

Abstract
Sexually transmitted infections (STIs) increase the likelihood of HIV transmission. Defensins are part of the innate mucosal immune response to STIs and therefore we investigated their role in HIV infection. We found that human defensins 5 and 6 (HD5 and HD6) promoted HIV infection, and this effect was primarily during viral entry. Enhancement was seen with primary viral isolates in primary CD4+ T cells and the effect was more pronounced with R5 virus compared with X4 virus. HD5 and HD6 promoted HIV reporter viruses pseudotyped with vesicular stomatitis virus and murine leukemia virus envelopes, indicating that defensin-mediated enhancement was not dependent on CD4 and coreceptors. Enhancement of HIV by HD5 and HD6 was influenced by the structure of the peptides, as loss of the intramolecular cysteine bonds was associated with loss of the HIV-enhancing effect. Pro-HD5, the precursor and intracellular form of HD5, also exhibited HIV-enhancing effect. Using a cervicovaginal tissue culture system, we found that expression of HD5 and HD6 was induced in response to Neisseria gonorrhoeae (GC, for gonococcus) infection and that conditioned medium from GC-exposed cervicovaginal epithelial cells with elevated levels of HD5 also enhanced HIV infection. Introduction of small interfering RNAs for HD5 or HD6 abolished the HIV-enhancing effect mediated by GC. Thus, the induction of these defensins in the mucosa in the setting of GC infection could facilitate HIV infection. Furthermore, this study demonstrates the complexity of defensins as innate immune mediators in HIV transmission and warrants further investigation of the mechanism by which defensins modulate HIV infection.
Sexual contact is the most common route of HIV transmission. Women comprise nearly 50% of those infected worldwide and >70% in sub-Saharan Africa (1, 2). Prevention strategies using multiple approaches are urgently needed to reduce the probability of transmission. Epidemiological and clinical studies indicate that sexually transmitted infections (STIs)3 significantly increase the likelihood of HIV transmission (3–7), although the underlying mechanisms are not clear. STIs not only increase HIV shedding in HIV-infected adults, they also increase susceptibility to HIV infection in those who are HIV negative (3, 4, 8). The adjusted odds ratios for HIV acquisition range from 2.2 to 11.3 for individuals with ulcerative diseases, and from 3 to 4 for nonulcerative STIs (3, 8). Although the contribution of STIs to the increase in HIV transmission is multifaceted (reviewed in Refs. 3 and 4), understanding these mechanisms will contribute to the development of new strategies to reduce the spread of HIV.
Gonorrhea, caused by the Gram-negative diplococcus Neisseria gonorrhoeae, remains a major global health problem; >60 million new cases are diagnosed worldwide each year (9). Men with gonorrhea frequently have acute urethritis, an inflammatory response resulting from gonococcal (GC) infection (10). In contrast, 50 –80% of women with lower genital tract GC infection are asymptomatic (11, 12) and 45% of women with GC cervicitis will develop an ascending infection, a prerequisite for pelvic inflammatory disease (13, 14). Thus, individuals with asymptomatic GC infection are particularly at risk for acquiring HIV.
GC can adhere to and penetrate mucosal epithelial cells, access submucosal sites, and interact with immune cells (15). GC infection of epithelial cells involves the phase-variable expression of several outer membrane components, including pili, opacity-associated (Opa) proteins, lactosyl lipooligosaccharide (LOS), and the PorB porin protein. Recently, LOS, which interacts with TLR4 (16), has been shown to induce the secretion of TNF-α, IL-1β, IL-6, and IL-8 in male primary urethral epithelial cells (17). These are the same cytokines that are prevalent in the urethral lumen in active GC infection. Studies of cytokine induction in response to GC infection in women have not been consistent (14); IL-1, IL-6, and IL-8 levels are not increased in genital tract secretions from women with GC cervicitis (18) but are elevated in GC-exposed human cervical and vaginal epithelial cell lines (19). In men with GC urethritis, HIV RNA is decreased in semen following treatment for GC (5). GC infection is reported to enhance HIV infection in vitro by activating the HIV long terminal repeat (LTR) in transformed T cells (20) or in vivo by increasing the number of endocervical CD4+ T lymphocytes in GC-infected women (21). Recently, GC infection has also been shown to enhance HIV infection in monocyte-derived dendritic cells by activating TLR2 (22).
Mammalian defensins are antimicrobial peptides important to innate host defense and are thought to play a role in mucosal immunity (23–26). Indeed, in humans, defensin levels in the mucosa are frequently elevated in response to mucosal infection (27–29), suggesting a potential role in modulating STIs, including HIV. Human neutrophil peptides (HNPs) 1– 4 from neutrophils and human β-defensins (HBDs) 2 and 3 from epithelial cells exhibit anti-HIV-1 activity (30, 31), whereas HBD1 displays little anti-HIV activity (32, 33). Defensins with anti-HIV activity appear to inhibit HIV through multiple mechanisms; different defensins may have similar or distinct antiviral actions (30, 31). For example, HNP1 plays a dual role in anti-HIV innate immunity (34) by acting on HIV virions and target cells. It also blocks HIV infection of macrophages by up-regulation of CC chemokines (35). On the target cell, HNP1 interferes with HIV nuclear import and transcription after entry. At least one of the cellular effects associated with HIV inhibition by HNP1 is interference with protein kinase C signaling in primary CD4+ T cells (34). Similarly as HNPs, HBDs exhibit multiple anti-HIV functions (32, 33). Unlike HNP-1, HBDs only display the direct effect on a HIV virion under a low salt condition (10 mM phosphate buffer) (32). After viral entry, HBD2 inhibits the formation of early reverse-transcribed HIV products (32, 33). In addition, HBDs 2 and 3 down-regulate the HIV coreceptor CXCR4 in PBMCs in the absence of serum (32), and HBD3 competes with stro-mal-derived factor 1, the natural ligand for CXCR4 (36).
Human α-defensins HD5 and HD6 are constitutively expressed by intestinal Paneth cells, playing an important role in gut mucosal immunity (23–26). HD5 is also found in cervical lavage fluid as well as in the epithelium of the vagina and the ectocervix (37, 38). A recent report indicates that HD5 is induced in the male urethra during Chlamydia trachomatis and N. gonorrhoeae infection (29), further supporting a role for defensins in patients with STIs. Considering the clinically observed association of GC infection and increased HIV incidence and the up-regulation of HD5 in gonococcal urethritis, we investigated the role of defensins in STI-mediated HIV enhancement. Our study demonstrated, for the first time, that HD5 and HD6 actually enhanced HIV infection and therefore may contribute to GC-mediated HIV enhancement. These studies highlight the complexity of the interactions between an innate immune response and HIV transmission and may offer insights into novel approaches to prevent HIV infection by targeting HD5 and HD6.
Materials and Methods
HD5, HD6, and pro-HD5 peptides
HD5 and HD6 as well as linear unstructured forms of HD5 and HD6, [Abu]HD5 and [Abu]HD6, in which the six cysteine residues were replaced by isosteric α-aminobutyric acid (Abu), were chemically synthesized and folded as described previously (39). The molecular mass of the peptides was verified by electrospray ionization mass spectrometry as described previously (39). Both synthetic HD5 and HD6 are correctly folded as indicated through structural analysis by x-ray crystallography (40). Recombinant HD5 propeptide (aa 20 to 94) was biosynthesized using the baculovirus/insect cell culture system as previously described (41).
Cell culture
PBMCs from normal healthy blood donors were isolated by Ficoll-Hypaque gradient centrifugation. CD4+ T cells were isolated from PBMCs by negative selection using a CD4+ T cell isolation kit from Miltenyi Biotech. The purity of cells is 98% based on flow cytometry analysis. CD4+ T cells were stimulated with PHA at 5 μg/ml and maintained in RPMI 1640 medium supplemented with 10% FBS and IL-2 at 25 U/ml for 3 days at 37°C before viral infection. HeLa-CD4-CCR5 cells were provided by D. Kabat (University of Oregon, Portland, OR) and maintained in DMEM containing 10% FBS. U87-CD4-CCR5 cells were obtained from AIDS Research and Reference Reagent Program (ARRRP), National Institute of Allergy and Infectious Disease (NIAID), National Institutes of Health, Bethesda, MD) and maintained in DMEM containing 10% FBS. Immortalized vaginal, endocervical, and ectocervical epithelial cells were provided by R. Fichorova (Brigham and Women's Hospital, Boston, MA) and maintained as described previously (42, 43).
HIV-1 infection
Replication-defective HIV-1 HxB2, JR-FL, and vesicular stomatitis virus (VSV) envelope-pseudotyped, luciferase-expressing reporter viruses for a single-cycle infection assay were produced as described previously (44, 45). Briefly, HEK293T cells were cotransfected with a plasmid encoding the envelope-deficient HIV-1 NL4-3 virus with the luciferase reporter gene inserted into nef (pNL4-3.Luc.R-E-; ARRRP, from N. Landau) and a pSV plasmid expressing the HIV-1 HxB2 and JR-FL glycoprotein (gift of D. Littman, New York University, New York, NY) or the VSV-G glycoprotein (gift of D. Trono, University of Geneva, Geneva, Switzerland). The supernatant medium was collected 48 h after transfection and filtered. Virus stocks were analyzed for HIV-1 p24 Ag concentration by ELISA (SAIC-Frederick, Frederick, MD). To produce HIV-1JR-FL-pseudotyped viruses in the absence of serum, transfected cells were incubated for 24 h, washed with PBS, and cultured in medium without serum for additional 24 h before collecting viruses.
To determine the effect of defensins on HIV infection using a single-cycle infection assay, serum-free HIV-1JR-FL-pseudotyped luciferase reporter virus was incubated with HD5, HD6, or medium only at varying concentrations at 37°C for 1 h. FBS at a final concentration of 10% (v/v) was added to the defensin-virus mixture before the addition to activated CD4+ T cells at 1 × 106 per well or HeLa-CD4-CCR5 at 5 × 104 per well in a 48-well plate for 2 h at 37°C. Unbound virus was removed by washing and infected cells were incubated at 37°C for 48 h before lysis with luciferase substrate buffer (Promega). Luciferase activity (in relative light units) reflecting viral infection was measured on an EG & G (Berthold) Mini-Lumat LB9506 luminometer. To determine the post-entry effect, CD4+ T cells or HeLa-CD4-CCR5 were infected with HIV-1vsv-or HIV-1JR-FL-pseudotyped luciferase reporter viruses at 37°C for 2 h. After washing off unbound virus, infected cells were treated with defensins at varying concentrations for 48 h before the measurement of luciferase activity.
The effect of HD5 and HD6 on the infection of HIV-1 primary isolates was determined in PHA-activated primary CD4+ T cells or TZM-bl indicator cells (from ARRRP; gift of Drs. J.C. Kappes, and X. Wu). For a multiple-round infection assay, HIV-1 primary isolates (ARRRP, the UNAIDS Network for HIV Isolation and Characterization, and the Division of Acquired Immunodeficiency Syndrome, NIAID), prepared from PBMCs, were incubated with HD5 and HD6 at varying concentrations in medium with 1% FBS at 37°C for 1 h. FBS was added to virusdefensin mixtures to a final concentration of 10% before exposure to PHA-activated primary CD4+ T cells at 37°C for 2 h. Virus was washed out with medium and infected cells were incubated with RPMI 1640 containing 10% FBS and IL-2. HIV-1 p24 levels in the medium were measured at different time points after viral infection by HIV p24 ELISA as described above.
The effect of defensins on various subtypes of HIV primary isolates was also determined using TZM-bl indicator cells that are HeLa cell lines expressing CD4, CXCR4, and CCR5 coreceptors. TZM-bl cells also contain HIV LTR-driven luciferase reporter genes that are activated by HIV Tat proteins during productive HIV infection. HIV primary isolates from PBMCs were incubated with HD5 and HD6 at 20 mg/ml in medium with 1% FBS at 37°C for 1 h before addition to TZM-bl indicator cells as described above. After washing off unbound virus, infected cells were incubated in DMEM with 10% FBS and luciferase activity in TZM-bl cells was measured at 48 h after infection as described above.
N. gonorrhoeae infection
N. gonorrhoeae infection of human cervical and vaginal epithelial cells was performed as described by Fichorova et al. (43) with minor modifications. N. gonorrhoeae (American Type Culture Collection (ATTC) 43069) was grown on chocolate agar for 48 h in 5% CO2. Colonies were scraped and resuspended in PBS. After bacteria were washed once with PBS, bacterial cell density was adjusted to an OD600 equal to 0.1, corresponding to 108 CFU/ml bacteria. Human cervical and vaginal epithelial cells (1 × 106) were cultured overnight in 6-well plates and then exposed to N. gonorrhoeae at a multiplicity of infection (MOI) of 10. Total RNA or conditioned medium from cells without or with exposure to GC was collected at the indicated time for RT-PCR analysis or HIV infection assay, respectively.
Immunoblotting analysis
HD5 proteins were analyzed by immunoblotting as described previously (29) with modifications. Samples (300 μl) from conditioned medium without or with GC exposure were concentrated by lyophilization for protein analysis. Whole cell extracts were prepared by lysis of cells in 20 mM HEPES buffer (pH 7.9) with 0.2% Nonidet P-40, 10% glycerol, 200 mM NaCl, 0.1 mM EDTA, 1 mM DTT, 1 mM sodium orthovanadate, 0.5 mM PMSF, and a protease inhibitor cocktail (Roche Applied Science). The protein concentration in the extracts was determined by the Bradford method using the Bio-Rad protein assay (Bio-Rad). Proteins were separated by NuPAGE 4–12% gel followed by transference to polyvinylidene fluoride membranes (Millipore). Membranes were blocked and probed with rabbit polyclonal Abs against HD5 (41) at a dilution 1/2000 and washed as described previously (29). The polyvinylidene difluoride membrane was further incubated with HRP-linked goat anti-rabbit Ab (Kirkegaard & Perry Laboratories) at a dilution 1/10,000 for 1 h at room temperature and then washed and specific bands were visualized using the ECL kit (Amersham Biosciences). Immunoblotting for HD6 could not be performed due to the lack of an Ab.
RNA interference
Small interfering RNAs (siRNA) targeting HD5 or HD6 were introduced into human vaginal epithelial cells by transfection using HiPerFect trans-fection reagent (Qiagen) according to the manufacturer's suggestions. All-Stars negative control siRNA and siRNAs for HD5 or HD6 were purchased from Qiagen. The target sequences for HD5 and HD6 were 5′-AGCTTC CTAGATAGAAACCAA-3′ and 5′-CAGAGAGAAATATATTCATAA-3′, respectively. Using the Qiagen fast-forward transfection protocol, cells were seeded at 7 × 105 in a 6-well plate followed by transfection with various siRNAs at 10 nM. Transfection efficiency was 90% by FACS analysis using Alexa Fluor 488-labled siRNA at 10 nM. Transfected cells were incubated overnight, washed with PBS, and cultured in fresh medium before GC exposure as described above. After exposure to GC for 48 h, conditioned medium and total RNA from GC-exposed cells were prepared for HIV infection assay and RT-PCR analysis, respectively.
RT-PCR analysis of HD5 and HD6 gene expression
Total RNA was isolated using a Qiagen RNeasy total RNA mini kit with treatment by RNase-free DNase I. To synthesize first-strand cDNA, total RNA (200–500 ng), oligo d(T)16 (Invitrogen) at 2.5 μg/ml, and 0.5 mM dNTP were incubated at 65°C for 5 min and quick chilled on ice. Reverse transcription was performed at 42°C for 50 min using SuperScript II reverse transcriptase (Invitrogen) according to the manufacture's protocol. Primers for RT-PCR with HD5 and HD6 were 5′-ACCTCAGGTTCTCAGGCAAGAGC-3′ (HD5 forward) and 5′-GACACAAGGTACACAGAGTAAAATGT-3′ (HD5 reverse) and 5′-GCTTTGGGCTCAACAAGGGCTTTC-3′ (HD6 forward) and 5′-GACACACGACAGTTTCCTTCTAGGTCATA-3′ (HD6 reverse) (46). Primers for GAPDH were 5′-ACCACAGTCCATGCCATCAC-3′ (forward) and 5′-TCCACCACCCTGTTGCTGTA-3′ (reverse). The PCR contained Qiagen Tag master mix, 200 nM each of primer sets, and 3 μl of reverse transcriptase reaction. After an initial incubation at 94°C for 3 min, 35 cycles of amplification were performed as follows: denaturation for 30 s at 94°C, annealing for 30 s at 55°C and 30 s at 72°C, and a final extension cycle of 7 min at 72°C in a PerkinElmer 480 DNA thermal cycler. PCR products were separated by electrophoresis on a 2% agarose gel and analyzed with the Fluro-Chem 8800 imaging system (Alpha Innotech).
Results
HD5 and HD6 enhance HIV infection
We first determined whether HD5 and HD6 displayed any effect on HIV infection using a single-cycle infection assay. Serum-free HIV-1JR-FL-pseudotyped luciferase reporter virus was incubated with varying concentrations of HD5 or HD6 for 1 h at 37°C before exposure to PHA-activated primary CD4+ T cells or HeLa-CD4-CCR5 cells for 2 h at 37°C. After washing off unbound virus, infected cells were incubated for 48 h before measurement of luciferase activity. Both HD5 and HD6 promoted HIV infection (Fig. 1, A and B) in contrast to previous findings showing that HNPs and HBDs block HIV infection (reviewed in Refs. 30 and 31). HIV-1 replication was enhanced 41- and 67-fold in HeLa-CD4-CCR5 cells following treatment of virus with HD5 at 10 and 50 μg/ml, respectively. Similarly, HD6 at 10 and 50 μg/ml enhanced HIV-1 replication by 64- and 116-fold, respectively (Fig. 1A). HD5 and HD6 also significantly enhanced HIV infection observed in PHA-activated primary CD4+ T cells (15- or 78-fold with treatment of HD5 and HD6 at 50 μg/ml, respectively) and U87-CD4-CCR5 cells (Fig. 1B; data not shown for U87 cells). In addition, we found that HD5 and HD6 enhanced HIV infection in the presence of 10% FBS or human serum, albeit at lower levels (e.g., 5- to 20-fold, depending on the concentrations of defensins; data not shown).
FIGURE 1.
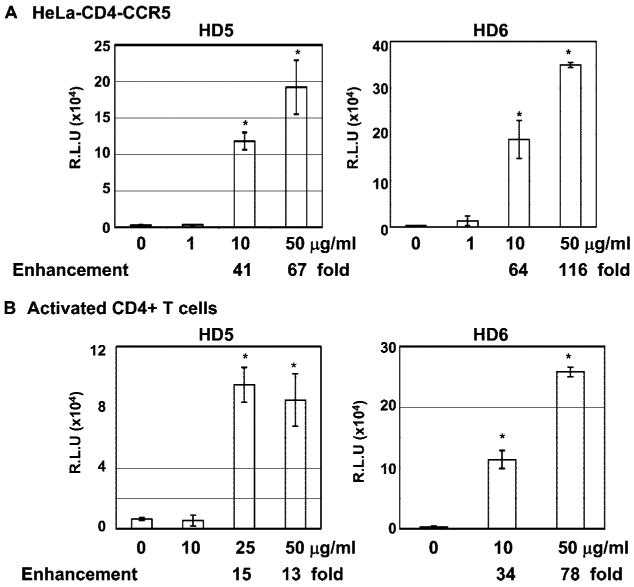
HD5 and HD6 enhanced HIV infection. To examine the effect of defensins on HIV infection, serum-free pseudo-typed HIVJR-FL virus were incubated with HD5 or HD6 at various concentrations at 37°C for 1 h followed by infection of HeLa-CD4-CCR5 cells (A) or PHA-activated CD4+ T cells (B) for 2 h. Cells were washed and incubated in complete medium for 48 h before measuring luciferase activity. Difference between control (HIV-1 infected, no treatment) and samples with treatment of HD5 or HD6 at 10 μg/ml and above is significant as calculated by the two-tailed, paired Student's t test; *, p < 0.05. Data are means ± SD of triplicate sample and represent four independent experiments. R.L.U., reflective light units.
HD5 and HD6 do not promote cell proliferation or inhibit HIV infection after viral entry
To determine whether the defensin-mediated HIV enhancement was due to a mitogenic effect on the cell, we measured the effect of HD5 and HD6 on cell proliferation using an 3-(4,5-dimethyl-thiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt (MTS) assay. PHA-activated PBMCs or HeLa-CD4-CCR5 cells were treated with HD5 and HD6 at varying concentrations for 24 h at 37°C before measurement of cell proliferation. HD5 and HD6 at 50 μg/ml did not have a significant effect on cell proliferation (Fig. 2A and data not shown for HeLa-CD4-CCR5). In agreement with previous report demonstrating that HD5 is not cytotoxic in the absence of serum (41), we did not observe any effect on cell viability when HeLa-CD4-CCR5 were treated with HD5 or HD6 in the absence of serum (data not shown).
FIGURE 2.
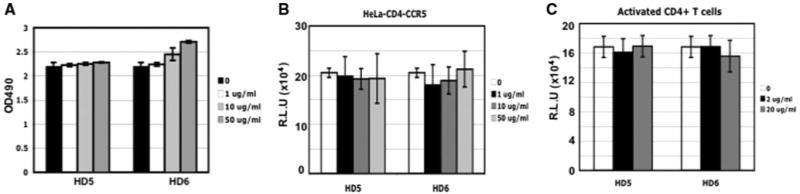
HD5 and HD6 did not affect cell proliferation or HIV infection after viral entry. A, PHA-activated CD4+ T cells were incubated with HD5 or HD6 at various concentrations for 24 h. Cell viability was measured by Promega CellTiter 96 aqueous MTS assay. No difference was observed in cells in the absence or presence of HD5 or HD6 (p > 0.05). (B and C) To determine the effect of HD5 and HD6 on HIV infection after viral entry, HeLa-CD4-CCR5 cells or PHA-activated primary CD4+ T cells were infected with pseudotyped HIVVSV reporter virus for 2 h. Cells were washed and treated with HD5 or HD6 at various concentrations for 48 h before measuring luciferase activity. No difference was found between the control (without treatment) and defensin-treated samples (p > 0.05). Data are means ± SD of triplicate samples and represent three independent experiments. R.L.U., reflective light units.
To further determine whether HD5 and HD6 exerted an effect on HIV-1 infection by acting on the target cells, PHA-activated primary CD4+ T cells, HeLa-CD4-CCR5 cells, or U87-CD4-CCR5 cells were exposed to HIV-1vsv luciferase reporter virus for 2 h, washed, and then treated with HD5 or HD6 at varying concentrations for 48 h before measurement of luciferase activity. We did not observe any post-entry effect of HD5 and HD6 on activated primary CD4+ T cells, HeLa-CD4-CCR5 cells, or U87-CD4-CCR5 (Fig. 2, B and C; data not shown for U87 cells). Similar results were obtained when HIV-1JR-FL pseudotyped virus was used (data not shown). Because HD5 and HD6 had no effect on HIV infection after viral entry (Fig. 2, B and C), we concluded that these peptides enhance HIV infection at the level of viral entry.
HD5 and HD6 enhance infection of HIV primary isolates
To determine the potential relevance of this enhancement, we assessed whether HD5 and HD6 also enhanced infection with HIV primary isolates. HIV R5 and X4R5 primary isolates produced in PBMCs were incubated with HD5 and HD6 at various concentrations at 37°C for 1 h before exposure to activated primary CD4+ T cells for an additional 2 h. Cells were then washed with PBS and incubated in medium with 10% FBS and IL-2. HIV production in the medium was monitored by HIV p24 assay. A laboratory-adapted X4 strain HIVIIIB was also included for comparison. When present only during the initial infection, HD5 and HD6 enhanced infection of primary isolates in a multiple-round infection assay (Fig. 3). HD5 and HD6 at concentrations >10 μg/ml promoted HIV-1 infection by 3- to 53-fold depending on the virus strain in activated primary CD4+ T cells.
FIGURE 3.
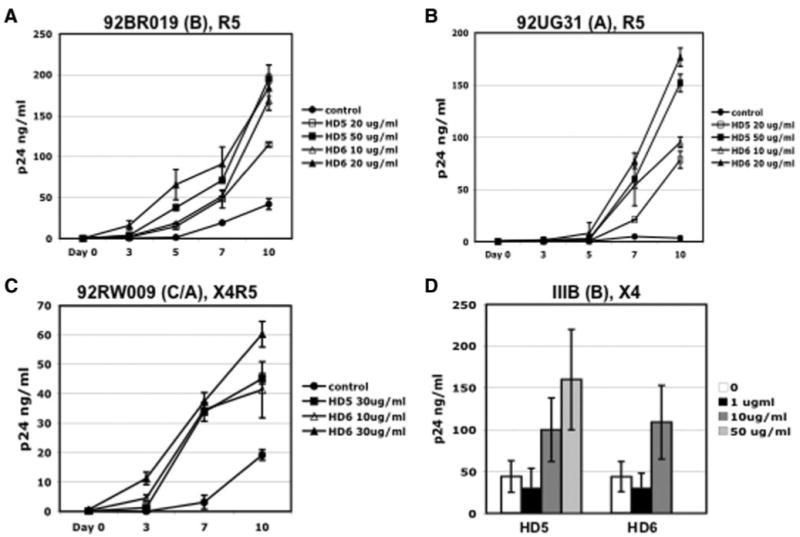
HD5 and HD6 promoted infection of HIV primary isolates in activated primary CD4+ T cells. HIV-1 primary isolates using R5 or X4R5 coreceptors (A–C) and a laboratory-adapted strain HIV-1IIIB (D) were treated with HD5 and HD6 at various concentrations at 37°C for 1 h before addition to PHA-activated primary CD4+ T cells for 2 h. Cells were washed and incubated in RPMI 1640 containing 10% FBS and IL-2 in the absence of defensins. HIV-1 production was measured and the levels of HIV-1 p24 at days 0 (baseline), 3, 5, 7, and 10 after viral infection. For HIV-1IIIB, the p24 level at day 10 is shown. The names and tropisms of virus isolates are indicated and viral genotypes are shown in parentheses. Difference between nontreated control and primary isolates with treatment of defensins at day 10 after viral infection is significant; p < 0.05. Data are means ± SD of triplicate samples and represent two independent experiments.
We further examined whether HD5 and HD6 enhanced viral entry of various subtypes of HIV-1 primary isolates using TZM-bl indicator cells. TZM-bl cells expressing CD4, CXCR4, and CCR5 coreceptors also contain an HIV LTR-driven luciferase reporter gene that is activated by HIV Tat protein during productive HIV infection. Replication-competent X4, R5, or X4R5 primary isolates were incubated with HD5 or HD6 at 37°C for 1 h before exposure to TZM-bl indicator cells. After incubation at 37°C for 2 h, cells were washed and incubated for 48 h before measurement of luciferase activity. HD5 and HD6 promoted infection of different subtypes of X4, R5, and dual tropic X4R5 HIV-1 primary isolates (Table I). The enhancing effect of HD5 was more pronounced on HIV R5 viruses as compared with X4 viruses, an important observation in light of the fact that R5 viruses are almost exclusively transmitted (47).
Table I. HD5 and HD6 enhance infection of HIV primary isolatesa.
| Envelope | HD5 | HD6 |
|---|---|---|
| 92UG029(A), X4 | 1.68 ± 0.12 fold | 5.2 ± 1.04 |
| 92HT599(B), X4 | 1.7 ± 1.3 | 6.4 ± 0.9 |
| 92UG024(D), R5 | 7.2 ± 0.2 | 25 ± 4.8 |
| 92IN905(C), R5 | 8.5 ± 0.8 | 13.8 ± 0.8 |
| 92BR019(B), R5 | 39 ± 1.31 | 157 ± 9.1 |
| 92UG031(C), R5 | 69 ± 2.88 | 42.2 ± 3.9 |
| 92RW009(C/A), X4R5 | 145 ± 34 | 117 ±62 |
HIV-1 primary isolates (10 ng of p24 per sample) with various subtypes and coreceptor usages were incubated with HD5 or HD6 at 20 μg/ml at 37°C for 1 h before addition to TZM- bl indicator cells for 2 h. Cells were washed and luciferase activities were measured 48 h after infection. The data are presented as fold increase compared to untreated samples in triplicate. Results represent two independent experiments (mean ± SD). The subtypes are shown in parentheses.
Disulfide bonding of defensins is required to enhance HIV infection
To determine whether the structure of HD5 or HD6 is required for HIV enhancement, we compared the effect on HIV entry of HD5 or HD6 with that of linear analogues with mutations in the Cys residues that disrupt three disulfide bridges in HD5 or HD6. Abu is considered isosteric to Cys, the -CH3 replaces the -SH group (48, 49), and is used to block the disulfide bridges in HD5 or HD6. [Abu]HD5 and [Abu]HD6, in which Abu replaces all six Cys residues, have been shown to be unstructured in aqueous solution by circular dichroism spectroscopy (Ref. 50 and data not shown for HD6). HD5 R9H, a known single nucleotide polymorphism, and HD5 R13H, an analog with Arg replaced by His, both contain intact disulfide bonds and both exhibited HIV-enhancing effects (Fig. 4A; p < 0.001 between untreated control and mutants). However, the HIV enhancing effects of HD5 R9H and HD5 R13H were less than that of wild type (p = 0.025 for HD5 R9H and p = 0.005 for HD5 R13H), suggesting that host polymorphisms may modulate defensin-mediated enhanced HIV infectivity. Importantly, in contrast to wild-type HD5 and HD6, [Abu]HD5 and [Abu]HD6 no longer enhanced HIV replication (Fig. 4, A and B). Because wild-type defensins and their linear analogues have the same charge, this result suggests that the HIV-enhancing effect requires a specific structural conformation and is not due to simple charge-charge interactions alone.
FIGURE 4.
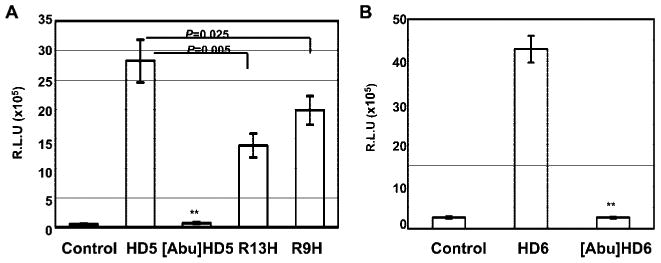
Disulfide bonding of defensins was required to enhance HIV infection. Pseudotyped HIVJR-FL luciferase reporter virus was incubated without or with HD5, HD6, and their analogues at 10 μg/ml at 37°C for 1 h before addition to HeLa-CD4-CCR5 cells in the presence of 10% FBS. After 2 h of incubation, cells were washed and incubated for 48 h before measurement of luciferase activity. Difference between HIV-infected, untreated control, and samples treated with linear analogues, [Abu]HD5 (A) or [Abu]HD6 (B), is not significant; **, p > 0.05. Data are means ± SD of triplicate samples and represent three independent experiments. R.L.U., reflective light units.
Viral enhancement by HD5 and HD6 is CD4 and coreceptor independent
To determine whether defensin-mediated viral enhancement was dependent on CD4 and coreceptors, HIV reporter viruses were pseudotyped with VSV or murine leukemia virus envelopes that do not use CD4 and CXCR4 or CCR5 coreceptors for viral entry. HIV luciferase reporter viruses pseudotyped with HIV-1HxB2(X4), HIV-1JR-FL(R5), and a primary isolate HIV-1RB23-1 (R5) envelope (51) were also included in this study. The direct effect of HD5 and HD6 (10 μg/ml) on the virion was determined using a single-cycle infection assay. Similar to the finding in Table I, HD5 and HD6 enhanced infection of both R5 and X4 HIV strains and the effect was more striking on R5 virus compared with X4 virus. HD5 and HD6 promoted HIV infection of pseudotyped virus with VSV and murine leukemia virus envelopes that do not use CD4 and core-ceptors for viral entry (Table II), indicating that the viral enhancement was not dependent on CD4 and coreceptors.
Table II. HD5 and HD6 enhance entry of virus with various envelopesa.
| Envelope | HD5 | HD6 |
|---|---|---|
| Vesicular stomatitis virus | 2.8 ± 0.7 fold | 23 ± 5.8 |
| Murine leukemia virus | 6.8 ± 0.1 | 99 ± 7.7 |
| HIVHxB2 (B), X4 | 2.7 ± 0.6 | 8.4 ± 1.2 |
| HIVRB231 (B), R5 | 17.8 ± 4.6 | 41 ± 6.9 |
| HIVJR-FL (B), R5 | 41 ± 0.4 | 64 ± 13.9 |
Pseudotyped luciferase reporter viruses with different envelopes were incubated with defensins at 10 μg/ml at 37°C for 1 h. Viral entry was then determined as described in Fig. 1 in HeLa-CD4-CCR5 cells containing endogenous CXCR4. Results are expressed as fold increase compared to untreated samples in triplicate and represent three independent experiments (mean ± S.D).
HD5 is expressed in vaginal epithelial cells as precursor molecules that enhance HIV infection
Expression of HD5 proteins has been demonstrated in tissues from normal vagina, ectocervix, and endocervix (52). HD5 is produced as pre-pro-peptides and the pro-peptide is released and processed extracellularly in the small intestine (53). HD5 is present as precursor molecules in cervicovaginal lavage from normal women (52) and in the male urethra during STIs (29). Similar to previous reports, HD5 precursor molecules were detected in vaginal epithelial cells by immunoblotting using Ab against HD5 (Fig. 5A). We accessed the effect of recombinant pro-HD5 (aa 22–94) on HIV infection using a single-cycle infection and observed that pro-HD5 protein at 50 μg/ml promoted HIV infection of HeLa-CD4-CCR5 cells by 11-fold (p < 0.05).
FIGURE 5.
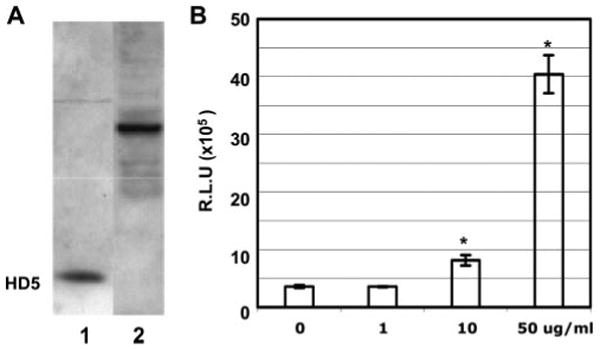
HD5 expresses in vaginal epithelial cells as precursor molecules that enhance HIV infection. A, Whole cell extracts from vaginal epithelial cells (lane 2) were prepared and analyzed by immunoblotting using polyclonal anti-HD5 Ab. Synthetic HD5 peptide (50 ng) was included in lane 1. B, Pseudotyped HIVJR-FL luciferase reporter virus was incubated with recombinant pro-HD5 proteins at different concentrations at 37°C for 1 h before addition to HeLa-CD4-CCR5 cells in the presence of 10% FBS. After 2 h of incubation, cells were washed and cultured in medium with 10% FBS for 48 h before measurement of luciferase activity. Difference between HIV-infected, untreated control, and samples with treatment of pro-HD5 at 10 or 50 μg/ml is significant; *, p < 0.05. Data are means ± SD of triplicate samples and represent two independent experiments. R.L.U., reflective light units.
N. gonorrhoeae infection induces gene expression of HD5 and HD6 and enhances HIV infection
The expression of HD5 has been shown in reproductive tract tissues (25, 52). In addition, elevated levels of HD5 are found in the urethral discharge during N. gonorrhoeae and C. trachomatis infection (29). Thus, we hypothesized that bacterial STIs could induce HD5 and HD6 expression in genital epithelial cells that, in turn, could enhance HIV infectivity. We exposed immortalized human vaginal, endocervical, and ectocervical epithelial cells to N. gonorrhoeae (ATCC no. 43069) and expression of HD5 and HD6 was measured by RT-PCR analysis at various time points after bacterial exposure. As a control, diluted cDNA from small intestine tissue, where HD5 and HD6 are highly abundant, was included in the PCR analysis. We observed that expression of HD5 and HD6 was induced in cervicovaginal epithelial cells in response to GC infection (Fig. 6A).
FIGURE 6.
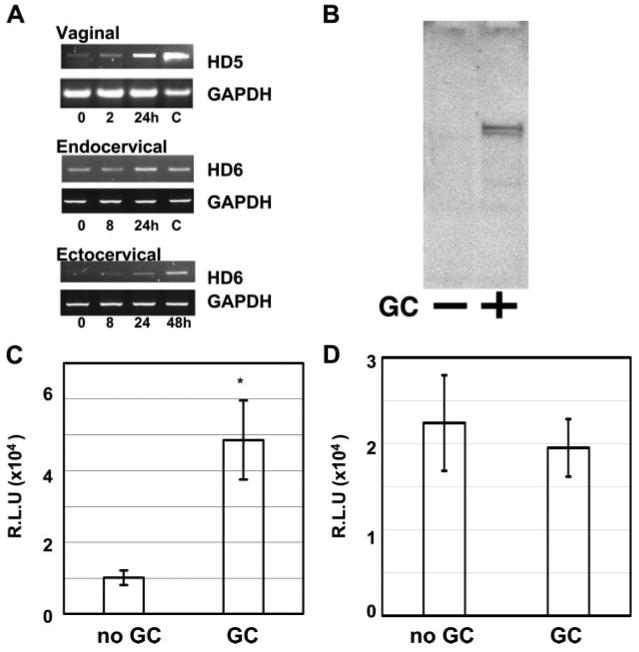
GC infection induces HD5 and HD6 gene expression and enhances HIV infection. A, Immortalized vaginal, endocervical, and ectocervical epithelial cells were infected with N. gonorrhoeae (ATCC no. 43069) at a MOI of 10 and cultured at 37°C. Total RNA was prepared at various time points for RT-PCR analysis. Diluted small intestine cDNA (C, as control; Clontech Laboratories) was included as a positive control for the size of PCR products. The identity of PCR products was confirmed by sequencing. B, Conditioned media from vaginal epithelial cells without (−) or with GC exposure (+) were concentrated and expression of HD5 was analyzed by immunoblotting using anti-HD5 Abs. C, Vaginal epithelial cells were exposed to N. gonorrhoeae at a MOI of 10 and cultured at 37°C for 48 h. Conditioned medium from cells without or with exposure to GC were incubated with pseudotyped HIVJR-FL luciferase reporter virus at 37°C for 1 h before addition to HeLa-CD4-CCR5 cells in the presence of 10% FBS. Difference between conditioned medium from cells without GC exposure and that from cells with GC exposure is significant; *, p < 0.05. Data are means ± SD of triplicate samples and represent two independent experiments. D, To determine the effect of conditioned medium from GC-exposed cells on HIV infection without preincubation with virus, pseudotyped HIVJR-FL luciferase reporter virus mixed with conditioned medium was added to HeLa-CD4-CCR5 cells in the presence of 10% FBS at 37°C for 2 h. Cells were then washed and cultured for 48 h before measurement of luciferase activity. Difference between conditioned medium from cells without GC exposure and that from cells with GC exposure is not significant; p > 0.05. R.L.U., reflective light units.
We then confirmed the presence of HD5 in conditioned medium from GC-exposed cervicovaginal epithelial cells by immunoblotting using polyclonal Abs against HD5 that react with both pro-HD5 and fully processed HD5 (41, 53). In agreement with a previous report demonstrating HD5 precursor molecules in cervicovaginal lavage from normal women (52), HD5 was produced and released as precursor molecules in vaginal and ectocervical epithelial cells (Fig. 6B and data not shown for ectocervical epithelial cells). Similar to HD5 precursors from intestine (53), HD5 precursors in cervicovaginal epithelial cells were sensitive to trypsin and processed into mature peptides in vitro (data not shown). In addition, the level of HD5 was increased in conditioned medium from GC-exposed epithelial cells compared with non-treated control samples (Fig. 6B).
To determine whether conditioned medium from GC-infected vaginal epithelial cells containing elevated level of HD5 (and possibly HD6) could enhance HIV infection, vaginal epithelial cells were exposed to GC for 48 h and conditioned medium was collected for the HIV entry assay. HIVJR-FL-pseudotyped reporter virus was preincubated with conditioned medium at 37°C for 1 h before the infection of HeLa-CD4-CCR5 cells (Fig. 6C). As a control, virus and conditioned medium were added directly to cells without preincubation (Fig. 6D). Conditioned medium from GC-exposed cells had little effect on the target cells during the 2-h incubation time (Fig. 6D). However, HIV infectivity was enhanced when viruses were preincubated with conditioned medium from GC-infected cells before exposure to the target cells (Fig. 6C). Similar results were observed in conditioned medium from GC-exposed ectocervical epithelial cells. Taken together, these results indicated that GC infection induced HD5 and HD6, and this was associated with HIV enhancement.
siRNAs targeting HD5 or HD6 diminished GC-mediated enhanced HIV infectivity
To obtain direct evidence for an involvement of HD5 or HD6 in GC-mediated enhanced HIV infectivity, we transfected siRNAs targeting HD5 or HD6 and negative control siRNAs into vaginal epithelial cells. Transfected cells were incubated overnight and exposed to GC for 48 h before collection of conditioned medium and total RNAs for HIV infection assay and RT-PCR analysis, respectively. While transfection of negative control siRNAs did not have a significant effect on enhanced HIV infectivity by GC, introduction of siRNAs targeting HD5 or HD6 significantly diminished GC-mediated enhanced HIV infectivity (Fig. 7A). In addition, we did not observe induction of HD5 or HD6 by GC in cells transfected with siRNAs for HD5 or HD6, whereas GC infection induced HD5 and HD6 gene expression in cells transfected with negative control siRNAs (Fig. 7B). Taken together, these results indicated that HD5 and HD6 play a role in GC-mediated enhanced HIV infection.
FIGURE 7.
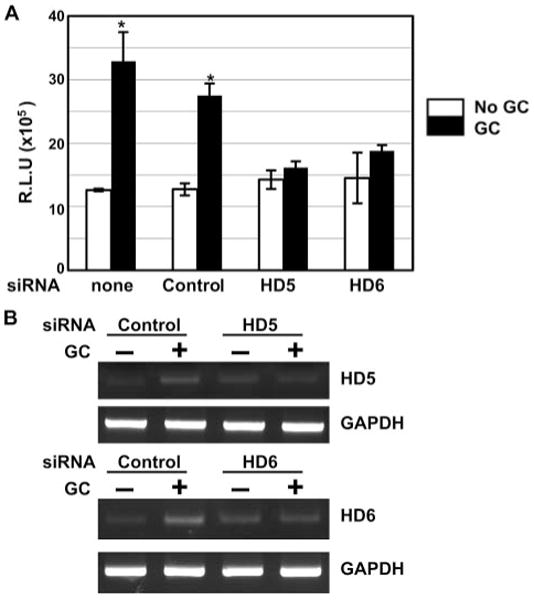
A and B, siRNAs targeting HD5 or HD6 diminished GC-mediated enhanced HIV infectivity. Vaginal epithelial cells were transiently transfected with siRNAs targeting HD5 or HD6. Cells without transfection or with transfection of control siRNAs (control) were also included. After overnight incubation, cells were washed, exposed to GC, and cultured in fresh medium for 48 h. Conditioned medium were collected for HIV infection assay, whereas total RNAs were prepared for analyzing expression of HD5, HD6, and GAPDH by RT-PCR. When comparing the HIV enhancing effect of conditioned medium from untransfected cells or control siRNA-transfected cells, the difference between samples without and with GC exposure was significant (*, p < 0.05). However, when compared with the effect of conditioned medium from cells transfected with HD5 or HD6 siRNA, there was no difference between samples without or with GC exposure (p > 0.05). R.L.U., reflective light units.
Discussion
Previous studies have shown that human defensins inhibit infection of various viruses, including HIV (reviewed in Ref. 31). In this study we provide evidence that HD5 and HD6 can promote HIV infection in various experimental settings. HD5 and HD6 enhanced HIV infection at the step of viral entry, and these peptides promoted R5 virus, the predominant strain transmitted, to a greater extent than X4 virus. In light of the report demonstrating elevation of HD5 proteins by 10- to 30-fold in urethral fluid from men with STIs (29), we hypothesized that STIs may induce expression of HD5 and HD6 and, in turn, could enhance vaginal transmission of HIV. Indeed, we found that GC infection of cervicovaginal epithelial cells induced HD5 and HD6 expression (Fig. 6A). Importantly, conditioned medium from GC-exposed epithelial cells contained HD5 and promoted HIV infection (Fig. 6C). Furthermore, introduction of siRNAs targeting HD5 or HD6 blocked GC-mediated enhanced HIV infectivity. Because HD5 is normally present at a concentration of 1 μg/ml in vaginal fluid (52), an increase of HD5 and HD6 levels to 10 μg/ml, which is sufficient to enhance HIV infection in vitro, could promote HIV transmission in women with STIs.
Our findings are in contrast to those reported by Tanabe et al. (54), who demonstrated that recombinant HD5 had no effect on HIV replication in the transformed T cell line MT-2. This discrepancy is most likely due to the choice of target cells, as has been demonstrated with HNP1-mediated inhibition of HIV (34, 55). Importantly, we observed the enhancement of HIV in the primary target cells, CD4+ T cells. Additionally, we incubated defensins and virus in PBS rather than water, which may also account for the discrepancy, as the effect of HBDs on the HIV virion only occurs under a low salt conditions (10 mM phosphate buffer) (32). Interestingly, Tanabe et al. did found that cryptdin3, a mouse enteric defensin ortholog, enhanced HIV infection (54). The effects of defensins on virus replication, particularly HIV, are quite complex and defensin specific.
Our results suggest that interactions with HIV glycoproteins may play a role in defensin-mediated enhancement of HIV infectivity. The maximal HIV-enhancing effect of HD5 and HD6 was achieved when HIV was preincubated with defensins. In addition, the promoting effect of HD5 and HD6 was more pronounced on R5 virus compared with X4 virus. It is possible that the relatively modest enhancing effect of defensins on X4 viruses was due to the high positive charge of X4 gp120 proteins, which may inhibit the interactions with the positively charged defensins. As HD5 and HD6 appeared to enhance HIV entry, we are currently dissecting specific steps of the early HIV life cycle, including attachment and fusion modulated by these peptides, as well as determining the specific region(s) of HIV envelopes that interact with HD5 and HD6 and the influence of HIV gp120 charge to these interactions and to the HIV-enhancing effect.
Polycationic polymers, such as Polybrene, have been used to enhance the infection of HIV and other retroviruses (56 – 63). It is possible that defensins act like polycationic polymers to promote HIV infection as they are positively charged. The viral enhancement of Polybrene is thought to be mediated through increased viral adsorption as a consequence of decreased repulsive forces between virions and cells, both of which contain negatively charged lipid membranes. Davis et al. have shown that enhancement of retrovirus transduction by polybrene is receptor and envelope independent (59). In addition to a neutralizing membrane charge, positive charge polymers >15 kDa in size can promote infection through virus aggregation (58). We demonstrated that HD5 and HD6 increased HIV infection in a glycoprotein-dependent and HIV receptor-independent manner. Furthermore, HD5 and HD6 linear analogues with the same charge as their structured counterparts failed to promote HIV infection (Fig. 4). These results suggest that neutralizing negative charges on viral envelopes or target cell membranes cannot fully account for defensin-mediated HIV enhancement, although charge may be important in the context of proper conformation. It remains to be determined whether proper formation of hydrophobic domains facilitates membrane fusion upon viral entry.
HD5 and HD6 have been shown to exhibit antiviral activity against other sexually transmitted viruses such as human papillomavirus (a nonenveloped virus) and HSV-2 (64, 65), although their antiviral mechanisms are distinct. HD5 does not affect binding and endocytosis of human papillomavirus but prevents virion release from the endosome (64). HD5 and HD6 appear to block HSV-2 infection via different mechanisms (65). Although HD6 inhibits HSV-2 attachment and penetration, HD5 blocks later stages of the HSV-2 life cycle. HD6 binds to heparan sulfate, required for HSV-2 attachment on the target cells, whereas HD5 binds to HSV-2 gB, which is essential for viral penetration but does not bind to heparan sulfate. These interactions are quite specific and distinct for each defensin.
We observed differential HIV enhancing effects of HD5 and HD6 between HeLa-CD4-CCR5 and primary CD4+ T cells, implicating factors on the target cell that influence the effect. The levels of glycosaminoglycans and ICAM vary between adherent cells (HeLa cells) and suspension cells (CD4+ T cells) and can modulate HIV attachment (reviewed in Ref. 66). It is possible that the defensin-mediated effect alters HIV envelope interactions with glycosaminoglycans, including heparan sulfate and chondroitin sulfate on the cell surface, as glycosaminoglycans influence HIV attachment in an envelope-dependent and coreceptor-independent manner (56, 66).
In response to an invasion of pathogens, HBDs are known to be induced through TLR2 and TLR4 activation as well as via proinflammatory cytokines such as IL-1 (23, 31, 67). Activation of TLR2 or TLR4 induces HBD2 in immortalized vaginal epithelial cells via the NF-κB pathway (68). Although it has been suggested that TNF-α and LPS can induce HD5 (52), the HD5 gene contains an IFN response element in the promoter region (29) and can be induced in response to IFN-γ in HeLa cells (E. Porter, unpublished data). With respect to induction of HD5 and HD6 by GC infection, it remains to be determined whether TLR activation via bacterial LOS or porins directly modulates expression of HD5 and HD6 or via the production of cytokines such as IFNs and TNF-α.
Gonococcal pili and colony Opa proteins play a role in GC adherence to and invasion of epithelial cells (69 –71), although GC can invade epithelial cells via a LOS-dependent and Opa-independent manner (72). Fichorova et al. have demonstrated that both piliated and nonpiliated gonococci can induce proinflammatory host cytokine responses despite their different abilities to invade epithelial cells (19). It remains to be determined whether the presence of pili or Opa affects GC-mediated induction of defensin gene expression and enhanced HIV infectivity as we did not characterize the expression of pili and Opa by the gonococcal strain used in these studies.
HD5 is primarily secreted as precursor molecules in the lumen of the normal small intestine and female vaginal tract and the inflamed male urethra (29, 53, 73–75). In contrast to HNPs that are processed intracellularly in neutrophils (23), HD5 is cleaved upon secretion by trypsin in the small intestine and neutrophil protease in inflamed penile urethra (29, 53). We observed that cervicovaginal epithelial cells produced HD5 precursor molecules in response to GC infection and that pro-HD5 enhanced HIV infection at similar molar levels as HD5. The concentration of HD5 precursor molecules in the conditioned medium from GC-exposed vaginal epithelial cells that enhanced HIV infection was ∼1 μg/ml by Western blot analysis using synthetic HD5 as a standard, ∼10-fold less than that required to enhance HIV infection in vitro. However, considering the much larger medium volume covering cultured epithelial cells compared with the much smaller volume of mucus covering vaginocervical cells in vivo, it is likely that HD5 concentrations affecting HIV infectivity are reached in vivo. Furthermore, native HD5 appears to be in part glycosylated (53), affecting its antibacterial activity, and it needs to be determined whether vaginocervical cells release glycosylated HD5 with stronger HIV-inducing activity. Venkataraman et al. demonstrated that the anti-HIV activity of cation-depleted vaginal fluid can be fully restored by adding back the cationic polypeptide fraction, but the activity is only restored partially by using a mixture of different recombinant cationic polypeptides (76). It is also likely that native HD5 proteins at a lower concentration could promote HIV infectivity by interacting with other proteins in conditioned medium from GC-exposed epithelial cells. In support of these possibilities, introduction of siRNAs targeting HD5 and HD6 blocked the HIV-enhancing effect of the conditioned medium from GC-exposed cells, demonstrating that HD5 and HD6 play a direct role in GC-mediated enhanced HIV infectivity.
Induction of HNPs and HD5 has been reported in the male urethra during C. trachomatis and N. gonorrhoeae infection (29). Although neutrophil infiltration accompanied by elevation of HNPs is the major vaginal immune response to most STIs (27, 77, 78), induction of other defensins such as HBDs, HD5, and HD6 in women with C. trachomatis and N. gonorrhoeae infection has not been reported. N. gonorrhoeae is resistant to HNPs but not to HD5 and its precursor (29, 79 – 81). Pro-HD5, processed by neutrophil proteases in the urogenital mucosa in men with STIs, also exerts antibacterial activity against N. gonorrhoeae (29). It remains to be determined whether HD5 and HD6 are elevated in women with C. trachomatis and N. gonorrhoeae infection and whether elevation of HD5 and HD6 by GC results in controlling bacterial growth but at the same time enhancing HIV infection in vivo. Furthermore, as GC can induce anti-HIV factors such as HBDs, RANTES, MIP-1α, and MIP-1β, which may moderate the HIV enhancing effect of HD5, the interaction between these anti-HIV factors and HD5/HD6 and their net effect on HIV infection may be complex and dynamic. Thus, further studies are warranted to determine how different defensins inhibit or enhance HIV entry as well as affect the overall outcome of HIV infection in the presence of various peptides at physiologic concentrations.
In summary, we report a novel mechanism by which STIs, specifically GC, could enhance HIV transmission through up-regulation of the defensins HD5 and HD6. In addition to promoting HIV entry, HD5 can induce cytokines such as IL-8 (50) that may increase HIV transmission within the cervicovaginal mucosa (82). Understanding the mechanism of HD5- and HD6-mediated HIV enhancement and the complex contribution of these host factors to transmission is critically important for the development of new strategies for HIV prevention, particularly those that target or alter the vaginal mucosa.
Acknowledgments
We thank Flora Samaroo for providing Neisseria gonorrhoeae, Bernadette Benito for technical assistance, and Viviana Simon and members of the Klotman laboratories for helpful discussions.
Footnotes
This work was supported by National Institutes of Health Grants AI062430, AI073205 (to T.L.C.), AI061482 (to W.L.), and AI063927 (to G.A.J.). E.P. was in part supported by National Institutes of Health Grant 1P20 MD001824.
Abbreviations used in this paper: STI, sexually transmitted infection; Abu, α-ami-nobutyric acid; GC, gonococcus/gonococcal; HBD, human β-defensin; HNP, human neutrophil peptide; LOS, lipooligosaccharide; LTR, long terminal repeat; MOI, multiplicity of infection; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphe-nyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt; Opa, opacity-associated (protein); siRNA, small interfering RNA; VSV, vesicular stomatitis virus.
Disclosures: The authors have no financial conflict of interest.
References
- 1.Quinn TC, Overbaugh J. HIV/AIDS in women: an expanding epidemic. Science. 2005;308:1582–1583. doi: 10.1126/science.1112489. [DOI] [PubMed] [Google Scholar]
- 2.Simon V, Ho DD, Abdool Karim Q. HIV/AIDS epidemiology, pathogenesis, prevention, and treatment. Lancet. 2006;368:489–504. doi: 10.1016/S0140-6736(06)69157-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galvin SR, Cohen MS. The role of sexually transmitted diseases in HIV transmission. Nat Rev Microbiol. 2004;2:33–42. doi: 10.1038/nrmicro794. [DOI] [PubMed] [Google Scholar]
- 4.Plummer FA. Heterosexual transmission of human immunodeficiency virus type 1 (HIV): interactions of conventional sexually transmitted diseases, hormonal contraception, and HIV-1. AIDS Res Hum Retroviruses. 1998;14(Suppl 1):S5–S10. [PubMed] [Google Scholar]
- 5.Cohen MS, Hoffman IF, Royce RA, Kazembe P, Dyer JR, Daly CC, Zimba D, Vernazza PL, Maida M, Fiscus SA, Eron JJ., Jr Reduction of concentration of HIV-1 in semen after treatment of urethritis: implications for prevention of sexual transmission of HIV-1. AIDSCAP Malawi Research Group. Lancet. 1997;349:1868–1873. doi: 10.1016/s0140-6736(97)02190-9. [DOI] [PubMed] [Google Scholar]
- 6.Chesson HW, Pinkerton SD. Sexually transmitted diseases and the increased risk for HIV transmission: implications for cost-effectiveness analyses of sexually transmitted disease prevention interventions. J Acquired Immune Defic Syndr. 2000;24:48–56. doi: 10.1097/00126334-200005010-00009. [DOI] [PubMed] [Google Scholar]
- 7.Mabey D. Interactions between HIV infection and other sexually transmitted diseases. Trop Med Int Health. 2000;5:A32–A36. doi: 10.1046/j.1365-3156.2000.00595.x. [DOI] [PubMed] [Google Scholar]
- 8.Laga M, Manoka A, Kivuvu M, Malele B, Tuliza M, Nzila N, Goeman J, Behets F, Batter V, Alary M, et al. Non-ulcerative sexually transmitted diseases as risk factors for HIV-1 transmission in women: results from a cohort study. AIDS. 1993;7:95–102. doi: 10.1097/00002030-199301000-00015. [DOI] [PubMed] [Google Scholar]
- 9.Centers for Disease Control Surveillance for gonorrhea and primary and secondary syphilis among adolescents, United States-1981–1991. Morbid Mortal Wkly Rep. 1998;42:55–63. [PubMed] [Google Scholar]
- 10.Ramsey KH, Schneider H, Cross AS, Boslego JW, Hoover DL, Staley TL, Kuschner RA, Deal CD. Inflammatory cytokines produced in response to experimental human gonorrhea. J Infect Dis. 1995;172:186–191. doi: 10.1093/infdis/172.1.186. [DOI] [PubMed] [Google Scholar]
- 11.Korenromp EL, Sudaryo MK, de Vlas SJ, Gray RH, Sewankambo NK, Serwadda D, Wawer MJ, Habbema JD. What proportion of episodes of gonorrhoea and chlamydia becomes symptomatic? Int J STD AIDS. 2002;13:91–101. doi: 10.1258/0956462021924712. [DOI] [PubMed] [Google Scholar]
- 12.Farley TA, Cohen DA, Elkins W. Asymptomatic sexually transmitted diseases: the case for screening. Prev Med. 2003;36:502–509. doi: 10.1016/s0091-7435(02)00058-0. [DOI] [PubMed] [Google Scholar]
- 13.Bolan G, Ehrhardt AA, Wasserheit JN. Gender Perspectives and STDs. McGraw-Hill; New York: 1999. [Google Scholar]
- 14.Edwards JL, Apicella MA. The molecular mechanisms used by Neisseria gonorrhoeae to initiate infection differ between men and women. Clin Microbiol Rev. 2004;17:965–981. doi: 10.1128/CMR.17.4.965-981.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGee ZA, Johnson AP, Taylor-Robinson D. Pathogenic mechanisms of Neisseria gonorrhoeae: observations on damage to human fallopian tubes in organ culture by gonococci of colony type 1 or type 4. J Infect Dis. 1981;143:413–422. doi: 10.1093/infdis/143.3.413. [DOI] [PubMed] [Google Scholar]
- 16.Pridmore AC, Jarvis GA, John CM, Jack DL, Dower SK, Read RC. Activation of toll-like receptor 2 (TLR2) and TLR4/MD2 by Neisseria is independent of capsule and lipooligosaccharide (LOS) sialylation but varies widely among LOS from different strains. Infect Immun. 2003;71:3901–3908. doi: 10.1128/IAI.71.7.3901-3908.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harvey HA, Post DM, Apicella MA. Immortalization of human urethral epithelial cells: a model for the study of the pathogenesis of and the inflammatory cytokine response to Neisseria gonorrhoeae infection. Infect Immun. 2002;70:5808–5815. doi: 10.1128/IAI.70.10.5808-5815.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hedges SR, Sibley DA, Mayo MS, Hook EW, III, Russell MW. Cytokine and antibody responses in women infected with Neisseria gonorrhoeae: effects of concomitant infections. J Infect Dis. 1998;178:742–751. doi: 10.1086/515372. [DOI] [PubMed] [Google Scholar]
- 19.Fichorova RN, Desai PJ, Gibson FC, III, Genco CA. Distinct proinflammatory host responses to Neisseria gonorrhoeae infection in immortalized human cervical and vaginal epithelial cells. Infect Immun. 2001;69:5840–5848. doi: 10.1128/IAI.69.9.5840-5848.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen A, Boulton IC, Pongoski J, Cochrane A, Gray-Owen SD. Induction of HIV-1 long terminal repeat-mediated transcription by Neisseria gonorrhoeae. AIDS. 2003;17:625–628. doi: 10.1097/00002030-200303070-00019. [DOI] [PubMed] [Google Scholar]
- 21.Levine WC, Pope V, Bhoomkar A, Tambe P, Lewis JS, Zaidi AA, Farshy CE, Mitchell S, Talkington DF. Increase in endocervical CD4 lymphocytes among women with nonulcerative sexually transmitted diseases. J Infect Dis. 1998;177:167–174. doi: 10.1086/513820. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, Li G, Bafica A, Pantelic M, Zhang P, Broxmeyer H, Liu Y, Wetzler L, He JJ, Chen T. Neisseria gonorrhoeae enhances infection of dendritic cells by HIV type 1. J Immunol. 2005;174:7995–8002. doi: 10.4049/jimmunol.174.12.7995. [DOI] [PubMed] [Google Scholar]
- 23.Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. 2003;3:710–720. doi: 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
- 24.Fellermann K, Stange EF. Defensins: innate immunity at the epithelial frontier. Eur J Gastroenterol Hepatol. 2001;13:771–776. doi: 10.1097/00042737-200107000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Svinarich DM, Wolf NA, Gomez R, Gonik B, Romero R. Detection of human defensin 5 in reproductive tissues. Am J Obstet Gynecol. 1997;176:470–475. doi: 10.1016/s0002-9378(97)70517-9. [DOI] [PubMed] [Google Scholar]
- 26.Frye M, Bargon J, Dauletbaev N, Weber A, Wagner TO, Gropp R. Expression of human α-defensin 5 (HD5) mRNA in nasal and bronchial epithelial cells. J Clin Pathol. 2000;53:770–773. doi: 10.1136/jcp.53.10.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simhan HN, Anderson BL, Krohn MA, Heine RP, Martinez de Tejada B, Landers DV, Hillier SL. Host immune consequences of asymptomatic Trichomonas vaginalis infection in pregnancy. Am J Obstet Gynecol. 2007;196:59 e1–e5. doi: 10.1016/j.ajog.2006.08.035. [DOI] [PubMed] [Google Scholar]
- 28.Valore EV, Wiley DJ, Ganz T. Reversible deficiency of antimicrobial polypeptides in bacterial vaginosis. Infect Immun. 2006;74:5693–5702. doi: 10.1128/IAI.00524-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porter E, Yang H, Yavagal S, Preza GC, Murillo O, Lima H, Greene S, Mahoozi L, Klein-Patel M, Diamond G, et al. Distinct defensin profiles in Neisseria gonorrhoeae and Chlamydia trachomatis urethritis reveal novel epithelial cell-neutrophil interactions. Infect Immun. 2005;73:4823–4833. doi: 10.1128/IAI.73.8.4823-4833.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang TL, Klotman ME. Defensins: natural anti-HIV peptides. AIDS Rev. 2004;6:161–168. [PubMed] [Google Scholar]
- 31.Klotman ME, Chang TL. Defensins in innate antiviral immunity. Nat Rev Immunol. 2006;6:447–456. doi: 10.1038/nri1860. [DOI] [PubMed] [Google Scholar]
- 32.Quinones-Mateu ME, Lederman MM, Feng Z, Chakraborty B, Weber J, Rangel HR, Marotta ML, Mirza M, Jiang B, Kiser P, et al. Human epithelial β-defensins 2 and 3 inhibit HIV-1 replication. AIDS. 2003;17:F39–F48. doi: 10.1097/00002030-200311070-00001. [DOI] [PubMed] [Google Scholar]
- 33.Sun L, Finnegan CM, Kish-Catalone T, Blumenthal R, Garzino-Demo P, La Terra Maggiore GM, Berrone S, Kleinman C, Wu Z, Abdelwahab S, et al. Human β-defensins suppress human immunodeficiency virus infection: potential role in mucosal protection. J Virol. 2005;79:14318–14329. doi: 10.1128/JVI.79.22.14318-14329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang TL, Vargas J, Jr, DelPortillo A, Klotman ME. Dual role of α-defensin-1 in anti-HIV-1 innate immunity. J Clin Invest. 2005;115:765–773. doi: 10.1172/JCI200521948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo CJ, Tan N, Song L, Douglas SD, Ho WZ. α-Defensins inhibit HIV infection of macrophages through upregulation of CC-chemokines. AIDS. 2004;18:1217–1218. doi: 10.1097/00002030-200405210-00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng Z, Dubyak GR, Lederman MM, Weinberg A. Cutting edge: human β defensin 3: a novel antagonist of the HIV-1 coreceptor CXCR4. J Immunol. 2006;177:782–786. doi: 10.4049/jimmunol.177.2.782. [DOI] [PubMed] [Google Scholar]
- 37.Jones DE, Bevins CL. Paneth cells of the human small intestine express an antimicrobial peptide gene. J Biol Chem. 1992;267:23216–23225. [PubMed] [Google Scholar]
- 38.Jones DE, Bevins CL. Defensin-6 mRNA in human Paneth cells: implications for antimicrobial peptides in host defense of the human bowel. FEBS Lett. 1993;315:187–192. doi: 10.1016/0014-5793(93)81160-2. [DOI] [PubMed] [Google Scholar]
- 39.Wu Z, Ericksen B, Tucker K, Lubkowski J, Lu W. Synthesis and characterization of human α-defensins 4 – 6. J Pept Res. 2004;64:118–125. doi: 10.1111/j.1399-3011.2004.00179.x. [DOI] [PubMed] [Google Scholar]
- 40.Szyk A, Wu Z, Tucker K, Yang D, Lu W, Lubkowski J. Crystal structures of human α-defensins HNP4, HD5, and HD6. Protein Sci. 2006;15:2749–2760. doi: 10.1110/ps.062336606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Porter EM, Liu L, Oren A, Anton PA, Ganz T. Localization of human intestinal defensin 5 in Paneth cell granules. Infect Immun. 1997;65:2389–2395. doi: 10.1128/iai.65.6.2389-2395.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fichorova RN, Rheinwald JG, Anderson DJ. Generation of papillomavirus-immortalized cell lines from normal human ectocervical, endo-cervical, and vaginal epithelium that maintain expression of tissue-specific differentiation proteins. Biol Reprod. 1997;57:847–855. doi: 10.1095/biolreprod57.4.847. [DOI] [PubMed] [Google Scholar]
- 43.Fichorova RN, Cronin AO, Lien E, Anderson DJ, Ingalls RR. Response to Neisseria gonorrhoeae by cervicovaginal epithelial cells occurs in the absence of toll-like receptor 4-mediated signaling. J Immunol. 2002;168:2424–2432. doi: 10.4049/jimmunol.168.5.2424. [DOI] [PubMed] [Google Scholar]
- 44.Chen BK, Saksela K, Andino R, Baltimore D. Distinct modes of human immunodeficiency virus type 1 proviral latency revealed by superinfection of nonproductively infected cell lines with recombinant luciferase-encoding viruses. J Virol. 1994;68:654–660. doi: 10.1128/jvi.68.2.654-660.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR. Change in coreceptor use coreceptor use correlates with disease progression in HIV-1-infected individuals. J Exp Med. 1997;185:621–628. doi: 10.1084/jem.185.4.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fahlgren A, Hammarstrom S, Danielsson A, Hammarstrom ML. Increased expression of antimicrobial peptides and lysozyme in colonic epithelial cells of patients with ulcerative colitis. Clin Exp Immunol. 2003;131:90–101. doi: 10.1046/j.1365-2249.2003.02035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meng G, Wei X, Wu X, Sellers MT, Decker JM, Moldoveanu Z, Orenstein JM, Graham MF, Kappes JC, Mestecky J, et al. Primary intestinal epithelial cells selectively transfer R5 HIV-1 to CCR5+ cells. Nat Med. 2002;8:150–156. doi: 10.1038/nm0202-150. [DOI] [PubMed] [Google Scholar]
- 48.Lu W, Apostol I, Qasim MA, Warne N, Wynn R, Zhang WL, Anderson S, Chiang YW, Ogin E, Rothberg I, et al. Binding of amino acid side-chains to S1 cavities of serine proteinases. J Mol Biol. 1997;266:441–461. doi: 10.1006/jmbi.1996.0781. [DOI] [PubMed] [Google Scholar]
- 49.Bigler TL, Lu W, Park SJ, Tashiro M, Wieczorek M, Wynn R, Laskowski M., Jr Binding of amino acid side chains to preformed cavities: interaction of serine proteinases with turkey ovomucoid third domains with coded and noncoded P1 residues. Protein Sci. 1993;2:786–799. doi: 10.1002/pro.5560020509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Leeuw E, Burks SR, Li X, Kao JP, Lu W. Structure-dependent functional properties of human defensin 5. FEBS Lett. 2007;581:515–520. doi: 10.1016/j.febslet.2006.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zerhouni-Layachi B, Husain M, Ross MJ, Marras D, Sunamoto M, Liu X, Klotman PE, Klotman ME. Dual tropism of HIV-1 envelopes derived from renal tubular epithelial cells of patients with HIV-associated nephropathy. AIDS. 2006;20:621–624. doi: 10.1097/01.aids.0000210618.68083.8e. [DOI] [PubMed] [Google Scholar]
- 52.Quayle AJ, Porter EM, Nussbaum AA, Wang YM, Brabec C, Yip KP, Mok SC. Gene expression, immunolocalization, and secretion of human defensin-5 in human female reproductive tract. Am J Pathol. 1998;152:1247. [PMC free article] [PubMed] [Google Scholar]
- 53.Ghosh D, Porter E, Shen B, Lee SK, Wilk D, Drazba J, Yadav SP, Crabb JW, Ganz T, Bevins CL. Paneth cell trypsin is the processing enzyme for human defensin-5. Nat Immunol. 2002;3:583–590. doi: 10.1038/ni797. [DOI] [PubMed] [Google Scholar]
- 54.Tanabe H, Ouellette AJ, Cocco MJ, Robinson WE., Jr Differential effects on human immunodeficiency virus type 1 replication by α-defensins with comparable bactericidal activities. J Virol. 2004;78:11622–11631. doi: 10.1128/JVI.78.21.11622-11631.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang TL, Francois F, Mosoian A, Klotman ME. CAF-mediated human immunodeficiency virus (HIV) type 1 transcriptional inhibition is distinct from α-defensin-1 HIV inhibition. J Virol. 2003;77:6777–6784. doi: 10.1128/JVI.77.12.6777-6784.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang YJ, Hatziioannou T, Zang T, Braaten D, Luban J, Goff SP, Bieniasz PD. Envelope-dependent, cyclophilin-independent effects of glycosaminoglycans on human immunodeficiency virus type 1 attachment and infection. J Virol. 2002;76:6332–6343. doi: 10.1128/JVI.76.12.6332-6343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.O'Doherty U, Swiggard WJ, Malim MH. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J Virol. 2000;74:10074–10080. doi: 10.1128/jvi.74.21.10074-10080.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davis HE, Rosinski M, Morgan JR, Yarmush ML. Charged polymers modulate retrovirus transduction via membrane charge neutralization and virus aggregation. Biophys J. 2004;86:1234–1242. doi: 10.1016/S0006-3495(04)74197-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davis HE, Morgan JR, Yarmush ML. Polybrene increases retrovirus gene transfer efficiency by enhancing receptor-independent virus adsorption on target cell membranes. Biophys Chem. 2002;97:159–172. doi: 10.1016/s0301-4622(02)00057-1. [DOI] [PubMed] [Google Scholar]
- 60.Le Doux JM, Landazuri N, Yarmush ML, Morgan JR. Complexation of retrovirus with cationic and anionic polymers increases the efficiency of gene transfer. Hum Gene Ther. 2001;12:1611–1621. doi: 10.1089/10430340152528110. [DOI] [PubMed] [Google Scholar]
- 61.Pan LZ, Werner A, Levy JA. Detection of plasma viremia in human immunodeficiency virus-infected individuals at all clinical stages. J Clin Microbiol. 1993;31:283–288. doi: 10.1128/jcm.31.2.283-288.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Manning JS, Hackett AJ, Darby NB., Jr Effect of polycations on sensitivity of BALD-3T3 cells to murine leukemia and sarcoma virus infectivity. Appl Microbiol. 1971;22:1162–1163. doi: 10.1128/am.22.6.1162-1163.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rusert P, Fischer M, Joos B, Leemann C, Kuster H, Flepp M, Bonhoeffer S, Gunthard HF, Trkola A. Quantification of infectious HIV-1 plasma viral load using a boosted in vitro infection protocol. Virology. 2004;326:113–129. doi: 10.1016/j.virol.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 64.Buck CB, Day PM, Thompson CD, Lubkowski J, Lu W, Lowy DR, Schiller JT. Human α-defensins block papillomavirus infection. Proc Natl Acad Sci USA. 2006;103:1516–1521. doi: 10.1073/pnas.0508033103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hazrati E, Galen B, Lu W, Wang W, Ouyang Y, Keller MJ, Lehrer RI, Herold BC. Human α- and β-defensins block multiple steps in herpes simplex virus infection. J Immunol. 2006;177:8658–8666. doi: 10.4049/jimmunol.177.12.8658. [DOI] [PubMed] [Google Scholar]
- 66.Ugolini S, Mondor I, Sattentau QJ. HIV-1 attachment: another look. Trends Microbiol. 1999;7:144–149. doi: 10.1016/s0966-842x(99)01474-2. [DOI] [PubMed] [Google Scholar]
- 67.Yang D, Biragyn A, Hoover DM, Lubkowski J, Oppenheim JJ. Multiple roles of antimicrobial defensins, cathelicidins, and eosinophil-derived neurotoxin in host defense. Annu Rev Immunol. 2004;22:181–215. doi: 10.1146/annurev.immunol.22.012703.104603. [DOI] [PubMed] [Google Scholar]
- 68.Pivarcsi A, Nagy I, Koreck A, Kis K, Kenderessy-Szabo A, Szell M, Dobozy A, Kemeny L. Microbial compounds induce the expression of pro-inflammatory cytokines, chemokines and human β-defensin-2 in vaginal epithelial cells. Microbes Infect. 2005;7:1117–1127. doi: 10.1016/j.micinf.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 69.Griffiss JM, Lammel CJ, Wang J, Dekker NP, Brooks GF. Neisseria gonorrhoeae coordinately uses Pili and Opa to activate HEC-1-B cell microvilli, which causes engulfment of the gonococci. Infect Immun. 1999;67:3469–3480. doi: 10.1128/iai.67.7.3469-3480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gorby GL, Schaefer GB. Effect of attachment factors (pili plus Opa) on Neisseria gonorrhoeae invasion of human fallopian tube tissue in vitro: quantitation by computerized image analysis. Microb Pathog. 1992;13:93–108. doi: 10.1016/0882-4010(92)90070-5. [DOI] [PubMed] [Google Scholar]
- 71.Kupsch EM, Knepper B, Kuroki T, Heuer I, Meyer TF. Variable opacity (Opa) outer membrane proteins account for the cell tropisms displayed by Neisseria gonorrhoeae for human leukocytes and epithelial cells. EMBO J. 1993;12:641–650. doi: 10.1002/j.1460-2075.1993.tb05697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Song W, Ma L, Chen R, Stein DC. Role of lipooligosaccharide in Opa-independent invasion of Neisseria gonorrhoeae into human epithelial cells. J Exp Med. 2000;191:949–960. doi: 10.1084/jem.191.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cunliffe RN, Rose FR, Keyte J, Abberley L, Chan WC, Mahida YR. Human defensin 5 is stored in precursor form in normal Paneth cells and is expressed by some villous epithelial cells and by metaplastic Paneth cells in the colon in inflammatory bowel disease. Gut. 2001;48:176–185. doi: 10.1136/gut.48.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Porter EM, Poles MA, Lee JS, Naitoh J, Bevins CL, Ganz T. Isolation of human intestinal defensins from ileal neobladder urine. FEBS Lett. 1998;434:272–276. doi: 10.1016/s0014-5793(98)00994-6. [DOI] [PubMed] [Google Scholar]
- 75.Quayle AJ. The innate and early immune response to pathogen challenge in the female genital tract and the pivotal role of epithelial cells. J Reprod Immunol. 2002;57:61–79. doi: 10.1016/s0165-0378(02)00019-0. [DOI] [PubMed] [Google Scholar]
- 76.Venkataraman N, Cole AL, Svoboda P, Pohl J, Cole AM. Cationic polypeptides are required for anti-HIV-1 activity of human vaginal fluid. J Immunol. 2005;175:7560–7567. doi: 10.4049/jimmunol.175.11.7560. [DOI] [PubMed] [Google Scholar]
- 77.Hook EW. Sexual Transmitted Diseases. McGraw-Hill; New York: 1999. [Google Scholar]
- 78.Wiesenfeld HC, Heine RP, Krohn MA, Hillier SL, Amortegui AA, Nicolazzo M, Sweet RL. Association between elevated neutrophil defensin levels and endometritis. J Infect Dis. 2002;186:792–797. doi: 10.1086/342417. [DOI] [PubMed] [Google Scholar]
- 79.Qu XD, Harwig SS, Oren AM, Shafer WM, Lehrer RI. Susceptibility of Neisseria gonorrhoeae to protegrins. Infect Immun. 1996;64:1240–1245. doi: 10.1128/iai.64.4.1240-1245.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Qu XD, Harwig SS, Shafer WM, Lehrer RI. Protegrin structure and activity against Neisseria gonorrhoeae. Infect Immun. 1997;65:636–639. doi: 10.1128/iai.65.2.636-639.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shafer WM, Qu X, Waring AJ, Lehrer RI. Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc Natl Acad Sci USA. 1998;95:1829–1833. doi: 10.1073/pnas.95.4.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Narimatsu R, Wolday D, Patterson BK. IL-8 increases transmission of HIV type 1 in cervical explant tissue. AIDS Res Hum Retroviruses. 2005;21:228–233. doi: 10.1089/aid.2005.21.228. [DOI] [PubMed] [Google Scholar]