tyrosine kinases that could activate trkB. Conversely, the antagonist antibodies would induce a different twisted-shape conformation that is inactive by itself but that partially affects the ability of BDNF to activate trkB. It should be noted that the antibodies used in this study were either mAbs or pAbs obtained from commercial sources. Therefore, although reproducibility is not an issue for monoclonal production, it is very critical for pABs. The variability between immunological responses could affect pAbs' effects and selectivity over time. Moreover, pAbs are by definition a mixture of antibodies directed against different epitopes. Therefore, the effects described for pAbs are likely to be the result of overall effects of a combination of agonists, antagonists or neutral antibodies. The composition of this mixture is also subject to change with time and could add variability to the biological response. However, at the time this study was performed, no significant variability could be observed between the different batches of pAbs we used. Western blots (Figure 1) and KIRA-elisa concentration-response curves (Figure 2B,C) performed with every new batch showed only slight differences in effects and no major changes (such as changes in pharmacological class, significant shift of EC50/IC50, etc.). Although reproducibility was not affected in the present study, we cannot rule out the possibility of significant differences in the future, making the use of mAbs (mAb-BD5 and mAb-AC7) more consistent and therefore more interesting for further studies and/or for the development of new therapies.
For partial agonists, KIRA-elisa analyses showed that BDNF efficacy but not affinity is affected, while binding studies using radiolabelled BDNF showed that they prevent BDNF from interacting with trkB. Given the nature of antibody–antigen interactions, two mechanisms of action could be envisaged: (i) by interacting with trkB, both antibodies directly mask BDNF binding site(s) while leading to a partial activation of trkB; or (ii) binding of antibodies changes trkB into a semi-active conformation which does not allow BDNF to access its binding site(s) with equal potency and efficacy as to the unoccupied receptor. These antibodies could therefore be defined as ‘strong modulators’ as they prevent the effects of the endogenous ligand to elicit their own activity and cannot be overcome by BDNF. It must be noted that these antibodies were designed against the whole extracellular domain of trkB which certainly contains multiple epitopes. Because the antibodies may recognize and bind one (mAbs) or several (pAbs) distinct epitopes, it is probable that they exert their effects through different mechanisms and/or binding sites. For antagonists, KIRA-elisa assays showed that BDNF efficiency was not affected but affinity was shifted rightward, while binding studies showed that BDNF binding to trkB was unaltered. Therefore, the following mechanism can be proposed: binding of the two antibodies to trkB does not affect the interaction of BDNF with trkB, but partially prevents the ligand from activating the receptor by inducing an inactive conformation of trkB which could be overcome by BDNF. These antibodies could therefore be defined as ‘weak modulators’, as they affect the apparent affinity of BDNF without altering its binding and their effects can be overcome. It is noteworthy that both antagonist antibodies were raised against a restricted portion of trkB, namely subdomains D4 and D5. In trk receptors, the D5 subdomain has been shown to be critical for the activity of the receptor but not for the interaction with its ligand (Perez et al., 1995; Banfield et al., 2001; Pattarawarapan and Burgess, 2003). Therefore, the data obtained with pAb-BD5 and mAb-BD5 are in line with these seminal studies, as their binding to subdomains D4 and D5 inhibits trkB activity without preventing the binding of BDNF to the receptor.
It is generally thought that bivalent agents are able to dimerize and activate tyrosine kinase receptors (Clary et al., 1994). In this study, we demonstrated that despite their bivalency, only three antibodies were agonists of trkB (pAb-UB1, pAb-SA3 and mAb-AC7), while the other two were antagonists (mAb-BD5 and pAb-BD5). Therefore, it is possible that the latter antibodies dimerized two trkB monomers in a configuration in which the transactivation of the kinase domains did not occur. The use of a monovalent version of these antibodies (Fab fragment) will certainly help to understand if bivalency is a requirement for their effects on trkB activity.
Overall, this study shows that trkB-selective antibodies obtained from commercial sources can constitute a broad-range class of pharmacological tools for the study of the trkB receptor (see summary in Table 2). At present, there are very few TrkB-selective compounds with such diverse properties and no effect on p75NTR. This study further demonstrates that ready-to-use antibodies are available to test the therapeutic interest of pharmacological intervention on trkB in animal models of diseases, particularly for new anti-cancer strategies. As antibodies are unlikely to cross the blood-brain barrier, central effects will be easily avoided which will facilitate their use for the treatment of metastasis and tumour formation in cancer.
Acknowledgments
We are grateful to P. Facchinetti, S. Jacquier and A. Madeira for the cortical neurone dissections. The authors would like to thank G.M. Brodeur for the gift of the full human trkB sequence and M.V. Chao for providing the nnr5 PC12 cells. We also thank E. Urizar for the critical reading of the manuscript.
This work was supported by Institut National de la Santé et de la Recherche Médicale.
Glossary
Abbreviations
- BDNF
brain-derived neurotrophic factor
- KIRA-ELISA
kinase receptor activation enzyme-linked immunosorbent assay
- mAb
monoclonal antibody
- MAPK
mitogen-activated protein kinase
- NGF
nerve-growth factor
- NT
neurotrophin
- pAb
polyclonal antibody
Conflict of interest
None.
Supplemental material
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. 4th edn. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfield MJ, Naylor RL, Robertson AG, Allen SJ, Dawbarn D, Brady RL. Specificity in Trk receptor: neurotrophin interactions: the crystal structure of TrkB-d5 in complex with neurotrophin-4/5. Structure (Camb) 2001;9:1191–1199. doi: 10.1016/s0969-2126(01)00681-5. [DOI] [PubMed] [Google Scholar]
- Benedetti M, Levi A, Chao MV. Differential expression of nerve growth factor receptors leads to altered binding affinity and neurotrophin responsiveness. Proc Natl Acad Sci USA. 1993;90:7859–7863. doi: 10.1073/pnas.90.16.7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo A, Capsoni S, Margotti E, Righi M, Kontsekova E, Pavlik P, et al. Functional blockade of tyrosine kinase A in the rat basal forebrain by a novel antagonistic anti-receptor monoclonal antibody. J Neurosci. 1999;19:9687–9697. doi: 10.1523/JNEUROSCI.19-22-09687.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazorla M, Jouvenceau A, Rose C, Guilloux JP, Pilon C, Dranovsky A, et al. Cyclotraxin-B, the first highly potent and selective TrkB inhibitor, has anxiolytic properties in mice. PLoS One. 2010;5:e9777. doi: 10.1371/journal.pone.0009777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Chao MV, Hempstead BL. p75 and Trk: a two-receptor system. Trends Neurosci. 1995;18:321–326. [PubMed] [Google Scholar]
- Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clary DO, Weskamp G, Austin LR, Reichardt LF. TrkA cross-linking mimics neuronal responses to nerve growth factor. Mol Biol Cell. 1994;5:549–563. doi: 10.1091/mbc.5.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet CJ, Peeper DS. The neurotrophic receptor TrkB: a drug target in anti-cancer therapy? Cell Mol Life Sci. 2006;63:755–759. doi: 10.1007/s00018-005-5490-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douma S, Van Laar T, Zevenhoven J, Meuwissen R, Van Garderen E, Peeper DS. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature. 2004;430:1034–1039. doi: 10.1038/nature02765. [DOI] [PubMed] [Google Scholar]
- Durany N, Thome J. Neurotrophic factors and the pathophysiology of schizophrenic psychoses. Eur Psychiatry. 2004;19:326–337. doi: 10.1016/j.eurpsy.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Geiger TR, Peeper DS. The neurotrophic receptor TrkB in anoikis resistance and metastasis: a perspective. Cancer Res. 2005;65:7033–7036. doi: 10.1158/0008-5472.CAN-05-0709. [DOI] [PubMed] [Google Scholar]
- Green SH, Rydel RE, Connolly JL, Greene LA. PC12 cell mutants that possess low- but not high-affinity nerve growth factor receptors neither respond to nor internalize nerve growth factor. J Cell Biol. 1986;102:830–843. doi: 10.1083/jcb.102.3.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemard V, Ivanisevic L, Garcia AG, Scholten V, Lazo OM, Bronfman FC, et al. An agonistic mAb directed to the TrkC receptor juxtamembrane region defines a trophic hot spot and interactions with p75 coreceptors. Dev Neurobiol. 2009;70:150–164. doi: 10.1002/dneu.20776. [DOI] [PubMed] [Google Scholar]
- Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF, Chao MV. High-affinity NGF binding requires coexpression of the trk proto-oncogene and the low-affinity NGF receptor. Nature. 1991;350:678–683. doi: 10.1038/350678a0. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Ilag LL, Lonnerberg P, Persson H, Ibanez CF. Role of variable beta-hairpin loop in determining biological specificities in neurotrophin family. J Biol Chem. 1994;269:19941–19946. [PubMed] [Google Scholar]
- Ip NY, Stitt TN, Tapley P, Klein R, Glass DJ, Fandl J, et al. Similarities and differences in the way neurotrophins interact with the Trk receptors in neuronal and nonneuronal cells. Neuron. 1993;10:137–149. doi: 10.1016/0896-6273(93)90306-c. [DOI] [PubMed] [Google Scholar]
- Klein R, Conway D, Parada LF, Barbacid M. The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell. 1990;61:647–656. doi: 10.1016/0092-8674(90)90476-u. [DOI] [PubMed] [Google Scholar]
- Kramer K, Gerald W, LeSauteur L, Saragovi HU, Cheung NK. Monoclonal antibody to human Trk-A: diagnostic and therapeutic potential in neuroblastoma. Eur J Cancer. 1997;33:2090–2091. doi: 10.1016/s0959-8049(97)00210-4. [DOI] [PubMed] [Google Scholar]
- Lafont F, Rouget M, Triller A, Prochiantz A, Rousselet A. In vitro control of neuronal polarity by glycosaminoglycans. Development. 1992;114:17–29. doi: 10.1242/dev.114.1.17. [DOI] [PubMed] [Google Scholar]
- LeSauteur L, Maliartchouk S, Le Jeune H, Quirion R, Saragovi HU. Potent human p140-TrkA agonists derived from an anti-receptor monoclonal antibody. J Neurosci. 1996;16:1308–1316. doi: 10.1523/JNEUROSCI.16-04-01308.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nat Rev Neurosci. 2005;6:603–614. doi: 10.1038/nrn1726. [DOI] [PubMed] [Google Scholar]
- McNamara JO, Huang YZ, Leonard AS. Molecular signaling mechanisms underlying epileptogenesis. Sci STKE. 2006;2006:re12. doi: 10.1126/stke.3562006re12. [DOI] [PubMed] [Google Scholar]
- Mojsilovic-Petrovic J, Jeong GB, Crocker A, Arneja A, David S, Russell DS, et al. Protecting motor neurons from toxic insult by antagonism of adenosine A2a and Trk receptors. J Neurosci. 2006;26:9250–9263. doi: 10.1523/JNEUROSCI.1856-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muragaki Y, Timothy N, Leight S, Hempstead BL, Chao MV, Trojanowski JQ, et al. Expression of trk receptors in the developing and adult human central and peripheral nervous system. J Comp Neurol. 1995;356:387–397. doi: 10.1002/cne.903560306. [DOI] [PubMed] [Google Scholar]
- Murer MG, Yan Q, Raisman-Vozari R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer's disease and Parkinson's disease. Prog Neurobiol. 2001;63:71–124. doi: 10.1016/s0301-0082(00)00014-9. [DOI] [PubMed] [Google Scholar]
- Nakagawara A, Azar CG, Scavarda NJ, Brodeur GM. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol Cell Biol. 1994;14:759–767. doi: 10.1128/mcb.14.1.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- Pattarawarapan M, Burgess K. Molecular basis of neurotrophin-receptor interactions. J Med Chem. 2003;46:5277–5291. doi: 10.1021/jm030221q. [DOI] [PubMed] [Google Scholar]
- Perez P, Coll PM, Hempstead BL, Martin-Zanca D, Chao MV. NGF binding to the trk tyrosine kinase receptor requires the extracellular immunoglobulin-like domains. Mol Cell Neurosci. 1995;6:97–105. doi: 10.1006/mcne.1995.1010. [DOI] [PubMed] [Google Scholar]
- Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006;29:507–538. doi: 10.1146/annurev.neuro.29.051605.112929. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Qian MD, Zhang J, Tan XY, Wood A, Gill D, Cho S. Novel agonist monoclonal antibodies activate TrkB receptors and demonstrate potent neurotrophic activities. J Neurosci. 2006;26:9394–9403. doi: 10.1523/JNEUROSCI.1118-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadick MD, Galloway A, Shelton D, Hale V, Weck S, Anicetti V, et al. Analysis of neurotrophin/receptor interactions with a gD-flag-modified quantitative kinase receptor activation (gD.KIRA) enzyme-linked immunosorbent assay. Exp Cell Res. 1997;234:354–361. doi: 10.1006/excr.1997.3614. [DOI] [PubMed] [Google Scholar]
- Shaltiel G, Chen G, Manji HK. Neurotrophic signaling cascades in the pathophysiology and treatment of bipolar disorder. Curr Opin Pharmacol. 2007;7:22–26. doi: 10.1016/j.coph.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.