

Abstract
Obesity is a complex genetic and behavioral disorder arising from improper integration of peripheral signals at central autonomic centers. For the hypothalamus to respond to dynamic physiological alterations, it must retain a degree of plasticity throughout life. Evidence is mounting that an intricate balance between matrix metalloproteinase (MMP)-mediated extracellular matrix proteolysis and tissue inhibitor of metalloproteinase (TIMP)-mediated proteolysis inhibition contributes to tissue remodeling. However, few studies have examined the role of MMPs/TIMPs in hypothalamic remodeling and energy homeostasis. To determine TIMP-2’s contribution to hypothalamic regulation of feeding, body mass and food consumption was monitored in TIMP-2 knockout (KO) mice fed a standard chow or high fat diet (HFD). TIMP-2 KO mice of both sexes gained more weight than wild-type (WT) mice even when fed the chow diet. Prior to obesity onset, TIMP-2 KO mice were hyperphagic, without increased orexigenic or decreased anorexigenic neuropeptide expression, but leptin resistant (i.e. reduced leptin-induced anorexigenic response and STAT3 activation). HFD exacerbated weight gain and hyperleptinemia. In addition, proteolysis was increased in the arcuate nucleus of TIMP-2 KO mice. These data suggest a role for TIMP-2 in hypothalamic control of feeding and energy homeostasis.
Keywords: gelatinase assay, hypothalamus, in situ zymography, MMP, obesity
Maintenance of body mass depends largely on the perfect coupling of caloric intake and energy expenditure, with both functions under tight control of specialized hypothalamic neurons. Since the discovery of leptin (1, 2), much attention has been focused on its role in obesity. Leptin, which is secreted mainly by white adipocytes, acts through its receptor, ObR, to provide a critical signal to hypothalamic neurocircuits on the relative state of adipose stores to accordingly modulate food intake as well as energy expenditure. Of the six ObR isoforms, primarily generated by alternative splicing of the RNA transcript of the db gene, ObRb represents the full-length isoform (3). Since ObR does not possess intrinsic tyrosine kinase activity, it associates with cytoplasmic kinases, predominantly Janus kinase 2 and signal transducer and activator of transcription 3 (STAT3). This signalling pathway can be inhibited by suppressor of cytokine signalling 3 (SOCS3). To exert its negative effects on food intake, leptin activates anorexigenic (i.e., satiety promoting) proopiomelanocortin (POMC) and cocaine- and amphetamine-regulated transcript (CART) neurons and inhibits orexigenic (i.e., feeding promoting) agouti-related peptide (AgRP) and neuropeptide Y (NPY) neurons within the arcuate nucleus (4). In addition to acting as a powerful satiety signal, leptin increases energy expenditure via hypothalamic projections to brainstem autonomic centers that promote thermogenesis. Monogenic forms of obesity are rare (i.e., melanocortin-4, POMC, or leptin receptor) (5). Rather, the increased prevalence of obesity is mainly attributable to lifestyle – greater consumption of foods high in fats and sugars and decreased physical activity. Humans and rodents that consume a HFD exhibit obesity, hyperphagia, and hyperleptinemia, implicating leptin resistance as a contributing factor.
Similar to the hippocampus, a site of learning-based synaptic plasticity, the hypothalamus must display life-long plasticity to respond to the dynamic physiological alterations required to maintain homeostasis. MMPs, and the closely related ADAM (a disintegrin and metalloproteinase) and ADAMTS (ADAM proteases with thrombospondin motifs) proteases (6, 7), regulate physiological (8) as well as pathological (9) tissue remodeling. MMPs regulate synaptic plasticity not only via proteolysis of extracellular matrix (ECM) proteins, but activation of growth factors and cytokines, and “shedding” of ECM receptors (10). The relevance of proteolysis to obesity has been demonstrated in MMP/TIMP-deficient mice fed a HFD. While MMP-3 and MMP-11 KO mice gained more weight than WT mice (11, 12), MMP-2 and TIMP-1 KO mice gained less weight (13, 14) when fed a HFD. However, these studies did not examine the regulation of leptin signalling or other hypothalamic alterations in the mice.
MMP activity is inhibited by forming tight, but relatively low selectivity, complexes with one of four TIMPs (15). TIMP-2 not only inhibits MMP activity, but also, via interaction with MT1-MMP (MMP-14), is required for proMMP-2 activation (16, 17). Although traditionally recognized for their MMP inhibitory activity, TIMPs are multifunctional molecules with diverse MMP-independent functions (e.g., cell cycle arrest, angiogenesis inhibition) (18).
Given the pleiotropic roles of TIMP-2, chow- and HFD-fed TIMP-2 KO mice were examined to elucidate TIMP-2’s contribution to hypothalamic regulation of energy homeostasis. Unlike most MMP or TIMP KO mice examined thus far, TIMP-2 KO mice gained more weight than WT mice even when fed a standard chow diet. Weight gain was exacerbated when mice were fed a HFD. TIMP-2 KO mice were hyperphagic before obesity onset, yet displayed decreased leptin-mediated signaling and anorexigenic response, indicative of leptin resistance. Furthermore, TIMP-2 KO mice showed increased proteolysis in the hypothalamic arcuate nucleus. Taken together, our data suggest a role for TIMP-2 in energy metabolism, possibly via hypothalamic proteolytic remodeling.
Materials and Methods
Animal Care
All procedures involving animals were in accordance with approved University of Vermont Animal Care and Use Committee protocols. Mice bearing a targeted disruption of the TIMP-2 gene have been described elsewhere (16). After more than ten backcrosses on a C57Bl/6 background (Charles Rivers), WT and KO offspring of heterozygous breeders underwent PCR-based genome scans to determine the extent of 129 genomic sequence flanking the TIMP-2 targeting locus. Offspring from at least five independent heterozygous matings were then used to establish homozygous breeding colonies. Mice were then maintained as in-bred homozygous matings. One week prior to the study start, at 2 months of age, average food consumption of group housed mice was determined daily for 5 days. Littermates were then singly housed with a standard chow diet (ProLab RMH 3000, 4.1 kcal/g with 26% protein, 60% carbohydrate, 14% fat; Purina LabDiet) for an additional 2 days; then, either remained on the chow diet or switched to a HFD (5.24 kcal/g with 20% protein, 20% carbohydrate, 60% fat which is 37.1% saturated, 46.0% monounsaturated, and 16.9% polyunsaturated fat; Research Diets, Inc.) for 90 days. In both cases, mice had ad libitum access to food and water. Food consumption and animal weight were measured daily within 1 hour of the light (sleep) cycle.
At the termination of the 3 month study, mice were humanely euthanized either via exsanguination by transcardial perfusion (for immunohistochemistry) or via decapitation (all other studies). Following euthanasia, brains were removed from calvaria and placed on their superior surface. A 2 mm-thick coronal block containing the hypothalamus was obtained with a rostral cut at the optic chiasm and a caudal cut anterior to the brainstem. For immunohistochemical studies, this block was post-fixed and processed as described below. For in situ zymography, this block was rapidly frozen on dry ice and stored at -80°C until sectioned. For all other studies, the hypothalamus was micro-dissected further by placing the block on its anterior surface and making two oblique cuts from the top of the third ventricle; thus, creating a triangular wedge containing the hypothalamus. The hypothalami were immediately frozen on dry ice and stored at −80°C until used.
Gelatinase Assay
Net gelatinolytic activity was determined using the EnzCheck Gelatinase Assay (Molecular Probes; Carlsbad, CA) as previously described (19) with minor modifications. Hypothalami were homogenized in lysis buffer (20 mM Tris pH 7.4, 137 mM NaCl, 25 mM β-glycerolphosphate pH 7.4, 2 mM sodium pyrophosphate, 0.5% Triton X-100, 10% glycerol) without EDTA or protease inhibitors. Proteolytic activity, in 250 μg protein, is reported as the rate of fluorescence increase over 3 hours normalized to assay reagent containing DQ gelatin alone. Fluorescence was measured at 515 nm (Excitation wavelength 485 nm) using a FLUOstar Galaxy microplate reader (BMG Labtechnologies; Bristol, RI). The specificity of the assay was determined as previously described (19).
In situ zymography
Coronal cryosections (12 μm thick) of fresh frozen brains were direct mounted on gelatin-coated slides and stored at −80°C until used. Slides were hydrated with PBS then coated with 1% low melting temperature agarose containing 75 μg/ml DQ Gelatin (EnzCheck, Molecular Probes) in 1X Reaction Buffer (50 mM Tris-HCl, 150 mM NaCl, 5 mM CaCl2, 0.2 mM sodium azide, pH 7.6). Slides were coverslipped and incubated at 37°C for 5 minutes to allow the agarose to set. The inclusion of agarose enhanced cellular localization of fluorescence. Slides were incubated at room temperature with images captured every 5 minutes. Peak fluorescence was detected after 15 minutes. Images were captured using a manually set exposure time identical for all four conditions. For inhibition of MMP activity, sections were incubated with 1,10 phenanthroline (200 mM) for 1 hour prior to DQ Gelatin application.
Quantitative real-time PCR (qPCR)
Total cellular RNA was prepared using 1 ml STAT-60 per hypothalamus with minor modification of manufacturer’s instructions (Tel-Test B; Friendswood, TX). Following homogenization and centrifugation, the aqueous phase was not precipitated with isopropanol; rather, it was treated with DNase to eliminate contaminating genomic DNA (SV Total RNA Isolation System; Promega; Madison, WI). Total RNA was quantitated and integrity verified using a NanoDrop spectrophotometer (Thermo Scientific; Wilmington, DE). cDNA was synthesized from 2 μg of total RNA using Superscript II reverse transcriptase (Invitrogen; Carlsbad, CA) and random hexamers in a total volume of 20 μl according to the manufacturer's instructions. Relative quantification of genes was performed using the ABI 7500 Fast Real-Time PCR System (Applied Biosystems; Carlsbad, CA) in accordance with the manufacturer's instructions. PCR reactions contained 5 ng of reverse transcribed RNA (1 ng for 18S rRNA analyses), 100 nM of each primer, 200 nM of probe and 50% TaqMan 2X Universal PCR Master Mix No AmpErase UNG or Power SYBR Green (Applied Biosystems; Carlsbad, CA) in a total volume of 25 μl. Primer and probe sequences for MMPs, TIMPs (20), AgRP, NPY, POMC (21), CART, melanin-concentrating hormone (MCH), orexin (22), SOCS3 (23), STAT3 and leptin receptor isoforms (24) are detailed in Table 1. Conditions for the PCR reaction were as follows: 2 minutes at 50°C, 10 minutes at 95°C, 40 cycles of 15 seconds at 95°C, and 1 minute at 60°C. Sybr-based amplifications included a terminal dissociation curve to verify a single amplicon. The threshold cycle (CT), the cycle number at which signal is detectable above the baseline, was manually determined for each gene. As a quality control, only cDNA falling within ± 1.5 CT of the median value for 18S rRNA for all samples were used in the study. To compare the expression of a single gene across samples, standard curves for each gene were generated using cDNA, pooled from one sample of each diet and genotype, that had undergone two-fold serial dilutions (e.g., 1 ng to 0.03125 ng for 18S and 20 ng to 0.625 ng for genes of interest). Relative input amounts of template cDNA were then calculated from CT using the standard curves. Data are presented as relative levels of mRNA normalized to 18S rRNA.
Table 1.
TaqMan Primers and Probes (Mus musculus).
| TIMP-1 | Forward Primer: | 5′-CATGGAAAGCCTCTGTGGATATG-3′ |
| Reverse Primer: | 5′-AAGCTGCAGGCACTGATGTG-3′ | |
| Probe: | 5′-FAM-CTCATCACGGGCCGCCTAAGGAAC-TAMRA-3′ | |
| TIMP-2 | Forward Primer: | 5′-CCAGAAGAAGAGCCTGAACCA-3′ |
| Reverse Primer: | 5′-GTCCATCCAGAGGCACTCATC-3′ | |
| Probe: | 5′-FAM-ACTCGCTGTCCCATGATCCCTTGC-TAMRA-3′ | |
| TIMP-3 | Forward Primer: | 5′-GGCCTCAATTACCGCTACCA-3′ |
| Reverse Primer: | 5′-CTGATAGCCAGGGTACCCAAAA-3′ | |
| Probe: | 5′-FAM-TGCTACTACTTGCCTTGTTTTGTGACCTCCA-TAMRA-3′ | |
| TIMP-4 | Forward Primer: | 5′-TGCAGAGGGAGAGCCTGAA-3′ |
| Reverse Primer: | 5′-GGTACATGGCACTGCATAGCA-3′ | |
| Probe: | 5′-FAM-CCACCAGAACTGTGGCTGCCAAATC-TAMRA-3′ | |
| MMP-2 | Forward Primer: | 5′-AACTACGATGATGACCGGAAGTG-3′ |
| Reverse Primer: | 5′-TGGCATGGCCGAACTCA-3′ | |
| Probe: | 5′-FAM-TCTGTCCTGACCAAGGATATAGCCTATTCCTCG-TAMRA 3′ | |
| MMP-14 | Forward Primer: | 5′-AGGAGACAGAGGTGATCATCATTG-3′ |
| Reverse Primer: | 5′-GTCCCATGGCGTCTGAAGA-3′ | |
| Probe: | 5′-FAM-CCTGCCGGTACTACTGCTGCTCCTG-TAMRA -3′ | |
| AgRP | Forward Primer: | 5′-CAGAAGCTTTGGCGGAGGT-3′ |
| Reverse Primer: | 5′-AGGACTCGTGCAGCCTTACAC-3′ | |
| CART | Forward Primer: | 5′-AAGTCCCCATGTGTGACGCT-3′ |
| Reverse Primer: | 5′-GACAGTCACACAGCTTCCCGA-3′ | |
| MCH | Forward Primer: | 5′-TTCAAAGAACACAGGCTCCAAA-3′ |
| Reverse Primer: | 5′-ACTCAGCATTCTGAACTCCATTCTC-3′ | |
| NPY | Forward Primer: | 5′-TCAGACCTCTTAATGAAGGAAAGCA-3′ |
| Reverse Primer: | 5′-GAGAACAAGTTTCATTTCCCATCA-3′ | |
| ObRa | Forward Primer: | 5′-GGAACACTGTTAATTTCACACCAGAG-3′ |
| Reverse Primer: | 5′-ATTCAAACCATAGTTTAGGTTTGTTTCC-3′ | |
| ObRb | Forward Primer: | 5′-TGTTCCTGGGCACAAGGACT-3′ |
| Reverse Primer: | 5′-TGGGTTCAGGCTCCAGAAGA-3′ | |
| ObRall | Forward Primer: | 5′-ACAGTGTGCTTCCTGGATCTTC-3′ |
| Reverse Primer: | 5′-ACAGTGCTTCCCACTAGTGATTGG-3′ | |
| Orexin | Forward Primer: | 5′-ATGAACTTTCCTTCTACAAAGGTTCC-3′ |
| Reverse Primer: | 5′-GGCAGCAGTAGCAGCAGCA-3′ | |
| POMC | Forward Primer: | 5′-CTGCTTCAGACCTCCATAGATGTG-3′ |
| Reverse Primer: | 5′-CAGCGAGAGGTCGAGTTTGC-3′ | |
| SOCS3 | Forward Primer: | 5′-GCGGGCACCTTTCTTATCC-3′ |
| Reverse Primer: | 5′-TCCCCGACTGGGTCTTGAC-3′ | |
| STAT3 | Forward Primer: | 5′-TCCTCTATCAGCACAACCTTCG-3′ |
| Reverse Primer: | 5′-AGCTGCTGCATCTTCTGTCTGG-3′ | |
Leptin Enzyme-immunoassay (EIA)
Mice were fasted for a maximum of 16 hours beginning at their dark (feeding) cycle. The next day, after weighing the mice, blood (~ 70 μl) was collected into a heparinized capillary tube from a small nick in the tail vein, transferred to chilled microcentrifuge tubes for at least 5 min on ice, spun at 4°C for 10 min at 1250X g, the serum collected and stored at −20°C until analyzed. Leptin levels at the study onset, midpoint, and termination were determined by leptin EIA according to manufacturer’s instructions (Alpco Diagnostics; Salem, NH) using an ELx800 absorbance microplate reader (BioTek; Winooski, VT). Samples, except study onset, were analyzed in duplicate and average optical density readings converted to pg/ml using kit provided standards and Prism software (GraphPad; San Diego, CA). Correlation of leptin concentration to body weight was determined using Excel software (Microsoft; Redmond, WA).
Leptin sensitivity
To test leptin-mediated STAT3 activation, two-month old weight-matched chow-fed WT and TIMP-2 KO mice were fasted overnight (maximum of 16 hours), injected intraperitoneally (i.p.) with 200 μl of sterile phosphate-buffered saline (PBS) or recombinant mouse leptin (5 μg/g in PBS; R & D Systems; Minneapolis, MN) and euthanized 30 minutes later (25) since leptin reaches maximal concentration in the brain 30 minutes after i.p. injection (26). Hypothalami were micro-dissected, as described above, for western blot analysis or immunohistochemistry.
To assess the effect of leptin on food consumption and weight loss, littermates were randomly assigned to receive PBS or leptin injections at the onset of the study. Body weight and food consumption were monitored daily in weight-matched group-housed mice for 3 days and an additional 4 days while singly housed to acclimate mice to daily weighing. Mice were then injected with 200 μl of sterile PBS or leptin (1 μg/g) twice daily (i.e., within 1 hour of light and dark cycle) for 3 days (27) and body weight and food consumption monitored for an additional 4 days.
Western Blot
Western blots were performed as previously described (28). Briefly, proteins (50 μg) were resolved by 12% SDS-PAGE, transferred to nitrocellulose, and probed with monoclonal rabbit anti-mouse phospho-STAT3 (Tyr 705) antibody (1:500, #9145; Cell Signaling Technology; Beverly, MA). Blots were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody (1:3,000; Jackson ImmunoResearch; West Grove, PA) and immunocomplexes visualized by enhanced chemiluminescence (PerkinElmer Life Sciences, Boston, MA). Immunocomplexes were stripped from the membrane by incubation with Restore Plus Western Blot Stripping Buffer (Pierce; Rockford, IL) and stripping solution (0.2 M glycine, 0.5 M NaCl, pH 2.5) for 10 minutes each followed by 15-minute washes in TBS and TBST, and blocked as described (28). Blots were then incubated in monoclonal mouse anti-mouse STAT3 antibody (1:1,500, #9139; Cell Signaling), and immunocomplexes detected as before. Finally, after stripping as above, blots were re-probed with polyclonal goat anti-human actin (1:1,000, sc-1616; Santa Cruz Biotechnology, Santa Cruz, CA). Densitometry was performed using Quantity One software (Bio-Rad; Hercules, CA). Relative expression of pSTAT3 was either normalized to total STAT3 or actin expression.
Immunohistochemistry
Immunohistochemistry with rabbit anti-mouse pSTAT3 (Ser727) (1:300, #9134; Cell Signaling) and visualization with Cy3-conjugated secondary antibody (1:500, Jackson ImmunoResearch) containing 1 μg/ml bisBenzimide (Sigma-Aldrich) was performed as described (19).
Statistics
Data are presented as mean ± standard error of the mean (SEM). Significant interactions were identified by student’s t-test at the study start and two-way analysis of variance (ANOVA) with Bonferroni’s multiple comparison tests at the study termination using Prism software. Statistical significance was assigned at probability of p < 0.05. Unless otherwise indicated, symbols designate statistically significant differences relative to WT mice on the same diet.
Results
Hyperphagia contributes to diet-induced weight gain in TIMP-2 KO mice
TIMP-2 deficiency is associated with an age-dependent increase in body weight. The body weight of WT and TIMP-2 KO mice fed a standard chow diet (i.e., 4.1 kcal/g) was monitored from weaning at postnatal day 21 (P21) to 1 year of age. TIMP-2 KO mice of both sexes had weights comparable to WT mice at P21, but by 3 months of age, TIMP-2 KO mice exhibited significantly greater body weights than WT mice (Jaworski, unpublished observation).
Furthermore, TIMP-2 KO mouse weight continued to increase unabated to 1 year of age. Since mice were maintained as homozygous breeders and perturbations in maternal nutritional status (e.g., undernutrition, obesity, diabetes) can irreversibly alter structures responsible for the control of ingestive behavior and energy expenditure (29), weight gain in offspring from heterozygous pairings was determined. Even with the small sample size (n = 3), due to reduced frequency of sex-matched WT and TIMP-2 KO littermates, TIMP-2 KO mice weighed more than their WT littermates at 3 months of age (M: WT = 25.45 ± 0.33 g, KO = 29.50 ± 0.69 g, p = 0.006; F: WT = 19.14 ± 0.25 g, KO = 23.01 ± 0.37 g, p = 0.001), suggesting that altered gestational metabolic programming is not the basis for greater TIMP-2 KO body weight. Thus, studies were undertaken to determine the basis for excessive weight gain in the absence of TIMP-2.
To determine the effect of diet on TIMP-2 KO obesity, 2-month-old (i.e., prior to the onset of TIMP-2 KO obesity) weight-matched WT and TIMP-2 KO mice either were maintained on the standard chow diet or switched to a HFD containing 60% fat (i.e., 5.24 kcal/g) and their body weights monitored daily for 3 months. Similar to our previous observations, chow-fed TIMP-2 KO mice of both sexes gained more weight than WT mice during the 3-month study (Fig. 1A). In fact, female TIMP-2 KO mice weighed as much as male WT mice at the study termination. The weight gain in HFD-fed TIMP-2 KO mice was even more pronounced (Fig. 1B). Male TIMP-2 KO mice weighed significantly more than male WT mice after only 6 days on the HFD. TIMP-2 KO mice maintained on the HFD for 5 months (e.g., ~60 g male weight) failed to show plateau of weight gain (data not shown), suggesting a defect in central recognition of adequate energy stores.
Fig. 1.
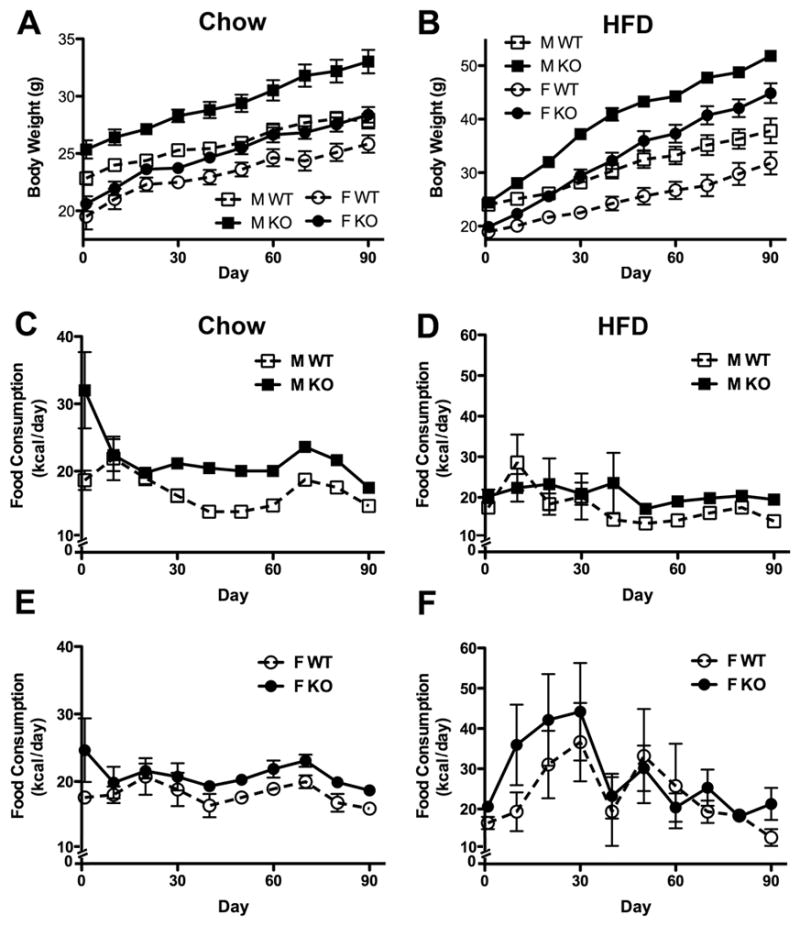
TIMP-2 KO mice gain more weight than WT mice regardless of diet. Both sexes of two-month-old TIMP-2 KO mice fed a chow (A) or HFD (B) for 3 months gained significantly more weight than WT mice (as determined by weight slope; chow: M p = 0.03, F p = 0.04; HFD: M p = 0.0001, F p = 0.0003). (C-F) Examination of average daily food consumption revealed that chow- (C, E) and HFD-fed (D, F) TIMP-2 KO mice failed to modify food intake despite increased body weight. Decreased food consumption in chow-fed mice after day 68 was due to overnight starvation and insulin tolerance testing (data not shown). Data are mean ± SEM. n = 8 per sex, genotype, and diet.
Since over-eating is a logical explanation for the profound TIMP-2 KO weight gain, food consumption was measured. To accurately assess food intake per mouse, animals were individually housed. Since TIMP-2 KO mice exhibit increased anxiety (30) and social isolation is a stressor, average food consumption was first monitored daily for 5 days in group-housed mice prior to individual housing. Even though TIMP-2 KO mice were of comparable weights to WT mice at the study onset, TIMP-2 KO mice of both sexes displayed hyperphagia, which was more pronounced in males (i.e., 8.37 kcal/day excess in males and 3.36 kcal/day excess in females). Regardless of diet (Figs. 1C-F), individually housed TIMP-2 KO mice of both sexes consumed more kcals/day throughout the 3-month study (genotype: p < 0.001). Hyperphagia in TIMP-2 KO mice was more pronounced in chow-fed male (Fig. 1C) than female (Fig. 1E) mice. Furthermore, while HFD-fed male TIMP-2 KO mice showed little variability in food intake (Fig. 1D), HFD-fed female mice of both genotypes displayed dramatic fluctuations in food intake (Fig. 1F). It should be noted that the decreased food intake after day 68 in chow-fed mice is a response to overnight starvation and insulin tolerance testing (data not shown). Taken together, our data demonstrate that TIMP-2 KO mice are hyperphagic prior to obesity onset and do not adjust caloric intake concomitant with increased body mass, suggesting the inability to detect adequate energy stores. Due to the less marked hyperphagia of TIMP-2 KO females and well-known sex differences in leptin responsiveness (31), all subsequent studies utilized only male mice.
Increased proteolysis in TIMP-2 KO mice may contribute to altered ECM remodeling
Because TIMP-2 plays a role in both MMP inhibition (15) and MMP activation (16, 17) one cannot predict a priori whether net proteolytic activity in a particular tissue will be increased or decreased in the absence of TIMP-2. Therefore, the effect of diet and TIMP-2 deficiency on hypothalamic protease expression (Fig. 2A) and activity (Figs. 2B, C) was examined.
Fig. 2.

Net proteolytic activity is increased in male TIMP-2 KO hypothalamus. (A) qPCR revealed that, at least at the mRNA level, there is no overall compensation by other TIMPs in response to TIMP-2 absence. There was no effect of diet at the study termination, but when compared to the study start TIMP-1 mRNA expression was increased in chow-fed (p = 0.03) and HFD-fed (p = 0.002) TIMP-2 KO mice while no increase was observed in WT mice. (B) Using a caged substrate that emits fluorescence when cleaved by proteases, it was revealed that net proteolytic activity was increased in TIMP-2 KO hypothalamus, but not globally increased throughout the entire TIMP-2 KO brain. Interestingly, while WT mice decreased proteolytic activity on the HFD, proteolysis remained elevated in HFD-fed TIMP-2 KO mice, suggesting that the inability to effectively inhibit proteolysis may contribute to defective energy homeostasis and obesity. (C) In situ zymography confirmed the results of the gelatinase assay and specifically revealed increased proteolysis within the arcuate nucleus of TIMP-2 KO mice. Data are mean ± SEM. n = 4 – 5 per genotype and diet. *p < 0.05, **p ≤ 0.01, #p≤0.001 relative to WT mice on the same diet, unless otherwise indicated.
TIMP-2 deficiency did not lead to a compensatory increase in the expression of the other TIMPs, relative to WT levels. In fact, in addition to the expected reduction of TIMP-2 mRNA (p = 0.0008), TIMP-1 (p = 0.04) and TIMP-4 (p = 0.003) mRNA expression were actually decreased in TIMP-2 KO mice at the study start. At the study termination, TIMP-2 (p < 0.0001), TIMP-3 (p = 0.039), and MMP-2 (p = 0.04) mRNA were reduced in TIMP-2 KO mice. No genotype effect on TIMP-1 (p = 0.49), TIMP-4 (p = 0.31) or MMP-14 (p = 0.27) mRNA was detected at the study termination. Two-way ANOVA at the study termination failed to reveal an effect of diet for any of the genes. However, when compared to the study start, TIMP-1 mRNA expression was increased in chow-fed (p = 0.03) and further increased in HFD-fed (p = 0.002) TIMP-2 KO mice, perhaps as a compensatory mechanism for TIMP-2’s absence.
The combination of reduced TIMP-2 and TIMP-3 mRNA expression in HFD-fed TIMP-2 KO mice would favor increased proteolytic activity. Therefore, rather than examine MMP and TIMP protein expression, net proteolytic activity within the hypothalamus was measured using a caged substrate (i.e., DQ gelatin) (Figs. 2B, C). This substrate is cleaved by a broad range of proteases (e.g., all proteases that cleave gelatin and collagen), not only the gelatinases MMP-2 and MMP-9. Cleavage yields highly fluorescent peptides, whose fluorescence is proportional to proteolytic activity. Proteolytic activity was not affected by diet (p = 0.79), but, as expected from the mRNA results, proteolysis was significantly increased in TIMP-2 KO mice (p = 0.003) (Fig. 2B). Moreover, while protease activity decreased in HFD-fed WT mice relative to chow-fed WT mice, an opposite trend was observed in TIMP-2 KO mice with a significant genotype by diet interaction (p = 0.01). To determine whether proteolysis was globally increased throughout the TIMP-2 KO brain, gelatinase activity was measured in several brain regions of 3-month-old chow-fed WT and TIMP-2 KO mice. Gelatinase activity in TIMP-2 KO mice was increased in the cerebral cortex, decreased in the hippocampus, and unaltered in the cerebellum, striatum, or thalamus. Thus, proteolysis is not indiscriminately increased throughout the TIMP-2 KO brain. Finally, in situ zymography to detect proteolytic activity specifically within the arcuate nucleus (Fig. 2C) recapitulated the gelatinase results (Fig. 2B). These data appear to support a more traditional MMP inhibitory role for TIMP-2 within the hypothalamus.
TIMP-2 KO mice exhibit altered leptin-mediated signal transduction
To determine the basis for TIMP-2 KO hyperphagia, qPCR was performed to assess the expression of orexigenic (i.e., arcuate-derived NPY, AgRP; lateral hypothalamus-derived orexin, MCH) and anorexigenic (i.e., arcuate-derived CART, POMC) hypothalamic neuropeptides (Fig. 3). Because feeding status modulates neurotransmitter expression, mRNA levels were measured in fasted mice. Contrary to what one might expect for orexigenic peptides, NPY (Fig. 3A) and AgRP (Fig. 3B) mRNA expression were decreased in hyperphagic TIMP-2KO mice at the study start (p = 0.0002 and p = 0.0173, respectively). However, at the study termination, neither NPY (p = 0.45) nor AgRP (p = 0.75) showed an effect of genotype, suggesting that to some extent these neuropeptides responded to excess caloric intake/positive energy balance in TIMP-2 KO mice. No significant genotype or diet difference in orexin (Fig. 3C), MCH (Fig. 3D), CART (Fig. 3E) or POMC (Fig. 3F) mRNA expression was observed. Therefore, at least at the mRNA level, TIMP-2 KO hyperphagia is not due to altered expression (i.e., increased orexigenic or decreased anorexigenic) of the most commonly regulated hypothalamic neuropeptides.
Fig. 3.

Alterations in hypothalamic neuropeptide mRNA expression are not the basis for increased TIMP-2 KO food consumption. Orexigenic (i.e., NPY, AgRP, orexin, MCH) and anorexigenic (i.e., CART, POMC) neuropeptide mRNA expression in micro-dissected male hypothalami was determined by qPCR. Both arcuate-derived NPY (A) and AgRP (B) mRNA expression was reduced in TIMP-2 KO mice at the study start, but not termination. No differences, either in genotype or diet, were observed for transcript levels of orexin (C) and MCH (D), most prevalent in the lateral hypothalamus, or arcuate-derived CART (D) and POMC (E). Data are mean ± SEM. n = 4 – 5 per genotype and diet. *p < 0.05, #p ≤ 0.001 relative to WT mice on the same diet.
Inasmuch as leptin modulates both food intake and energy expenditure, we sought to determine whether the hyperphagia and obesity in TIMP-2 KO mice could be due to defects in leptin production and/or signal transduction (Fig. 4). First, serum leptin levels were determined by EIA (Fig. 4A). At the start of the diet, when WT and TIMP-2 KO mice were of comparable weights (WT = 22.68 ± 0.20 g, KO = 23.20 ± 0.47 g, p = 0.31), no difference in serum leptin was observed (p = 0.39) (Fig. 4A), even though TIMP-2 KO mice were hyperphagic (Fig. 1C). After ~50 days, both chow-fed (p = 0.0004) and HFD-fed (p < 0.0001) TIMP-2 KO mice were hyperleptinemic. At the termination of the 90-day study, the effects of genotype (F1,19 = 53.06, p < 0.0001) and diet (F1,19 = 190.07, p < 0.0001) were further exacerbated. Since circulating leptin levels are proportionate to adipose tissue mass (2), the correlation of serum leptin relative to body mass was determined. The strong correlation (R2 = 0.85) suggests that the increased serum leptin in TIMP-2 KO mice is proportionate to their greater body weight (Figs. 1A, B).
Fig. 4.

TIMP-2 KO mice exhibit leptin-signalling deficiencies. (A) EIA of serum leptin revealed comparable leptin levels in weight-matched male WT and TIMP-2 KO mice at the study start (WT = 86.97 ± 33.47 pg/ml, KO = 131.1 ± 35.28 pg/ml, p = 0.39). While leptin levels increased greater in TIMP-2 KO mice than WT mice throughout the study, this increase was proportional to their body weight (R2 = 0.85). n = 6 per genotype and diet. (B-D) qPCR was used to analyze mRNA expression of components of the leptin signalling cascade in male hypothalami. n = 4 – 5 per genotype and diet. (E) Western blot analysis confirmed PCR findings of decreased STAT3 mRNA expression in chow-fed TIMP-2 KO mice. (F) Basal STAT3 activation was examined at the study termination. Since total STAT3 expression was decreased in chow-fed mice (E), pSTAT3 expression was normalized both to total STAT3 (left graph) and to actin (right graph). HFD increased STAT3 activation in TIMP-2 KO mice to a greater extent than in WT mice. n = 6 - 7 per genotype and diet. Data are mean ± SEM. *p < 0.05, **p ≤ 0.01, #p≤0.001 relative to WT mice on the same diet, unless otherwise indicated.
Since altered leptin production was not the basis for TIMP-2 KO hyperphagia, we next assessed whether defects in leptin signalling may underlie TIMP-2 KO hyperphagia. qPCR, with primers that recognize all isoforms, revealed significantly reduced ObR mRNA expression in TIMP-2 KO mice at the study start (p = 0.0008) (Fig. 4B), due to reduced transcription of ObRb, the full-length receptor (p = 0.014), but not ObRa. Similar to NPY and AgRP (Figs. 3A, B), no differences in ObR mRNA expression were observed at the study termination. Since increased MMP-mediated ObR proteolysis is a potential mechanism underlying TIMP-2 KO leptin resistance, ObR expression was examined. Despite the use of commercially available antibodies that purportedly recognized all ObR isoforms, western blot analysis proved inconclusive (data not shown). No change in SOCS3 mRNA expression was detected (Fig. 4C). Although STAT3 mRNA expression was not reduced in TIMP-2 KO mice at the study start (p = 0.11), expression was reduced at the study termination (p = 0.04) (Fig. 4D). Furthermore, an effect of diet was present at the study termination due to increased STAT3 mRNA in HFD-fed mice relative to chow-fed mice of the same genotype (p = 0.017) (Fig. 4D). Since STAT3 signalling is regulated by phosphorylation state, western blot analysis was performed for basal phospho-STAT3 (pSTAT3) as well as total STAT3 expression at the study termination. Decreased STAT3 mRNA (Fig. 4D) was reflected in reduced total STAT3 protein expression in chow-fed TIMP-2 KO mice (p = 0.014) (Fig. 4E). Usually, pSTAT3 is normalized to total STAT3, but because total STAT3 expression was reduced in chow-fed TIMP-2 KO mice (Fig. 4E), pSTAT3 was normalized against both total STAT3 (Fig. 4F, left) and, more appropriately, actin (Fig. 4F, right). Effects of genotype (p = 0.012) and diet (p = 0.0017) were observed for pSTAT3, primarily due to increased pSTAT3 in HFD-fed TIMP-2 KO mice (Fig. 4F). Our data are concordant with previous reports that basal STAT3 phosphorylation within the arcuate nucleus is increased upon high-fat feeding (25, 32). Since leptin can also act through components of the insulin signalling cascade, the phosphorylation state of Akt was also examined. Chow-fed TIMP-2 KO mice showed reduced pAktT308 expression (p = 0.007), while HFD-fed TIMP-2 KO mice showed reduced pAktS473 expression (p = 0.04) (data not shown). However, several signalling pathways converge to STAT3, as well as Akt, and analysis was performed with micro-dissected hypothalamus. Thus, we cannot ascertain whether the observed alterations only occurred within leptin-sensitive hypothalamic nuclei.
Leptin-mediated orexigenic responses are attenuated in TIMP-2 KO mice
To more accurately assess defects in leptin signalling, leptin-mediated STAT3 activation and anorectic responses were examined. First, weight-matched chow-fed 2-month-old mice were injected either with PBS or leptin (5 μg/g), euthanized 30 minutes later, and pSTAT3 expression examined by western blot analysis (Fig. 5A) or immunohistochemistry (Fig. 5B). This experiment was not conducted at the study termination because TIMP-2 KO mice are already hyperleptinemic. Since total STAT3 expression was not altered by genotype at 2 months of age (p = 0.10), pSTAT3 was normalized to total STAT3. Leptin-mediated STAT3 activation was dramatically reduced in TIMP-2 KO mice (p = 0.0097) (Fig. 5A). Histological examination confirmed fewer pSTAT3 immunoreactive nuclei within the arcuate nucleus of TIMP-2 KO mice (Fig. 5B). While reduced STAT3 activation is suggestive of leptin resistance, a more definitive demonstration of leptin resistance is an attenuated leptin-mediated reduction in food intake and weight loss. Food consumption (Fig. 5C) and weight (Fig. 5D) was monitored in weight-matched chow-fed 2-month-old mice for 7 days prior to and 4 days subsequent to 3 days of injection either with PBS or leptin (1 μg/g). In contrast to the minor transient decline in food consumption in response to PBS injection, which was comparable in WT and TIMP-2 KO mice (day 8 relative to day 9, p = 0.69), the leptin-mediated anorectic response was significantly reduced in TIMP-2 KO mice (WT = 33.81 ± 2.23%, KO = 15.89 ± 6.7%, p = 0.015 decline in food consumption at day 11 relative to the 2 days prior to leptin injection). The attenuated anorectic response in TIMP-2 KO mice was even more pronounced after only one day of leptin administration (WT = 42.92 ± 11.12%, KO = 9.24 ± 6.08%, p = 0.019). Given that PBS-injected mice displayed a mild anorectic response, leptin data were normalized to PBS. Both PBS normalized data and data normalized to starting food consumption (data not shown) showed a significantly (p < 0.0001) attenuated anorexigenic response in TIMP-2 KO mice. The attenuated leptin-mediated effect on weight loss was even more prominent in TIMP-2 KO mice (WT = 7.13 ± 0.60%, KO = 1.70 ± 0.73%, p < 0.001 decline in weight at day 11 relative to the 2 days prior to leptin injection). Furthermore, TIMP-2 KO mice also exhibited a more rapid increase in food consumption (Fig. 5C) and weight (Fig. 5D) upon termination of leptin administration. Taken together these data are supportive as leptin resistance as a contributor to TIMP-2 KO hyperphagia, even in the absence of increased body mass at 2 months of age. Furthermore, it likely contributes to their inability to auto-regulate food consumption even when faced with morbid obesity on the HFD.
Fig. 5.

Attenuated leptin-mediated anorectic responses in TIMP-2 KO mice. (A-B) STAT3 activation was assessed in weight-matched (WT = 24.06 ± 0.39 g, KO = 23.49 ± 0.41 g, p = 0.33) 8-week-old mice 30 minutes after a single injection of PBS or leptin (5 μg/g). (A) Western blot analysis showed decreased leptin-mediated STAT3 activation in TIMP-2 KO mice. n = 5. (B) Immunohistochemistry revealed fewer pSTAT3-labelled cells (red, pink in inset) in the arcuate nucleus of TIMP-2 KO mice (arrowheads indicate non-leptin responsive cells). Cell nuclei are counterstained with Hoechst (blue). Images representative of n = 4. Scale bars = 50 μm. (C) The effect of leptin (1 μg/g) administration (indicated by arrows) on food consumption. TIMP-2 KO mice exhibited an attenuated leptin-mediated anorectic response (p = 0.015, intake at day 11 relative to 2 days prior to leptin administration; p < 0.0001, 2-way ANOVA over entire study). Similar results are obtained when normalized to PBS injected mice. (D) Leptin-mediated weight loss was similarly attenuated in TIMP-2 KO mice (p < 0.0001, percent weight loss at day 11 relative to 2 days prior to leptin administration; p < 0.0001, 2-way ANOVA over entire study). Similar results are obtained when normalized to PBS injected mice. Data are mean ± SEM. **p≤0.01 relative to WT mice on the same diet, unless otherwise indicated.
Discussion
Herein, we demonstrated for the first time that, in contrast to most MMP or TIMP KO mice, TIMP-2 KO mice display hyperphagia and weight gain even when fed a chow diet. Given its pleiotropic MMP-dependent (i.e., MMP inhibition and activation) and MMP-independent (i.e., angiogenesis, cell cycle regulation) effects, it is likely that multiple mechanisms underlie TIMP-2’s contribution to metabolism.
Multiple factors contribute to obesity in TIMP-2 KO mice
Similar to ob/ob and db/db mutant mice (2, 33), chow-fed TIMP-2 KO mice show hyperphagia with early onset weight gain (Fig. 1) and delayed puberty onset (unpublished observation). However, chow-fed TIMP-2 KO mice are not hyperinsulinemic or hyperglycemic despite increased adiposity (unpublished observation). Accordingly, they do not display the morbid obesity of ob/ob or db/db mutant mice, unless they consume a fat-laden diet. Nonetheless, chow-fed TIMP-2 KO mice exhibit signs of leptin resistance, as revealed by reduced leptin-mediated STAT3 activation and anorectic response, prior to obesity onset. A number of mechanismshave been put forth to explain leptin resistance, including reduced leptin transport across the blood–brain barrier (BBB), ObR signalling defects, and perturbations in developmental programming (24, 34, 35).
Reduced BBB transport likely does not play a role in TIMP-2 KO leptin resistance. MMPs open the BBB by degradation of basal lamina proteins (9). Therefore, given the increased MMP activity in TIMP-2 KO hypothalamus, BBB integrity is likely compromised, thereby increasing, not decreasing, leptin transport.
Our data are consistent with intracellular leptin signalling defects in TIMP-2 KO mice, including decreased leptin-induced STAT3 activation (Fig. 4G) and anorectic response (Fig. 4I). Up-regulation of hypothalamic NPY elicits hyperphagia in response to physiological and pathophysiological states. Yet, neither increased AgRP/NPY nor decreased CART/POMC mRNA expression was observed in TIMP-2 KO mice (Fig. 3). Rather, a counter-intuitive reduction in AgRP/NPY mRNA was observed in TIMP-2 KO mice at the study start. Since the mRNA expression for none of the other neuropeptides was altered, it argues against some defect with these cDNA samples. One possible explanation for decreased orexigenic transcription is a leptin-mediated negative feedback since leptin inhibits NPY mRNA production (36). However, at the start of the diet, leptin levels in TIMP-2 KO mice were comparable to WT mice and when leptin levels were increased there was no difference in NPY mRNA, suggesting that NPY and AgRP are responding appropriately to excess caloric intake/positive energy balance. Increased NPY is not absolutely required for the occurrence of hyperphagia or obesity. In fact, several models of hyperphagia with low NPY abundance exist, including altered communication with hindbrain autonomic centers, as well as lesions within ventromedial or paraventricular (PVN) hypothalamic nuclei (37). At present, the mechanism by which NPY and AgRP mRNA are initially decreased in TIMP-2 KO mice remains unresolved and we do not exclude the possibility of defects in extrahypothalamic regions.
In addition to its satiety effects, leptin promotes energy expenditure, by increasing sympathetic neural activation to brown adipose tissue (BAT) and BAT-mediated thermogenesis (38, 39). Hypothalamic TIMP-2 mRNA expression is increased in mice with high heat loss (40), suggesting TIMP-2 promotes thermogenesis. Thus, its loss could be associated with lower core body temperature and the increased food consumption in TIMP-2 KO mice may be a compensatory behavioral thermoregulatory mechanism to promote heat production.
Finally, we cannot discount the effect of inactivity as a predisposing factor to TIMP-2 KO weight gain. Increased lean body mass is likely not the basis for weight gain in TIMP-2 KO mice since these mice display motor deficits (41) and exhibit decreased muscle mass (i.e., extensor digitorum longus, a predominantly fast-twitch muscle) (19). Thus, muscle weakness and a more sedentary lifestyle may contribute to TIMP-2 KO weight gain. Furthermore, if more oxidative slow-twitch muscle (e.g., soleus) were also lost in TIMP-2 KO mice, this would promote adipogenesis, as a prime glucose oxidizing tissue is lost. Because TIMP-2 is depleted globally, we also cannot exclude a peripheral effect on weight gain. Nonetheless, TIMP-2 KO mice exhibit defective central control of energy metabolism (e.g., hyperphagia and leptin resistance). The leptin resistance is not secondary to obesity because 8-week-old TIMP-2 KO mice were leptin resistant even when of comparable weight to WT mice.
Evidence is mounting that postnatal behavioral (e.g., hyperphagia, fat preference) and physiological (e.g. hyperlipidemia, obesity) patterns are established via epigenetic modifications during the prenatal and neonatal period (42). Although serum leptin levels were comparable in WT and TIMP-2 KO mice at 2 months of age, this does not preclude embryonic or neonatal alterations or changes below assay sensitivity. We cannot rule out an epigenetic contribution in TIMP-2 KO obesity; however, given that TIMP-2 KO offspring of chow-fed heterozygous pairings were also obese, relative to their WT littermates, it clearly suggests other contributing factors.
Proteolysis, ECM remodeling, and energy homeostasis
The MMP/TIMP enzymatic system plays a pivotal role in matrix remodeling underlying hippocampal plasticity (43), but there is a dearth of information regarding its contribution to hypothalamic remodeling. Hypothalamic TIMP-1 mRNA expression is decreased in ob/ob and db/db mice relative to WT mice (44). Similarly, TIMP-1 mRNA was initially decreased in TIMP-2 KO mice and was the only TIMP to display an effect of diet (Fig. 2A). Leptin promotes the expression of TIMP-1 mRNA within the hypothalamus (44) and, like TIMP-2 KO mice, female TIMP-1 KO mice display increased food intake prior to elevated weight gain (44). Thus, TIMP-1 and TIMP-2 may be the key TIMPs in the control of MMP-mediated hypothalamic ECM remodeling underlying energy homeostasis.
Because TIMP-2 plays a role in both MMP inhibition and activation, one cannot predict a priori whether proteolytic activity will be increased or decreased in its absence. In contrast to the decreased proteolysis observed in TIMP-2 KO muscle (19), proteolytic activity was increased in TIMP-2 KO hypothalamus; thus, excessive proteolysis may be the primary cause of the TIMP-2 KO phenotype. Proteolysis generates a soluble leptin receptor (45), but increased levels are associated with weight loss (46). Similarly, the shed syndecan-3 ectodomain inhibits AgRP’s ability to interact with its receptor; thereby, preventing its orexigenic effects (47). Thus, ObR or syndecans-3 proteolysis probably does not contribute to TIMP-2 KO obesity. Increased proteolysis could alter postnatal synaptic plasticity via cleavage of ECM proteins. This is an unexplored area of neuroscience and clearly deserves more attention. Because TIMP-2 is absent at all phases of development, the ECM cues required to guide neuronal migration from the ventricular zone to the arcuate nucleus or from the arcuate to extrahypothalamic sites might be lost due to increased proteolysis in TIMP-2 KO mice. For example, MT1-MMP KO mice die from cachexia at 3-4 weeks of age (48) even though neuronal perikarya in the arcuate nucleus display normal NPY and AgRP expression (49). However, neuropeptide expression at hypothalamic and extrahypothalamic projection areas is reduced, suggesting axonal mistargeting. Thus, developmental alterations (e.g., decreased PVN innervation from neurons in the arcuate) could be altered in young, pre-obese TIMP-2 KO mice and prime the mice for future weight gain. Hence, future studies are needed to determine the substrates whose proteolysis promotes hyperphagia in TIMP-2 KO mice.
TIMP-2's MMP-independent functions may also play a role in obesity. The potential contribution of neurogenesis in the regulation of energy balance has been a recent focus of considerable attention since the adult hypothalamus possesses neurogenic capacity (50) and leptin promotes neurogenesis (51). TIMP-2 promotes neurogenesis by acting as an anti-mitogenic signal independent of MMP activity (52). Interestingly, we observed fewer BrdU-positive cells in the neurogenic regions of the third ventricle and fewer NeuN-positive neurons in the TIMP-2 KO arcuate nucleus (unpublished observations). It remains unresolved whether this is due to decreased neurogenesis or increased apoptosis since TIMP-2 suppresses programmed cell death (53) and obesity promotes hypothalamic neuronal apoptosis (54). Interestingly, apoptosis appears to be a property of diet composition and not caloric intake since most of the pro-apoptotic activity of ad libitum HFD feeding was retained under isocaloric HFD pair feeding (54).
Obesity is not a simple phenomenon and no one molecule or organ system is solely responsible. If it were, its prevention/treatment would not be so elusive. As discussed above, inasmuch as TIMP-2 is deficient globally, multiple organs (e.g., brain, adipose tissue, muscle) likely play a role in TIMP-2 KO weight gain. Nonetheless, leptin resistance and hyperphagia appear to be prime contributors. The increased proteolysis in TIMP-2 KO hypothalamus suggests a more traditional MMP-inhibitory role for TIMP-2. Given the disparate HFD effects in MMP-deficient mice, future studies are required to identify proteases up-regulated by HFD. Future studies should also explore whether caloric restriction down-regulates hypothalamic MMP expression, thereby providing a plausible mechanism underlying the diverse beneficial effects of caloric restriction.
Acknowledgments
Thomm Buttolph and Ed Zelazney of the Neuroscience COBRE Cellular and Molecular Biology Core Facility are thanked for their assistance with qPCR and design-based stereology, respectively. We thank Dr. Tom Jetton, UVM Dept. of Medicine, for assistance with tissue procurement and generous use of his microplate reader, Drs. Felix Eckenstein and Rae Nishi, UVM Dept. of Anatomy & Neurobiology, for use of their confocal microscope and microplate reader, and Dr. Victor May, UVM Dept. of Anatomy & Neurobiology, for critical review of the manuscript. Finally, the authors are grateful to Garret Langlois for assistance with mouse weighing, Dr. Gentian Lluri for analyzing gelatinase activity levels in different brain regions, and Dr. Kendra K. Bence (Dept. of Animal Biology, Univ. of Pennsylvania School of Veterinary Medicine) for providing advice on the most appropriate leptin administration protocol for monitoring the effect on weight and food consumption.
Grants
This work was supported by R01NS045225 co-funded by NINDS and NCRR (DMJ) and a University of Vermont Undergraduate Research Endeavors Competitive Award (HMS). Facilities and equipment supported by the Neuroscience COBRE (NIH NCRR P20 RR016435) and Vermont Cancer Center DNA Analysis facility (NIH P30 CA22435) were instrumental to the completion of the study. Salary support for HMS was provided by an ARRA supplement to NIH NCRR P20 RR016435.
Abbreviations
- ADAM
a disintegrin and metalloproteinase
- ADAM-TS
ADAM with thrombospondin motif
- AgRP
agouti related peptide
- BAT
brown adipose tissue
- BBB
blood-brain barrier
- CART
cocaine- and amphetamine-regulated transcript
- EIA
enzyme immunoassay
- HFD
high fat diet
- HRP
horseradish peroxidase
- KO
knockout
- MCH
melanin-concentrating hormone
- MMP
matrix metalloproteinase
- NPY
neuropeptide Y
- ObR
leptin receptor
- PBS
phosphate buffered saline
- POMC
proopiomelanocortin
- PVN
paraventricular nucleus
- qPCR
quantitative real-time polymerase chain reaction
- SOCS3
suppressor of cytokine signalling-3
- STAT3
signal transducer and activator of transcription 3
- TBS
tris-buffered saline
- TBST
TBS with Tween
- TIMP
tissue inhibitor of metalloproteinase
- WT
wild-type
Footnotes
Disclosures
No con icts of interest, nancial or otherwise, are declared by the author(s).
References
- 1.Frederich RC, Lollmann B, Hamann A, Napolitano-Rosen A, Kahn BB, Lowell BB, Flier JS. Expression of ob mRNA and its encoded protein in rodents. Impact of nutrition and obesity. J Clin Invest. 1995;96:1658–63. doi: 10.1172/JCI118206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–32. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 3.Hegyi K, Fülöp K, Kovács K, Tóth S, Falus A. Leptin-induced signal transduction pathways. Cell Biol Int. 2004;28:159–69. doi: 10.1016/j.cellbi.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Valassi E, Scacchi M, Cavagnini F. Neuroendocrine control of food intake. Nutr Metab Cardiovasc Dis. 2008;18:158–68. doi: 10.1016/j.numecd.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Farooqi IS, O’Rahilly S. Monogenic obesity in humans. Annu Rev Med. 2005;56:443–58. doi: 10.1146/annurev.med.56.062904.144924. [DOI] [PubMed] [Google Scholar]
- 6.Rodríguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: What do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. 2010;1803:39–54. doi: 10.1016/j.bbamcr.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 7.Yang P, Baker KA, Hagg T. The ADAMs family: coordinators of nervous system development, plasticity and repair. Prog Neurobiol. 2006;79:73–94. doi: 10.1016/j.pneurobio.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8:221–33. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agrawal SM, Lau L, Yong VW. MMPs in the central nervous system: where the good guys go bad. Semin Cell Dev Biol. 2008;19:42–51. doi: 10.1016/j.semcdb.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Ethell IM, Ethell DW. Matrix metalloproteinases in brain development and remodeling: synaptic functions and targets. J Neurosci Res. 2007;85:2813–23. doi: 10.1002/jnr.21273. [DOI] [PubMed] [Google Scholar]
- 11.Maquoi E, Demeulemeester D, Vörös G, Collen D, Lijnen HR. Enhanced nutritionally induced adipose tissue development in mice with stromelysin-1 gene inactivation. Thromb Haemost. 2003;89:696–704. [PubMed] [Google Scholar]
- 12.Lijnen HR, Van HB, Frederix L, Rio MC, Collen D. Adipocyte hypertrophy in stromelysin-3 deficient mice with nutritionally induced obesity. Thromb Haemost. 2002;87:530–5. [PubMed] [Google Scholar]
- 13.Van Hul M, Lijnen HR. A functional role of gelatinase A in the development of nutritionally induced obesity in mice. J Thromb Haemost. 2008;6:1198–206. doi: 10.1111/j.1538-7836.2008.02988.x. [DOI] [PubMed] [Google Scholar]
- 14.Lijnen HR, Demeulemeester D, Van Hoef B, Collen D, Maquoi E. Deficiency of tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) impairs nutritionally induced obesity in mice. Thromb Haemost. 2003;89:249–55. [PubMed] [Google Scholar]
- 15.Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim Biophys Acta. 2010;1803:55–71. doi: 10.1016/j.bbamcr.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Z, Juttermann R, Soloway PD. TIMP-2 is required for efficient activation of proMMP-2 in vivo. J Biol Chem. 2000;275:26411–5. doi: 10.1074/jbc.M001270200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caterina JJ, Yamada S, Caterina NCM, Longenecker G, Holmbäck K, Shi J, Yermovsky AE, Engler JA, Birkedal-Hansen H. Inactivating mutation of the mouse Tissue Inhibitor of Metalloproteinases-2 (Timp-2) gene alters proMMP-2 activation. J Biol Chem. 2000;275:26416–22. doi: 10.1074/jbc.M001271200. [DOI] [PubMed] [Google Scholar]
- 18.Stetler-Stevenson WG. Tissue inhibitors of metalloproteinases in cell signaling: metalloproteinase-independent biological activities. Sci Signal. 2008:re6. doi: 10.1126/scisignal.127re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lluri G, Langlois GD, McClellan B, Soloway PD, Jaworski DM. Tissue inhibitor of metalloproteinase-2 (TIMP-2) regulates neuromuscular junction development via a β1 integrin-mediated mechanism. J Neurobiol. 2006;66:1365–77. doi: 10.1002/neu.20315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pennington CJ, Edwards DR. Real-time PCR expression profiling of MMPs and TIMPs. Methods Mol Biol. 2010;622:159–73. doi: 10.1007/978-1-60327-299-5_9. [DOI] [PubMed] [Google Scholar]
- 21.Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, Myers MG., Jr STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–9. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- 22.Stutz AM, Staszkiewicz J, Ptitsyn A, Argyropoulos G. Circadian expression of genes regulating food intake. Obesity. 2007;15:607–15. doi: 10.1038/oby.2007.564. [DOI] [PubMed] [Google Scholar]
- 23.Steppan CM, Wang J, Whiteman EL, Birnbaum MJ, Lazar MA. Activation of SOCS-3 by resistin. Mol Cell Biol. 2005;25:1569–75. doi: 10.1128/MCB.25.4.1569-1575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.El-Haschimi K, Pierroz DD, Hileman SM, Bjørbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest. 2000;105:1827–32. doi: 10.1172/JCI9842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ernst MB, Wunderlich CM, Hess S, Paehler M, Mesaros A, Koralov SB, Kleinridders A, Husch A, Münzberg H, Hampel B, Alber J, Kloppenburg P, Bruning JC, Wunderlich FT. Enhanced Stat3 activation in POMC neurons provokes negative feedback inhibition of leptin and insulin signaling in obesity. J Neurosci. 2009;29:11582–93. doi: 10.1523/JNEUROSCI.5712-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Heek M, Mullins DE, Wirth MA, Graziano MP, Fawzi AB, Compton DS, France CF, Hoos LM, Casale RL, Sybertz EJ, Strader CD, Davis HRJ. The relationship of tissue localization, distribution and turnover to feeding after intraperitoneal 125I-leptin administration to ob/ob and db/db mice. Horm Metab Res. 1996;28:653–8. doi: 10.1055/s-2007-979872. [DOI] [PubMed] [Google Scholar]
- 27.Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, Kahn BB. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. 2006;12:917–24. doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- 28.Lluri G, Jaworski DM. Regulation of TIMP-2, MT1-MMP, and MMP-2 expression during C2C12 differentiation. Muscle Nerve. 2005;32:492–9. doi: 10.1002/mus.20383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levin BE. Metabolic imprinting: critical impact of the perinatal environment on the regulation of energy homeostasis. Philos Trans R Soc Lond B Biol Sci. 2006;361:1107–21. doi: 10.1098/rstb.2006.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaworski DM, Boone J, Caterina J, Soloway P, Falls WA. Prepulse inhibition and fear-potentiated startle are altered in tissue inhibitor of metalloproteinase-2 (TIMP-2) knockout mice. Brain Res. 2005;1051:81–9. doi: 10.1016/j.brainres.2005.05.057. [DOI] [PubMed] [Google Scholar]
- 31.Harris RB, Bowen HM, Mitchell TD. Leptin resistance in mice is determined by gender and duration of exposure to high-fat diet. Physiol Behav. 2003;78:543–55. doi: 10.1016/s0031-9384(03)00035-0. [DOI] [PubMed] [Google Scholar]
- 32.Martin TL, Alquier T, Asakura K, Furukawa N, Preitner F, Kahn BB. Diet-induced obesity alters AMP kinase activity in hypothalamus and skeletal muscle. J Biol Chem. 2006;281:18933–41. doi: 10.1074/jbc.M512831200. [DOI] [PubMed] [Google Scholar]
- 33.Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379:632–5. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 34.Munzberg H. Differential leptin access into the brain--a hierarchical organization of hypothalamic leptin target sites? Physiol Behav. 2008;94:664–9. doi: 10.1016/j.physbeh.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 35.Djiane J, Attig L. Role of leptin during perinatal metabolic programming and obesity. J Physiol Pharmacol. 2008;59:55–63. [PubMed] [Google Scholar]
- 36.Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. J Clin Invest. 1996;98:1101–6. doi: 10.1172/JCI118891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dube MG, Kalra SP, Kalra PS. Low abundance of NPY in the hypothalamus can produce hyperphagia and obesity. Peptides. 2007;28:475–9. doi: 10.1016/j.peptides.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haynes WG, Morgan DA, Walsh SA, Mark AL, Sivitz WI. Receptor-mediated regional sympathetic nerve activation by leptin. J Clin Invest. 1997;100:270–8. doi: 10.1172/JCI119532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hwa JJ, Fawzi AB, Graziano MP, Ghibaudi L, Williams P, Van Heek M, Davis H, Rudinski M, Sybertz E, Strader CD. Leptin increases energy expenditure and selectively promotes fat metabolism in ob/ob mice. Am J Physiol. 1997;272:R1204–9. doi: 10.1152/ajpregu.1997.272.4.R1204. [DOI] [PubMed] [Google Scholar]
- 40.Wesolowski SR, Allan MF, Nielsen MK, Pomp D. Evaluation of hypothalamic gene expression in mice divergently selected for heat loss. Physiol Genomics. 2003;13:129–37. doi: 10.1152/physiolgenomics.00184.2002. [DOI] [PubMed] [Google Scholar]
- 41.Jaworski DM, Soloway P, Caterina J, Falls WA. Tissue inhibitor of metalloproteinase-2 (TIMP-2)-deficient mice display motor deficits. J Neurobiol. 2006;66:82–94. doi: 10.1002/neu.20205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gallou-Kabani C, Junien C. Nutritional epigenomics of metabolic syndrome: new perspective against the epidemic. Diabetes. 2005;54:1899–906. doi: 10.2337/diabetes.54.7.1899. [DOI] [PubMed] [Google Scholar]
- 43.Dzwonek J, Rylski M, Kaczmarek L. Matrix metalloproteinases and their endogenous inhibitors in neuronal physiology of the adult brain. FEBS Lett. 2004;567:129–35. doi: 10.1016/j.febslet.2004.03.070. [DOI] [PubMed] [Google Scholar]
- 44.Gerin I, Louis GW, Zhang X, Prestwich TC, Kumar TR, Myers MGJ, Macdougald OA, Nothnick WB. Hyperphagia and obesity in female mice lacking tissue inhibitor of metalloproteinase-1. Endocrinology. 2009;150:1697–704. doi: 10.1210/en.2008-1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maamra M, Bidlingmaier M, Postel-Vinay MC, Wu Z, Strasburger CJ, Ross RJ. Generation of human soluble leptin receptor by proteolytic cleavage of membrane-anchored receptors. Endocrinology. 2001;142:4389–93. doi: 10.1210/endo.142.10.8442. [DOI] [PubMed] [Google Scholar]
- 46.Ogier V, Ziegler O, Mejean L, Nicolas JP, Stricker-Krongrad A. Obesity is associated with decreasing levels of the circulating soluble leptin receptor in humans. Int J Obes Relat Metab Disord. 2002;26:496–503. doi: 10.1038/sj.ijo.0801951. [DOI] [PubMed] [Google Scholar]
- 47.Reizes O, Benoit SC, Clegg DJ. The role of syndecans in the regulation of body weight and synaptic plasticity. Int J Biochem Cell Biol. 2008;40:28–45. doi: 10.1016/j.biocel.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 48.Holmbeck K, Bianco P, Caterina J, Yamada S, Kromer M, Kuznetsov SA, Mankani M, Robey PG, Poole AR, Pidoux I, Ward JM, Birkedal-Hansen H. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 1999;99:81–92. doi: 10.1016/s0092-8674(00)80064-1. [DOI] [PubMed] [Google Scholar]
- 49.Byrne LC, Zhou Z, Tryggvason K, Hökfelt T, Fetissov SO. Altered NPY and AgRP in membrane type-1 matrix metalloproteinase-deficient mice. Neuroreport. 2004;15:569–74. doi: 10.1097/00001756-200403010-00037. [DOI] [PubMed] [Google Scholar]
- 50.Kokoeva MV, Yin H, Flier JS. Evidence for constitutive neural cell proliferation in the adult murine hypothalamus. J Comp Neurol. 2007;505:209–20. doi: 10.1002/cne.21492. [DOI] [PubMed] [Google Scholar]
- 51.Garza JC, Guo M, Zhang W, Lu XY. Leptin increases adult hippocampal neurogenesis in vivo and in vitro. J Biol Chem. 2008;283:18238–47. doi: 10.1074/jbc.M800053200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pérez-Martínez L, Jaworski DM. Tissue inhibitor of metalloproteinase-2 promotes neuronal differentiation by acting as an anti-mitogenic signal. J Neurosci. 2005;25:4917–29. doi: 10.1523/JNEUROSCI.5066-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mannello F, Gazzanelli G. Tissue inhibitors of metalloproteinases and programed cell death: conundrums, controversies and potential implications. Apoptosis. 2001;6:479–82. doi: 10.1023/a:1012493808790. [DOI] [PubMed] [Google Scholar]
- 54.Moraes JC, Coope A, Morari J, Cintra DE, Roman EA, Pauli JR, Romanatto T, Carvalheira JB, Oliveira AL, Saad MJ, Velloso LA. High-fat diet induces apoptosis of hypothalamic neurons. PLoS One. 2009;4:e5045. doi: 10.1371/journal.pone.0005045. [DOI] [PMC free article] [PubMed] [Google Scholar]