

Abstract
Purpose
To identify the gene mutations causing X-linked infantile nystagmus in two Chinese families (NYS003 and NYS008), of which the NYS003 family was assigned to the FERM domain–containing 7 (FRMD7) gene linked region in our previous study, and no mutations were found by direct sequencing.
Methods
Two microsatellites, DXS1047 and DXS1001, were amplified using a PCR reaction for the linkage study in the NYS008 family. FRMD7 was sequenced and mutations were analyzed. Multiplex ligation-dependent probe amplification (MLPA) was used to detect FRMD7 mutations in the NYS003 family.
Results
The NYS008 family yielded a maximum logarithm of odds (LOD) score of 1.91 at θ=0 with DXS1001. FRMD7 sequencing showed a nucleotide change of c. 623A>G in exon7 of the patients’ FRMD7 gene, which was predicted to result in an H208R amino acid change. This novel mutation was absent in 100 normal Han Chinese controls. No FRMD7 gene mutations were detected by MLPA in the NYS003 family.
Conclusions
We identified a novel mutation, c. 623A>G (p. H208R), in a Han Chinese family with infantile nystagmus. This mutation expands the mutation spectrum of FRMD7 and contributes to the research on the molecular pathogenesis of FRMD7.
Introduction
Infantile nystagmus (IN), also referred to as congenital nystagmus (CN), congenital “motor” nystagmus (CMN), idiopathic congenital nystagmus (ICN), or congenital idiopathic nystagmus (CIN), is a relatively common ocular motor disorder characterized by rapid to-and-fro oscillations of the eyes. It usually presents at birth or develops within the first few months of life, and it is different from “sensory defect nystagmus” caused by inherited ocular diseases, including: albinism, achromatopsia, Leber congenital amaurosis, congenital cataract, and anterior segment dysgenesis [1,2]. The etiology of infantile nystagmus is unclear.
IN may be inherited as an autosomal dominant, autosomal recessive, or X-linked trait, while the X-linked inheritance is believed to be the most common mode [3]. Two IN loci have been mapped on chromosome Xp11.4-p11.3 [4] and Xq26-q27 [5], respectively, of which the FERM domain–containing 7 gene (FRMD7) was identified to be on the chromosome Xq26-q27 region [6], but no responsible genes have been cloned yet from the Xp11.4-p11.3 region.
FRMD7 is a newly identified member of the FERM family (F for 4.1 protein, E for ezrin, R for radixin and M for moesin), and consists of 12 exons spanning approximately 51 kb on chromosome Xq26-q27. FRMD7 encodes 714 residues and is expressed in the ventricular layer of the forebrain, midbrain, cerebellar primordium, spinal cord, and the developing neural retina in embryos 56 day post-ovulation. It is restrictively expressed in the mid- and hindbrain (where the center of the eye’s movement is located) in earlier embryos 37 day post-ovulation [6]. The FRMD7 protein has B41, FERM-N, FERM-M, FERM-C, and FA structural domains with the conserved domains concentrated at the B41 and FERM-C domains (Figure 1). In fact, the FERM domain is the same as the B41 domain since it was originally identified in band 4.1 (also known as protein 4.1) [7]. The B41 domain is located between residues 1–192, and the FERM-C domain is located between residues 186–279. The FRMD7 protein has close homology to two other FERM domain containing proteins: RhoGEF and pleckstrin domain protein 1 (FARP1; NM_005766) and FARP2 (NM_014808). FARP1 is known to promote the dendritic growth of spinal motor neuron subtypes, while FARP2 has been shown to modulate the length and degree of neurite branching in developing cortical neurons.
Figure 1.

Graphical structure of the FRMD7 domains.
We previously reported five mutations in FRMD7 in seven Chinese families with infantile idiopathic nystagmus [8]. However, no mutations were identified by directly sequencing FRMD7 in the NYS003 family (one of the seven reported families assigned to the FRMD7 gene linked region). In this study, we tried to detect FRMD7 mutations in the NYS003 family by using multiplex ligation-dependent probe amplification (MLPA). We also attempted to identify gene mutations in the other Han Chinese family (NYS008) with infantile nystagmus.
Methods
Patient ascertainment
Two IN families (NYS003 and NYS008) were recruited for this study (Figure 2). All participants underwent an ophthalmologic examination, including visual acuities, slit examination of the lens, examination of the vitreous, fundus, electroretinograms (ERGs), and visual evoked potentials (VEP) as well. Eye movements were recorded with the Eye Tracker system (Eyelink 2000; SR Research, Kanata, Ontario, Canada). After informed consent was obtained, blood samples were collected from the affected and unaffected members in the two IN families. DNA was extracted from blood lymphocytes according to the standard protocol (Roche Biochemical, Inc., Shanghai, China). Briefly, the white blood cells were separated from whole blood via a preferential red blood cell lysis and then lysed by a strong anionic detergent. After the proteins were removed by dehydration and precipitation, the purified DNA is subsequently recovered via ethanol precipitation. This study obtained IRB approval from the Tianjin Eye Hospital, Tianjin, China, and was conducted according to the declaration of Helsinki’s tenets.
Figure 2.

Two Chinese families with X-linked infantile nystagmus. The squares represent the males, the circles represent females, the shaded symbols signify the affected individuals, the dotted circles represent the female carriers, a diagonal line through a symbol indicates a deceased family member, and the arrow indicates the proband.
Genotyping and linkage analysis
A linkage analysis was performed with two fluorescently labeled microsatellite markers, DXS1001 and DXS1047, after PCR amplification in a 10 μl reaction mixture containing 50 ng of genomic DNA, 1× PCR buffer, 2.0 mM MgCl2, 0.2 mmol/l of each dNTP, 5 pmol/l of each forward and reverse primer, and 0.2 U of Ampli Taq Gold DNA polymerase (Applied Biosystems, Foster city, CA). The PCR products from each DNA sample were pooled and mixed with a loading cocktail containing HD-400 size standards (PE Applied Biosystems, Foster City, CA) and loading dye. The resulting PCR products were separated on an ABI3130 sequencer, and analyzed with GENEMAPPER 3.7 (PE Applied Biosystems).
Two-point logarithm of odds (LOD) scores were calculated with easyLinkage plus v4.0 beta software, assuming an X-linked dominant trait with an affected allele frequency of 0.001. The marker order and distance between DXS1001 and DXS1047 were obtained from the National Center for Biotechnology Information (NCBI).
Mutation analysis
Mutation screening of FRMD7 in family NYS008 was conducted using a direct DNA sequence analysis. Individual exons of FRMD7 were amplified by PCR as previously described [8]. Briefly, whole coding regions and exon-intron boundaries of FRMD7 were PCR-amplified in 50 μl of standard PCR buffer containing 1.5 mmol/l MgCl2, 0.2 mmol/l of each dNTP, 0.5 μmol/l of each primer (Table 1), 1 U of Taq polymerase , and 50 ng of DNA. The amplification program was an initial 2 min denaturation at 98 °C, followed by 30 cycles of 30 s at 94 °C, 30 s at 55 °C, 1 min at 72 °C, and a final 7 min extension step at 72 °C. The PCR products were extracted using the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA). DNA sequencing analysis was performed with the BigDye Terminator Cycle Sequencing V3.1 Kit on an ABI PRISM 3130 Genetic Analyzer (PE Applied Biosystems). Sequencing results were assembled and analyzed with DNASTAR software’s Seqman program (DNASTAR Inc., Madison, WI). A multiple sequence alignment was performed using the Clustal W algorithm in the software package. The modeled structures were built with SWISS-MODEL.
Table 1. Primers used to amplify the individual exons of FRMD7.
| Exon | Forward primer | Reverse primer | Tm (°C) | Product length (bp) |
|---|---|---|---|---|
| 1 |
AGGAGACTGCCCAGATGCTA |
GGGCTGTTCACAAATGACAA |
60 |
348 |
| 2 |
GAAACAGGGCTTGCAGAGAG |
GCAATGCTAGACACAAAGAACC |
60 |
246 |
| 3 |
TGCTCAATGCCATGCTCTAC |
AAAGCCCTTTTCTCCCCTTA |
58 |
199 |
| 4 |
TGGGTGTGTGTGTGTGTGTG |
GCCCCCAATAAATGGAGAAT |
58 |
237 |
| 5 |
TGGATCTGGGAGAAGGAAAA |
GCTCCTGTGCTTGGTTCTCT |
60 |
238 |
| 6 |
GAGGACAAGGGTATGCTGGA |
TCAGGTTTAAGGGCTTGCTC |
58 |
281 |
| 7 |
GAGCTCTCAGGGTGGAAATG |
ACACCCAAGTTTGAGCCAAG |
58 |
293 |
| 8 |
TGCACTGTCTTACAAGCCAACT |
CGATTTGCAGAAACAACCAA |
56 |
231 |
| 9 |
TTGGGATTTGAAGGTCTTTGA |
TCCTCCTAAGCCTCCTGTGTT |
60 |
300 |
| 10-11 |
AGCCTATTGGTTTATGGCTAGAAC |
GCAGAATCAATTCATGGAAGC |
56 |
393 |
| 12a |
GGCCTTTTCCTTCTTTCACC |
CTGGGGAGGCATAATACCAA |
58 |
472 |
| 12b |
AGCTCCTTCCAAACAAGCTG |
TGACTGAGAGCAGGACAAGG |
60 |
524 |
| 12c | GCGGTAGGAGCAACATCAAT | CCAAGAAAATGGTTTCTACAACTTC | 58 | 560 |
Multiplex ligation-dependent probe amplification (MLPA)
MLPA was conducted by using the supplied protocol from the SALSA P269 FRMD7-NYS1 probe kit of MRC-Holland (Amsterdam, Holland), which contains 28 MLPA probes with amplification products between 166 and 391 nucleotides. One-hundred ng of DNA (5 μl) was denatured at 98 °C for 5 min, and then was mixed with the probe set and MLPA buffer after cooling at 25 °C. The mixture was re-heated to 95 °C, and then was incubated for 16 h at 60 °C. Following probe hybridization, the DNA ligase and ligation buffer were added, and the ligation was proceeded for 15 min at 54 °C. One microliter of ligation products were amplified by PCR according to the manufacturer’s protocol. The PCR products were then mixed with a loading cocktail containing HD-400 size standards (PE Applied Biosystems) and de-ionized formamide. The resulting PCR products were separated on an ABI3130 sequencer, and analyzed with GENEMAPPER 3.7 (PE Applied Biosystems). Specific peaks corresponding to each exon were identified according to their migration (relative to the size standards), and were exported to a Microsoft Excel (Microsoft Corporation, Redmond, WA) spreadsheet. Peak heights of each fragment were compared to a control sample to obtain a dosage quotient (DQ) representing the gene dosage of each amplicon. For normal sequences, a dosage quotient of 1.0 is expected; if a deletion or duplication is present, the dosage quotient should be 0.7 and 1.3, respectively.
Results
The NYS008 family from Hebei province, China, included 4 affected males and 4 female carriers. Individual IV3 was a proband in this family. His parents were consanguineously married (Figure 2A). He developed nystagmus at the age of 3 to 4 months, and had a horizontal jerk ocular oscillation at a distance, with the best-corrected visual acuity of 0.5 in both eyes at the age of 32. His eye movement recordings in a primary gaze of the right and left eyes showed horizontal nystagmus with slow rightward drift and fast beats to the left (Figure 3). The ocular oscillation could be dampened at neither a left nor a right gaze of 10°, but could be achieved at a viewing distance of 33 cm (Figure 3). Other male patients in this family had various reduced visual acuity, ranging from 0.2 to 1.0 with a horizontal jerk ocular oscillation. None of the patients in this family had a compensatory face turn.
Figure 3.
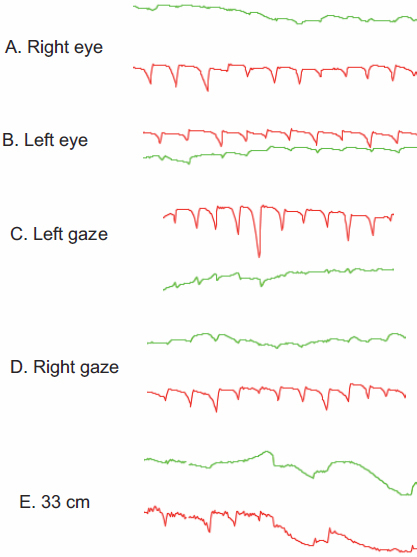
Eye movement recording of the proband in the NYS008 family. Eye movement recordings of the primary gaze of the right and left eyes of the proband showed horizontal nystagmus (A and B). The ocular oscillation could be dampened at neither the left (C) nor the right (D) gaze of 10°, but could be achieved at a viewing distance of 33 cm (E). The vertical component of the eye movement is denoted in green, and the horizontal movement is shown in red.
A linkage study showed that the maximum LOD score of 1.91 was yielded at the polymorphic marker DXS1001 (θ=0), and a positive LOD score of 1.31 was achieved at DXS1047. Sequencing of FRMD7 illustrated a single nucleotide change of c.623A>G in exon 7, which was predicted to result in a wild type amino acid of Histidine (H) substituted by a mutant type amino acid of Arginine (R) at codon 208 (Figure 4). This nucleotide change was absent in 100 normal controls after sequencing of FRMD7. The multiple sequence alignment of the FRMD7 protein indicated that H208R was conserved among Homo sapiens, Pongo abelii, Rattus norvegicus, Mus musculus, Canis familiaris, Gallus gallus, Xenopus lavis, and Danio rerio (Figure 5). A Position-Specific Independent Counts (PISC) score of 2.53 was yielded after using the POLYPHEN program to predict the functional and structural changes of the amino acid substitution. The modeled protein structure assessed by the ANOLEA program [9] would thus be unstable if the Histidine at codon 208 was substituted by the Arginine (Figure 6).
Figure 4.
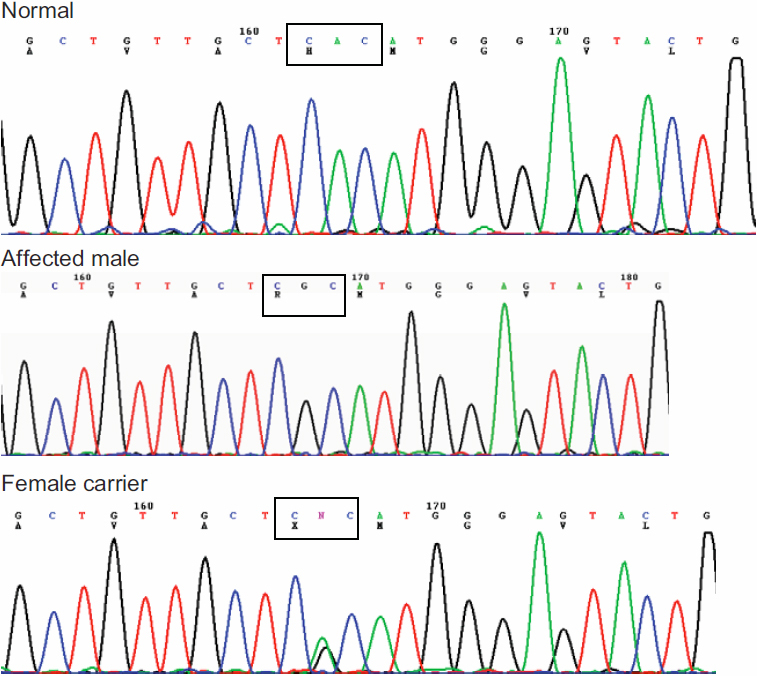
Identification of the 623A>G (p. H208R) in FRMD7. Sequencing chromatograms from a normal individual (top), an affected male (middle), and a female carrier (bottom), showing an A→G change in exon 7.
Figure 5.
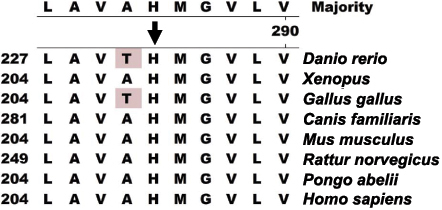
Alignment of FRMD7 amino acids. The alignment of amino acids around p.H208 (denoted by the black arrow) of FRMD7 revealed evolutionary conservation of the Histidine among Homo sapiens, Pongo abelii, Rattus norvegicus, Mus musculus, Canis familiaris, Gallus gallus, Xenopus lavis, and Danio rerio.
Figure 6.

The 3D model and the anolea mean force potential plot for the target protein of human FRMD7. A: The model rendered in 3-D for the target human FRMD7 protein. α-helices, coils, and β-sheets are depicted in red, gray, and green, respectively. Only two amino acids, H208 and M209, are located inside the region between the two β-sheets (denoted by an arrow). B: The anolea mean force potential plot for the target protein of the human FRMD7. Above is the mutant type of the FRMD7 protein; below is the wild type of the FRMD7 protein. The y-axis of the plot represents the energy for each amino acid of the protein chain. Negative energy values (in green) represent a favorable energy environment, whereas positive values (in red) indicate an unfavorable energy environment for a given amino acid. A favorable energy environment tends to be damaged because of an H to R change at codon208.
The NYS003 family reported in our previous study (the C family from the Liaoning province, China [8]), consisted of 6 male patients and 9 female carriers (Figure 2B). The results analyzed by MLPA using 28 probes on the patients are listed in Table 2, and the average dosage quotient (DQ) at each probe ranged from 0.8 to 1.1, which suggested that mutations were not detected by the MLPA method.
Table 2. FRMD7 gene dosage analysis by MLPA in the NYS003 family.
| Probe | Average DQ | SD | dosage |
|---|---|---|---|
| PDCD8 | 0.9 | 0.09 | Normal |
| FRMD7 exon1 | 1.07 | 0.07 | Normal |
| FRMD7 exon1 | 0.93 | 0.08 | Normal |
| FRMD7 exon2 | 0.81 | 0.15 | Normal |
| FRMD7 exon2 | 0.88 | 0.09 | Normal |
| FRMD7 exon4 | 0.97 | 0.07 | Normal |
| FRMD7 exon4 | 0.83 | 0.04 | Normal |
| FRMD7 exon5 | 0.86 | 0.07 | Normal |
| FRMD7 exon6 | 0.98 | 0.08 | Normal |
| FRMD7 exon7 | 0.98 | 0.07 | Normal |
| FRMD7 exon8 | 0.94 | 0.07 | Normal |
| FRMD7 exon9 | 0.91 | 0.08 | Normal |
| FRMD7 exon9 | 0.98 | 0.05 | Normal |
| FRMD7 exon11 | 1.02 | 0.03 | Normal |
| FRMD7 exon12 | 1.01 | 0.05 | Normal |
| FRMD7 exon12 | 1.03 | 0.06 | Normal |
| GPC4 | 0.99 | 0.08 | Normal |
| GPC3 | 1.01 | 0.08 | Normal |
| Control | 0.96 | 0.11 | Normal |
| Control | 1.03 | 0.08 | Normal |
| Control | 1.02 | 0.04 | Normal |
| Control | 1.03 | 0.07 | Normal |
| Control | 0.94 | 0.08 | Normal |
| Control | 0.93 | 0.06 | Normal |
| Control | 1.04 | 0.08 | Normal |
| Control | 1.02 | 0.07 | Normal |
| Control | 0.95 | 0.07 | Normal |
| Control | 1.09 | 0.06 | Normal |
For normal sequences, a dosage quotient (DQ) of 1.0 is expected; if a deletion or duplication is present, the dosage quotient should be 0.7 and 1.3, respectively.
Discussion
To date, 40 mutations in FRMD7 have been reported worldwide in families with X-linked congenital nystagmus from various ethnic backgrounds [6,8,10-16]. These mutations include missense, nonsense, and splicing mutations, as well as insertions and deletions according to the Human Gene Mutations Database. However, the underlying molecular mechanisms of these mutations in CIN are still not fully understood. FRMD7 may play an important role in neuronal development, particularly in brain regions that are associated with ocular motor control [17-20]. It may also be involved in signal transduction between the plasma membrane and cytoskeleton because it contains a FERM domain at the NH2-terminus [21].
In this study, we found a novel nucleotide change of c.623A>G in exon7 of the FRMD7 gene in the NYS008 family, but we were unable to detect FRMD7 mutations in the NYS003 family. The heterozygote change of c.623A>G in FRMD7 in the NYS008 family should be regarded as a novel gene mutation rather than a polymorphism nucleotide change due to the following reasons: 1) It is absent in 100 normal controls. 2) We predict that the c.623A>G change would result in a Histidine to Arginine change at codon 208 where it is located within highly conserved regions that are invariant in Homo sapiens, Pongo abelii, Rattus norvegicus, Mus musculus, Canis familiaris, Gallus gallus, Xenopus lavis, and Danio rerio. This suggests that the Histidine at codon 208 is critical to the protein’s normal functioning. 3) A PISC score of 2.53 predicted by POLYPHEN suggests that the substitution of Histidine to Arginine at codon 208 would most likely damage the protein structure and/or the protein function.
Further analysis of the FRMD7 protein structure in the model shows that the Histidine at codon 208 is one of two amino acids located inside the region between the two β-sheets, and substituting it with Arginine is likely to destabilize the protein by introducing a larger amino acid within restricted areas of the protein. This has been demonstrated by the ANOLEA program, which found that a favorable energy environment tends to be damaged by amino acid substitutions in that region (Figure 6).
It has been reported that nearly all missense mutations, except for G296R, Y301C, and S340L, occurred in the B41 and the FERM-C domains of FRMD7 [6,12]. H208R occurred in the FERM-C domain which seems to be a mutation-rich region [22], and was close to two other known mutations (Q201X and N221D [6]) in the same domain.
The NYS003 family’s responsible gene was previously localized to the region linked with the microsatellite marker DXS1047 (Zmax=2.42, θmax=0.1). No mutations were detected in FRMD7 by direct sequencing, although all coding regions and exon-intron boundaries were sequenced [8]. The method of direct sequencing based on a single exon's PCR amplification from genomic DNA has several shortcomings in detecting most exon deletions and duplications [23]. MLPA is believed to be a robust assay that offers several advantages for gene mutation detection, particularly for detecting deletions/duplications of one or more exons in the gene [23,24]. It is a variation of the PCR reaction that permits multiple targets to be amplified with only a single primer pair [23]. Each MLPA probe consists of two halves of the oligonucleotides which recognize adjacent target sites on the DNA and can be ligated into a complete probe when both of them are hybridized to their respective targets. Only the ligated oligonucleotides can be amplified, not the unbound probe oligonucleotides. Each complete probe has a unique length so that its resulting amplicons can be separated and identified by electrophoresis. By comparing the peak pattern obtained on a given sample with that obtained on various reference samples, the relative quantity of each amplicon can be determined. Because the amount of ligated probes is dependent on the number of specific primer binding sites, this method is suitable for the detection of chromosomal deletions or duplications.
To exclude the possibility of FRMD7 gene rearrangements in the NYS003 family, the MLPA method was used in this study. However, we have not yet identified FRMD7 mutations in this family. The fact that we detected no mutations from either direct sequencing or MLPA, illustrated that our experiments’ mutation screening techniques might be limited in their ability to detect unknown regions (e.g., the promoter or other regulatory regions including those within introns) in FRMD7. Further study on the patients’ RNA might find mutations in the unknown regions in FRMD7.
Recently, investigations of the molecular pathogenesis to some ocular movement diseases develop very quickly, especially to a group of the congenital cranial dysinnervation disorders (CCDDs), including congenital fibrosis of the extraocular muscles (CFEOM), Duane’s retraction syndrome (DRS), and horizontal gaze palsy with progressive scoliosis (HGPPS) and MÖbius syndrome [25]. Some responsible genes for CCDDs (kinesin family member 21A [KIF21A] for CFEOM1 [26], paired-like homeobox 2a [ARIX] for CFEOM2 [27], roundabout, axon guidance receptor, homolog 3 [Robo3] for HGPPS [28], and chimerin 1 [CHN1] for DRS [29]) have been identified and are believed to be involved either in the development of brainstem motor nuclei or in axon guidance. Some advanced neuroimaging techniques, such as diffusion tensor imaging (DTI) and diffusion spectrum imaging (DSI) [30], have also been used to accurately recognize new patterns of aberrant axon connectivity and phenotypically confirm the findings in genetic studies. The combination of advanced neuroimaging techniques and genetic studies is likely to advance our understanding of the molecular mechanisms of congential nystagmus.
In summary, we investigated the gene mutations in two Chinese families with infantile nystagmus and identified a novel gene mutation, c. 623A>G (p. H208R), in FRMD7 in one of the families. This mutation will expand the mutation spectrum of FRMD7 and contribute to the study of the FRMD7 gene’s molecular pathogenesis.
Acknowledgments
The authors thank all participants for their enthusiastic participation. This research is partially supported by the National Natural Science Foundation of China (Grant No. 30940081) and the Xuzhou Science and Technology Foundation (Grant No. XF10C050). Dr. Ningdong Li and Dr. Kanxing Zhao contributed equally to the conduct of the research and are to be considered as co-corresponding author.
References
- 1.Abadi RV, Bjerre A. Motor and sensory characteristics of infantile nystagmus. Br J Ophthalmol. 2002;86:1152–60. doi: 10.1136/bjo.86.10.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hertle RW, Maldanado VK, Maybodi M, Yang D. Clinical and ocular motor analysis of the infantile nystagmus syndrome in the first 6 months of life. Br J Ophthalmol. 2002;86:670–5. doi: 10.1136/bjo.86.6.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oetting WS, Armstrong CM, Holleschau AM, DeWan AT, Summers GC. Evidence for genetic heterogeneity in families with congenital motor nystagmus (CN). Ophthalmic Genet. 2000;21:227–33. [PubMed] [Google Scholar]
- 4.Cabot A, Rozet JM, Gerber S, Perrault I, Ducroq D, Smahi A, Souied E, Munnich A, Kaplan J. A gene for X-linked idiopathic congenital nystagmus (NYS1) maps to chromosome Xp11.4-p11.3. Am J Hum Genet. 1999;64:1141–6. doi: 10.1086/302324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kerrison JB, Vagefi MR, Barmada MM, Maumenee IH. Congenital motor nystagmus linked to Xq26-q27. Am J Hum Genet. 1999;64:600–7. doi: 10.1086/302244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tarpey P, Thomas S, Sarvananthan N, Mallya U, Lisgo S, Talbot CJ, Roberts EO, Awan M, Surendran M, McLean RJ, Reinecke RD, Langmann A, Lindner S, Koch M, Jain S, Woodruff G, Gale RP, Degg C, Droutsas K, Asproudis I, Zubcov AA, Pieh C, Veal CD, Machado RD, Backhouse OC, Baumber L, Constantinescu CS, Brodsky MC, Hunter DG, Hertle RW, Read RJ, Edkins S, O'Meara S, Parker A, Stevens C, Teague J, Wooster R, Futreal PA, Trembath RC, Stratton MR, Raymond FL, Gottlob I. Mutations in FRMD7, a newly identified member of the FERM family, cause X-linked idiopathic congenital nystagmus. Nat Genet. 2006;38:1242–4. doi: 10.1038/ng1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chishti AH, Kim AC, Marfatia SM, Lutchman M, Hanspal M, Jindal H, Liu SC, Low PS, Rouleau GA, Mohandas N, Chasis JA, Conboy JG, Gascard P, Takakuwa Y, Huang SC, Benz EJ, Jr, Bretscher A, Fehon RG, Gusella JF, Ramesh V, Solomon F, Marchesi VT, Tsukita S, Tsukita S, Hoover KB. The FERM domain: a unique module involved in the linkage of cytoplasmic proteins to the membrane. Trends Biochem Sci. 1998;23:281–2. doi: 10.1016/s0968-0004(98)01237-7. [DOI] [PubMed] [Google Scholar]
- 8.Li N, Wang L, Cui L, Zhang L, Dai S, Li H, Chen X, Zhu L, Hejtmancik JF, Zhao K. Five novel mutations of the FRMD7 gene in Chinese families with X-linked infantile nystagmus. Mol Vis. 2008;14:733–8. [PMC free article] [PubMed] [Google Scholar]
- 9.Melo F, Feytmans E. Assessing protein structures with a non-local atomic interaction energy. J Mol Biol. 1998;277:1141–52. doi: 10.1006/jmbi.1998.1665. [DOI] [PubMed] [Google Scholar]
- 10.Fingert JH, Roos B, Eyestone ME, Pham JD, Mellot ML, Stone E. Novel intragenic FRMD7 deletion in a pedigree with congenital X-linked nystagmus. Ophthalmic Genet. 2010;31:77–80. doi: 10.3109/13816810903584989. [DOI] [PubMed] [Google Scholar]
- 11.Shiels A, Bennett TM, Prince JB, Tychsen L. X-linked idiopathic infantile nystagmus associated with a missense mutation in FRMD7. Mol Vis. 2007;13:2233–41. [PubMed] [Google Scholar]
- 12.Zhang B, Liu Z, Zhao G, Xie X, Yin X, Hu Z, Xu S, Li Q, Song F, Tian J, Luo W, Ding M, Yin J, Xia K, Xia J. Novel mutations of the FRMD7 gene in X-linked congenital motor nystagmus. Mol Vis. 2007;13:1674–9. [PubMed] [Google Scholar]
- 13.Zhang Q, Xiao X, Li S, Guo X. FRMD7 mutations in Chinese families with X-linked congenital motor nystagmus. Mol Vis. 2007;13:1375–8. [PubMed] [Google Scholar]
- 14.Schorderet DF, Tiab L, Gaillard MC, Lorenz B, Klainguti G, Kerrison JB, Traboulsi EI, Munier FL. Novel mutations in FRMD7 in X-linked congenital nystagmus. Mutation in brief #963. Online. Hum Mutat. 2007;28:525. doi: 10.1002/humu.9492. [DOI] [PubMed] [Google Scholar]
- 15.He X, Gu F, Wang Y, Yan J, Zhang M, Huang S, Ma X. A novel mutation in FRMD7 causing X-linked idiopathic congenital nystagmus in a large family. Mol Vis. 2008;14:56–60. [PMC free article] [PubMed] [Google Scholar]
- 16.Kaplan Y, Vargel I, Kansu T, Akin B, Rohmann E, Kamaci S, Uz E, Ozcelik T, Wollnik B, Akarsu NA. Skewed X inactivation in an X linked nystagmus family resulted from a novel, p.R229G, missense mutation in the FRMD7 gene. Br J Ophthalmol. 2008;92:135–41. doi: 10.1136/bjo.2007.128157. [DOI] [PubMed] [Google Scholar]
- 17.Toyofuku T, Yoshida J, Sugimoto T, Zhang H, Kumanogoh A, Hori M, Kikutani H. FARP2 triggers signals for Sema3A-mediated axonal repulsion. Nat Neurosci. 2005;8:1712–9. doi: 10.1038/nn1596. [DOI] [PubMed] [Google Scholar]
- 18.Zhuang B, Su YS, Sockanathan S. FARP1 promotes the dendritic growth of spinal motor neuron subtypes through transmembrane Semaphorin6A and PlexinA4 signaling. Neuron. 2009;61:359–72. doi: 10.1016/j.neuron.2008.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kubo T, Yamashita T, Yamaguchi A, Sumimoto H, Hosokawa K, Tohyama M. A novel FERM domain including guanine nucleotide exchange factor is involved in Rac signaling and regulates neurite remodeling. J Neurosci. 2002;22:8504–13. doi: 10.1523/JNEUROSCI.22-19-08504.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Betts-Henderson J, Bartesaghi S, Crosier M, Lindsay S, Chen HL, Salomoni P, Gottlob I, Nicotera P. The nystagmus-associated FRMD7 gene regulates neuronal outgrowth and development. Hum Mol Genet. 2010;19:342–51. doi: 10.1093/hmg/ddp500. [DOI] [PubMed] [Google Scholar]
- 21.Cho W, Stahelin RV. Membrane-protein interactions in cell signaling and membrane trafficking. Annu Rev Biophys Biomol Struct. 2005;34:119–51. doi: 10.1146/annurev.biophys.33.110502.133337. [DOI] [PubMed] [Google Scholar]
- 22.He X, Gu F, Wang Z, Wang C, Tong Y, Wang Y, Yang J, Liu W, Zhang M, Ma X. A novel frameshift mutation in FRMD7 causing X-linked idiopathic congenital nystagmus. Genet Test. 2008;12:607–13. doi: 10.1089/gte.2008.0070. [DOI] [PubMed] [Google Scholar]
- 23.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor CF, Charlton RS, Burn J, Sheridan E, Taylor GR. Genomic deletions in MSH2 or MLH1 are a frequent cause of hereditary non-polyposis colorectal cancer: identification of novel and recurrent deletions by MLPA. Hum Mutat. 2003;22:428–33. doi: 10.1002/humu.10291. [DOI] [PubMed] [Google Scholar]
- 25.Engle EC. The genetic basis of complex strabismus. Pediatr Res. 2006;59:343–8. doi: 10.1203/01.pdr.0000200797.91630.08. [DOI] [PubMed] [Google Scholar]
- 26.Yamada K, Andrews C, Chan WM, McKeown CA, Magli A, de Berardinis T, Loewenstein A, Lazar M, O'Keefe M, Letson R, London A, Ruttum M, Matsumoto N, Saito N, Morris L, Del Monte M, Johnson RH, Uyama E, Houtman WA, de Vries B, Carlow TJ, Hart BL, Krawiecki N, Shoffner J, Vogel MC, Katowitz J, Goldstein SM, Levin AV, Sener EC, Ozturk BT, Akarsu AN, Brodsky MC, Hanisch F, Cruse RP, Zubcov AA, Robb RM, Roggenkaemper P, Gottlob I, Kowal L, Battu R, Traboulsi EI, Franceschini P, Newlin A, Demer JL, Engle EC. Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type 1 (CFEOM1). Nat Genet. 2003;35:318–21. doi: 10.1038/ng1261. [DOI] [PubMed] [Google Scholar]
- 27.Nakano M, Yamada K, Fain J, Sener EC, Selleck CJ, Awad AH, Zwaan J, Mullaney PB, Bosley TM, Engle EC. Homozygous mutations in ARIX (PHOX2A) result in congenital fibrosis of the extraocular muscles type 2. Nat Genet. 2001;29:315–20. doi: 10.1038/ng744. [DOI] [PubMed] [Google Scholar]
- 28.Jen JC, Chan WM, Bosley TM, Wan J, Carr JR, Rub U, Shattuck D, Salamon G, Kudo LC, Ou J, Lin DD, Salih MA, Kansu T, Al Dhalaan H, Al Zayed Z, MacDonald DB, Stigsby B, Plaitakis A, Dretakis EK, Gottlob I, Pieh C, Traboulsi EI, Wang Q, Wang L, Andrews C, Yamada K, Demer JL, Karim S, Alger JR, Geschwind DH, Deller T, Sicotte NL, Nelson SF, Baloh RW, Engle EC. Mutations in a human ROBO gene disrupt hindbrain axon pathway crossing and morphogenesis. Science. 2004;304:1509–13. doi: 10.1126/science.1096437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyake N, Chilton J, Psatha M, Cheng L, Andrews C, Chan WM, Law K, Crosier M, Lindsay S, Cheung M, Allen J, Gutowski NJ, Ellard S, Young E, Iannaccone A, Appukuttan B, Stout JT, Christiansen S, Ciccarelli ML, Baldi A, Campioni M, Zenteno JC, Davenport D, Mariani LE, Sahin M, Guthrie S, Engle EC. Human CHN1 mutations hyperactivate alpha2-chimaerin and cause Duane's retraction syndrome. Science. 2008;321:839–43. doi: 10.1126/science.1156121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engle EC. Human genetic disorders of axon guidance. Cold Spring Harb Perspect Biol. 2010;2:a001784. doi: 10.1101/cshperspect.a001784. [DOI] [PMC free article] [PubMed] [Google Scholar]