

Abstract
Primary cultures of adult rat dorsal root ganglia (DRG) sensory neurons were used to determine whether bradykinin and prostaglandins E1 (PGE1), E2 (PGE2) or I2 (PGI2) stimulate long-term calcitonin gene-related peptide (CGRP) mRNA accumulation and peptide release. Treatment (24 hours) of neurons with either bradykinin or PGE1, significantly increased CGRP mRNA content and iCGRP release. However, PGE2 or PGI2 were without effect. Exposure of the cultured neurons to increasing concentrations of bradykinin or PGE1 demonstrated that the stimulation of CGRP expression was concentration-dependent, while time-course studies showed that maximal levels of CGRP mRNA accumulation and peptide release were maintained for at least 48 hours. Treatment of the neuronal cultures with a bradyknin B2 receptor antagonist significantly inhibited the bradykinin-induced increase in CGRP expression and release. In addition, preincubation of neuronal cultures with the cyclooxygenase inhibitor indomethacin did not alter the PGE1-mediated stimulation of CGRP but blocked completely the bradykinin-induced increase in CGRP production. Therefore, these data indicate that bradykinin and PGE1 can regulate the synthesis and release of CGRP in DRG neurons and that the stimulatory effects of bradykinin on CGRP are mediated by a cyclooxygenase product(s). Thus, these findings suggest a direct relationship between chronic alterations in bradykinin/prostaglandin production that may arise from pathophysiological causes and long-term changes in CGRP expression.
Keywords: neuropeptides, gene expression, inflammation, pain, sensory nervous system, cell signaling
Introduction
Bradykinin (BK) and prostaglandins are critical mediators of the inflammatory process including increased responsiveness to peripheral stimuli (hyperalgesia and allodynia) [1-4]. BK production is enhanced in tissues following activation of kallikrein by pathophysiological stimuli [1-4]. Newly generated BK acts locally near its site of production with effects ranging from alterations in local blood flow, vascular permeability, immune cell stimulation, to the activation of neuronal pathways [1-5]. Most actions of BK are mediated through the BK B2 receptor. Activation of this receptor generates multiple effects, including stimulation of protein kinase C and increases in intracellular calcium. BK also stimulates the production of arachidonic acid and can directly activate phospholipase A2 in many cell types, leading to the production of prostaglandins through increased cyclooxygenase activity. [1-6]. Prostaglandins act upon a family of pharmacologically distinct receptors including EP1, EP2, EP3, EP4 and IP that activate several different G protein-coupled signaling pathways [7]. DRG sensory neurons express several prostanoid receptor subtypes including IP, EP1, EP2, EP3, and EP4 (Moriyama).
Sensory neurons are an important target for BK/prostaglandins and activation of these neurons plays a critical role in mediating various aspects of the inflammatory response [1-3, 6, 8]. The cell bodies for sensory neurons are found in dorsal root ganglia (DRG). The peptidergic neurons synthesize an number of peptides, including calcitonin gene-related peptide (CGRP) and substance P (SP), and extend nerve fibers peripherally to all tissues innervated by the sensory nervous system and centrally to the dorsal horn of the spinal cord [8, 9]. The release of CGRP and SP from afferent nerve terminals in peripheral tissues plays a key role in neurogenic inflammation, while release from terminals in the dorsal horn of the spinal cord modulates pain transmission [8, 9]. Sensory neurons express functional BK2 and prostaglandin receptors and these agents can regulate the activity of this class of neurons [2, 10, 11]. BK and/or prostaglandins markedly stimulate the acute release of neuropeptides from sensory neurons, although there is some question as to whether prostanoids directly enhance CGRP and/or SP release or act indirectly by sensitizing these neurons to other agents [9-11]. A critical question that has not been answered is whether these factors can modulate CGRP biosynthesis in these cells. This is an important issue because significant alterations in the long-term synthesis of CGRP are likely to be related directly to the amount of this neuropeptide that is available for release from sensory nerve terminals, both in peripheral tissues and in the spinal cord. Several lines of indirect evidence suggest that BK and/or prostaglandins can enhance the synthesis of CGRP. In both acute and chronic rat models of arthritis there is a significant increase in the levels of BK and prostaglandins at the site of inflammation [1-6]. This inflammatory response is accompanied by marked increases in CGRP and SP mRNA and their peptides in the lumbar DRG that innervate the arthritic joints [12]. In one such study, treatment of the arthritic rats with anti-inflammatory agents reduced DRG iCGRP levels to near normal [13].
Previous studies in our laboratory employing cultured adult rat DRG neurons showed that activation of the protein kinase A and C signal transduction pathways, which have been implicated in mediating the responses of sensory neurons to prostaglandins and BK, increased both CGRP mRNA accumulation, as well as iCGRP release [14]. Taken together, these studies suggest that BK can either directly enhance the neuronal biosynthesis of CGRP and/or acts indirectly through the stimulation of prostaglandin synthesis. Therefore, to examine this question, we utilized primary cultures of adult rat DRG neurons to investigate the effects of BK and prostaglandins PGE1, PGE2, and PGI2 on CGRP mRNA accumulation and iCGRP release.
Methods and Materials
Materials
BK (arg-pro-pro-gly-phe-ser-pro-phe-arg), BKp (pglu-gly-leu-pro-pro-arg-pro-lys-ile-pro-pro), an inhibitor of angiotensin-converting enzyme and BK-destroying plasma kininases, PGE1, PGE2, PGI2, indomethacin, ibuprofen, and Hoe 140, a BK B2 receptor antagonist, were obtained from Sigma (St. Louis, MO, USA). Nerve growth factor (NGF) was obtained from Harlan Scientific (Indianapolis, IN, USA).
Cell Culture
DRG neurons were prepared following a modified protocol initially described by Lindsay et al [15]. DRG (cervical, thoracic and lumbar, 40-45/animal) were dissected from male Sprague-Dawley rats (125-150 g) and collected in Ham's F-12 medium supplemented with 10% horse serum (growth medium). Ganglia were dissociated in 0.125% collagenase with a constant flow of 5% CO2/95% oxygen, washed, and then treated with 0.25% trypsin. After another wash, the ganglia were transferred to growth medium containing DNase (80 μg/ml) and soybean trypsin inhibitor (100 μg/ml). Single cell suspensions were obtained by trituration of enzymatically treated ganglia. The dissociated neurons were plated in six-well culture dishes coated with polyornithine and maintained in growth medium at 37° C in 5% CO2. After 24 hours, the cells were placed under serum free conditions in Ham's F-12 supplemented with insulin (5 μg/ml), transferrin (100 μg/ml), progesterone (20 nM), selenium (30 nM) and putrescine (100 μM, N2 Supplement, Gibco-BRL). The yield of neurons was approximately 1.5 to 2.0 × 105 from 40-45 ganglia. The dissociated DRG cells were plated at a density of 10,000 to 20,000 neurons per well and were incubated at 37° C in a 5% C02, 95% air atmosphere.
Hybridization probes, RNA isolation and analysis, radioimmunoassay
The α-CGRP hybridization probe was a 1.4 kb Sau 3A rat genomic restriction fragment containing CGRP exons 5 and 6 [16]. The 18S rRNA hybridization probe was a 1.15 kb BamHI-EcoRI restriction fragment from the mouse 18S rDNA gene [17]. The DNA inserts were purified by agarose-gel electrophoresis and subsequently labeled with [α-32P]dCTP using a random hexanucleotide DNA labeling kit (Amersham, Piscataway, NJ, USA). After dissociation and plating (48 hours) the cultured cells were treated with the regulatory agents as indicated. Total cellular RNA was isolated by the guanidine-isothiocynate method and analyzed by Northern blot hybridization. The nylon membranes were initially hybridized with the 32P-labeled CGRP DNA probe. As a control, the CGRP probe was removed from the membrane that was then rehybridized with the 18S rDNA probe. The membranes were then washed and exposed to X-ray film at -70° C with an intensifying screen and relative levels of CGRP mRNA and 18S rRNA were quantified by computerized scanning laser densitometry. To quantify iCGRP levels in the medium we employed a commercially available rabbit anti-rat CGRP radioimmunoassay kit (Phoenix Laboratories, Belmont, CA, USA). All assays were performed under conditions recommended by the supplier. The total protein content in each sample was determined by the Bradford method (Bio-Rad, Hercules, CA, USA).
Statistical analysis
Statistical significance was determined by analysis of variance followed by the Tukery-Kramer multiple comparisons test. The acceptable level of significance was p<0.05. Data in the figures are depicted as the mean ± SEM (standard error of mean).
Results
Bradykinin and prostaglandin E1 stimulate CGRP mRNA accumulation and iCGRP release
As a first step in establishing that kinins or specific prostaglandins can stimulate CGRP mRNA production, cultured DRG neurons were treated for 24 hours with either BK (1 μM), PGE1 (1 μM), or a combination of both. DRG neurons were also treated with PGE2 (1 μM). The results of one such experiment are shown in figure 1. All of the lanes displayed a 1.2 kb band, which corresponds to CGRP mRNA (both α- and β- CGRP mRNA species), as well as the 18S rRNA hybridization signal. To quantify these results, densitometric analysis of autoradiographs from several experiments was performed. The ratios of CGRP mRNA to 18S rRNA in treated versus control cells were calculated and are represented as fold-increases over control. As shown in figure 2A, BK or PGE1 treatment significantly increased CGRP mRNA content 2.4±0.1- and 2.4±0.3-fold over control levels, respectively, while the combination of the two agents stimulated CGRP mRNA accumulation 3.8±0.4-fold. Moreover, this increase in CGRP mRNA produced by the combination of BK and PGE1 was statistically significant when compared to the increases induced by each agent alone. Treatment of the neurons with PGE2 or PGI2 did not significantly change CGRP expression and release.
Figure 1.
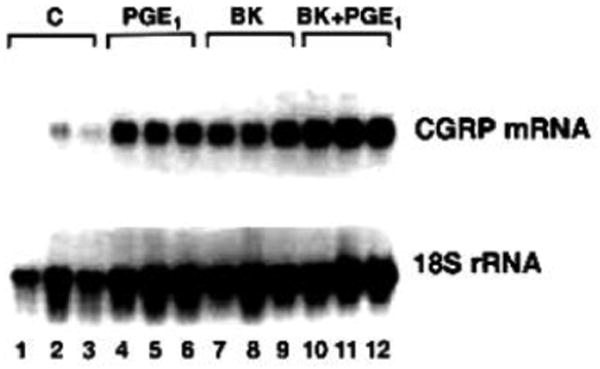
Northern blot analysis of CGRP mRNA isolated from primary cultures of DRG neurons. Following dissociation and plating (48 hours), cultured DRG neurons were treated for 24 hours with BK (1 μM) or PGE1 (1 μM) or a combination of both factors. The kininase inhibitor (1 μM) was included with the BK. Control cells were treated with vehicle. Total cellular RNA samples were fractionated on denaturing formaldehyde-agarose gels and transferred to a nylon membrane. The membrane was hybridized with the 32P-labeled CGRP genomic DNA insert (top) and subjected to autoradiography. The membrane was subsequently hybridized with the 32P-labeled 18S rDNA probe (bottom). RNA samples were: lanes 1-3, control; lanes 4-6, PGE1; lanes 7-9, BK; lanes 10-12 BK plus PGE1.
Figure 2.
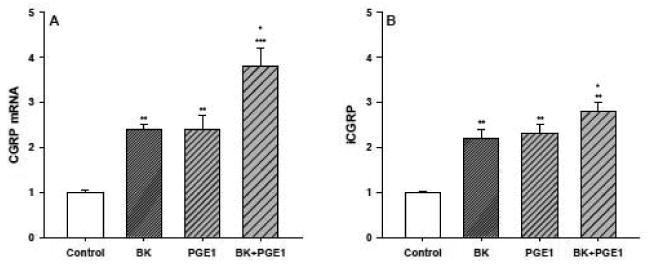
BK and PGE1 stimulate CGRP mRNA accumulation and iCGRP release in primary cultures of DRG neurons. (A) Densitometric analysis of the Northern blotting hybridization assays. The CGRP mRNA/18S ratios from the BK, PGE1, BK + PGE1 treated (24 hours) or control DRG neurons were determined by computer assisted laser densitometry and are shown here as fold-induction over control (n=6-10 in duplicate or triplicate for each treatment). * p < 0.05, BK + PGE1 vs BK and BK + PGE1 vs PGE1; ** p < 0.01, BK vs control and PGE1 vs control; *** p < 0.001, BK + PGE1 vs control. (B) Radioimmunoassay of iCGRP. Medium from the BK, PGE1, BK + PGE1 treated or control DRG neurons described above was tested to determine iCGRP levels. Values for iCGRP were in pg iCGRP/g total protein/0.1 ml medium and are shown here as fold-induction over control. * p < 0.05, BK + PGE1 vs BK and BK + PGE1 vs PGE1; ** p < 0.01, BK vs control, PGE1 vs control, and BK + PGE1 vs control.
In conjunction with the Northern hybridization experiments, we also used a CGRP-specific radioimmunoassay to determine iCGRP levels in the medium from the treated and control cultures of DRG neurons (Figure 2B). As anticipated, exposure of the neuronal cultures to either BK or PGE1 significantly enhanced the release of iCGRP 2.2±0.2- and 2.3±0.2-fold, respectively. Treatment of the cultures with a combination of BK and PGE1 produced a 2.8±0.2-fold increase in iCGRP in the medium that again was significantly greater than the increases observed with either agent alone. For all the experiments where BK was used, the kininase inhibitor described in the methods section was included at a concentration of 10-6 M. Even though the BK alone was able to stimulate CGRP expression to a similar extent, the inclusion of the kininase inhibitor reduced the variability and increased the reproducibility of the experiments. In addition, the kininase inhibitor, by itself, had no effect on either CGRP mRNA or iCGRP levels (unpublished observations).
Bradykinin and prostaglandin E1 enhance CGRP expression and release in a concentration- and time-dependent manner
To investigate further the effect of BK and PGE1 on CGRP and mRNA levels and iCGRP release, cultures of DRG neurons were treated for 24 hours with increasing concentrations of each agent. Exposing the neuronal cultures to 0.1 μM BK produced a significant 1.6±0.1-fold increase in CGRP mRNA content (Figure 3A). A similar 1.5±0.2-fold increase in iCGRP levels was also observed, but this did not achieve statistical significance. We observed a further increase in both CGRP mRNA (2.2±0.1-fold, p < 0.01) and iCGRP (1.9±0.2-fold, p < 0.05) at 0.5 μM BK, and 1.0 μM BK produced a 2.6±0.3-fold stimulation of CGRP mRNA without an additional increase in iCGRP release. Treatment of the neuronal cultures with 5 and 10 μM BK did not stimulate any additional elevation of CGRP mRNA or iCGRP levels.
Figure 3.
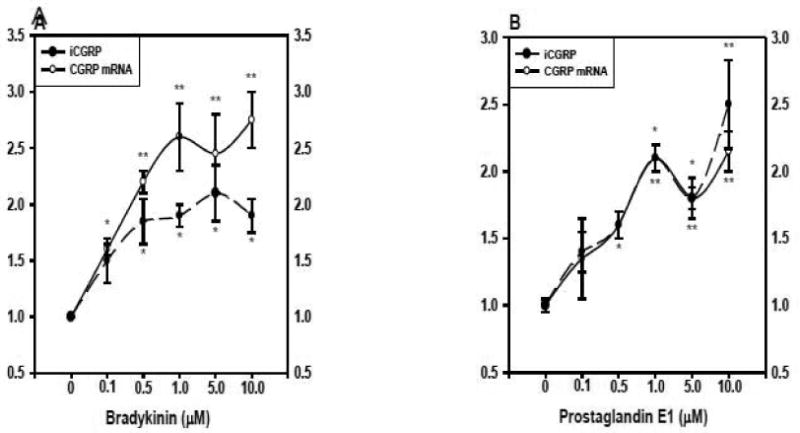
BK and PGE1 enhance CGRP expression in a concentration-dependent manner. After dissociation and plating (48 hours), cultures of DRG neurons (n=3 in triplicate/dose) were exposed to increasing concentrations of either BK (panel A) or PGE1 (panel B). The kininase inhibitor (1 μM) was added with the BK. Control cells were treated with vehicle only. RNA samples were analyzed as described in Figure 1. iCGRP levels in the medium (pg iCGRP/μg protein/0.1 ml) were quantified by radioimmunoassay. All values are shown here as fold induction over the controls. The circles and solid line represent CGRP mRNA content, whereas the triangles and dashed line represent iCGRP levels. * p < 0.05, indicated dose vs control; ** p < 0.01, indicated dose vs control.
A similar experimental protocol was used to test the concentration-dependent affects of PGE1 on neuronal CGRP expression (Figure 3B). When neuronal cultures were exposed to 0.1 μM PGE1 there was a nonsignificant stimulation of both CGRP mRNA and peptide release. The increase in CGRP mRNA (1.6±0.1-fold), but not iCGRP, achieved statistical significance at 0.5 μM PGE1, while both parameters were significantly elevated at 1.0 μM PGE1 (CGRP mRNA, 2.1±0.1-fold, p < 0.01; iCGRP, 2.0±0.1-fold, p < 0.05). However, at the two highest concentrations of PGE1 (5 and 10 μM), there were no further increases in either CGRP mRNA content or peptide release.
An experiment depicting the temporal expression of both CGRP mRNA and iCGRP is shown in figure 4. Basal levels of CGRP mRNA content and iCGRP release were not significantly changed at any of the time points studied (not shown). Treatment of the neurons with a single dose of BK (1 μM) produced relatively small, although not statistically significant increases in CGRP mRNA content and iCGRP release at 4 and 12 h (Figure 4A). By 24 hours, both parameters were significantly elevated (CGRP mRNA, 1.9±0.2-fold; iCGRP, 2.0±0.2-fold), while no further increases were observed at the 36- and 48-hour time points. Similarly, exposure of the neuronal cultures to a single dose of PGE1 (1 μM) caused an approximately 1.5-fold increase in CGRP mRNA and iCGRP levels at both the 4- and 12-hour time points (Figure 4B). By 24 hours, CGRP mRNA content was stimulated 1.9±0.1-fold and iCGRP release 1.6±1-fold. Again, CGRP mRNA and iCGRP remained elevated after 36 and 48 hours of incubation.
Figure 4.
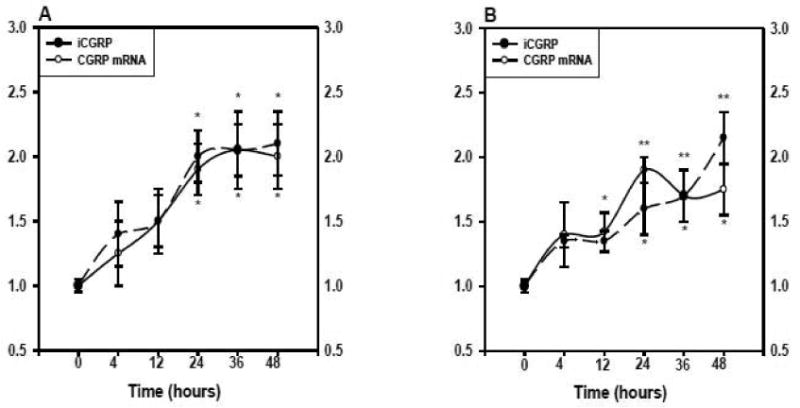
Time-dependent expression of CGRP in response to BK (panel A) or PGE1 (panel B). Cultured DRG neurons were treated for the indicated times (n=3 in triplicate/time point) with either BK (1 μM) or PGE1 (1 μM). The kininase inhibitor (1 μM) was added with the BK. Control cells for each time point were treated with vehicle only. The CGRP mRNA to 18S rRNA ratios and iCGRP levels in the medium were determined as described previously and are expressed as fold-induction over controls. The circles and solid line represent CGRP mRNA content, whereas the triangles and dashed line represent iCGRP levels. * p < 0.05, indicated time point vs control; ** p < 0.01, indicated time point vs control.
Indomethacin blocks the BK-stimulated increase in CGRP mRNA accumulation and iCGRP release
To assess the effects of the cyclooxygenase inhibitor indomethacin on CGRP expression DRG neurons were treated with BK or PGE1, with or without indomethacin pretreatment (30 μM for 30 minutes). The indomethacin was then left in the culture medium for the duration of the incubation. As shown in figure 5A, in the absence of indomethacin pretreatment exposure of the DRG neurons to BK (1 μM) or PGE1 (1 μM) for 24 hours resulted in 2.6±0.2- and 2.6±0.3-fold increases in CGRP mRNA content, respectively. Indomethacin on its own was without effect on CGRP mRNA production; however, it completely blocked the BK-stimulated increase in this mRNA species. The inhibition of the BK-induced increase in CGRP mRNA by indomethacin could be overcome by treating the neuronal cultures with PGE1 (2.3±0.2-fold increase). Similar results were obtained when iCGRP levels in the medium from the treated and control neuronal cultures described above were determined (1.8±0.2-fold increase). Without indomethacin pretreatment, BK and PGE1 stimulated 2.2±0.1 and 2.3±0.1-fold increases in peptide release. Incubation of the DRG cultures with indomethacin, by itself, did not alter iCGRP release, but it completely inhibited the BK-stimulated increase in peptide levels. Addition of exogenous PGE1 to the indomethacin treated cells was able to increase iCGRP release (1.8±0.3-fold) to the levels observed when the neuronal cultures were treated with BK or PGE1 alone. Virtually identical results were obtained when a second cyclooxygenase inhibitor, ibuprophen, was used instead of indomethacin (not shown).
Figure 5.
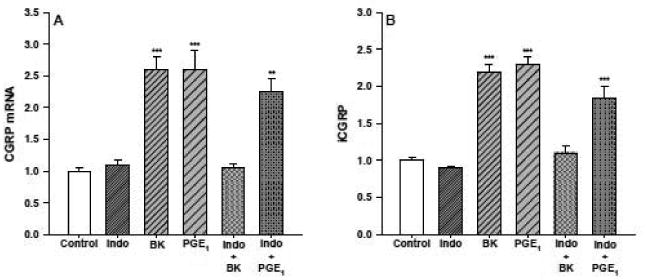
Indomethacin blocks the BK-stimulated increase in CGRP mRNA accumulation (panel A) and iCGRP release (panel B). Cultures of DRG neurons were treated for 24 hours with BK (1 μM), or PGE1 (1 μM, n=5 in triplicate/treatment). When indomethacin was used with either BK or PGE1 the neuronal cultures were pretreated with the cylooxygenase inhibitor (30 μM) 30 minutes prior to the addition of either BK or PGE1. The kininase inhibitor (1 μM) was added with the BK. Control cells were treated with vehicle alone. CGRP mRNA content and iCGRP levels in the medium were determined as described previously and the values obtained are expressed here as fold-induction over control. ** p < 0.01, BK, PGE1, and BK + Indo vs. control, Indo, and BK+ Indo.
The effects of BK on CGRP expression and release are mediated by the BK B2 receptor
To determine the effects of the BK B2 receptor antagonist Hoe 140 on CGRP expression DRG neurons were treated with BK, with or without Hoe 140 pretreatment (10 μM for 30 minutes). The antagonist was then left in the couture medium for the duration of the incubation. As shown in figure 6, in the absence of Hoe 140 pretreatment addition of BK (1 μM) for 24 hours produced 1.8±0.2 increases in CGRP mRNA content and iCGRP release. By itself, Hoe 140 treatment did not significantly change CGRP expression, however, it completely blocked the BK-stimulated increases in CGRP mRNA accumulation and iCGRP release.
Figure 6.
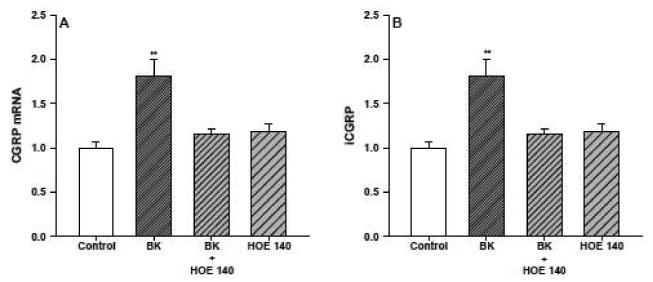
Hoe 140 blocks the BK-stimulated increase in CGRP mRNA accumulation (panel A) and iCGRP release (panel B). Neuronal cultures were treated for 24 hours with either Hoe 140 (10 μM) alone, BK (1 μM) alone, or a combination of BK and Hoe 140 (n=3 in triplicate). The BK2 receptor antagonist was added 30 minutes before the BK and was maintained in the medium throughout the incubation. Control cells were treated with vehicle. CGRP mRNA content and iCGRP levels in the medium were determined as described previously and the values are expressed as fold-induction over control. * p < 0.05, BK vs. control, BK + Hoe 140, Hoe 140.
Discussion
There are two new major findings of this study. First, BK and PGE1, but not PGE2 or PGI2, significantly enhance long-term (48 hr) CGRP mRNA accumulation and iCGRP release in primary cultures of adult rat DRG neurons. Second, the effect of BK on CGRP expression appears to be entirely mediated by the BK2 receptor via the synthesis of a cyclooxygenase product, presumably PGE1 which was able to completely reverse the effect of indomethacin on the BK-induced increase in CGRP mRNA and iCGRP levels. However, we cannot rule out that this effect was mediated by PGD2 or PGF2-alpha since these were not tested. Although we did not employ prostaglandin receptor antagonists in this study, based on the work from other investigators it is likely that PGE1 is acting through the EP2 and/or EP4 receptors which are coupled to the Gs protein and activate adenylate cyclase, increasing cAMP levels and signaling through the protein kinase A pathway, thereby increasing CGRP expression and release [18-20]. This is also consistent with our previous studies demonstrating that the adenylate cyclase activator, forskolin, and cAMP analogs significantly enhance long term CGRP expression and release in primary cultures of adult rat sensory neurons [14]. In regards to the source of the BK-enhanced liberation of arachadonic acid with the formation and release of prostaglandins, there is compelling evidence that numerous types of cells capable of synthesizing eicosanoids exist in tissues, including sensory neurons themselves [1-6, 19]. Therefore, in the DRG primary culture system used for these studies, the source of arachidonic acid metabolites in response to BK treatment are the sensory neurons, as well as the fibroblasts and glial cells present in these preparations.
The combined effect of BK and PGE1 on CGRP mRNA levels and CGRP release was higher than the effect of either agent alone, but smaller than the sum of the individual effects. This observation was unexpected since the concentration of PGE1 used (1 μM) is higher than the concentrations required for prostanoids to fully occupy and activate their receptors [10, 11, 20]. Therefore, the PGE1-induced increase in CGRP expression and release should be maximal and cannot be further enhanced. If this is true, than the potentiation of CGRP expression produced by the addition of BK cannot be mediated by prostaglandins. However, the experiments with the cyclooxygenase inhibitors indicate that the effects of BK are completely mediated by the prostaglandin pathway. Taken together, this data is conflicting. One possible explanation is that this result is a cyclooxygenase inhibitor artifact produced by the nonspecific inhibition of a prostaglandin-independent BK signaling pathway that stimulates CGRP expression and release. We do have indirect evidence that this is not the case. As a control for the specificity of indomethacin, we showed that this drug does not inhibit the stimulatory effects of nerve growth factor, an agent that does not involve the cyclooxygenase-dependent stimulation of prostaglandin synthesis, on CGRP expression and release (unpublished observations). Alternatively, BK could either directly or indirectly increase the abundance and/or efficacy of the relevant prostaglandin receptor(s) thereby increasing the magnitude of the PGE1 effects on CGRP. It has been reported that the prostaglandin receptors EP2 and EP4 are up-regulated in mast cells and joints in a murine model of arthritis/lupus [21]. Whether this occurs in DRG sensory neurons following BK treatment is not known.
The complete blockade of the BK effect by Hoe 140 indicates that during the long exposure time of the DRG neurons there was no detectable induction of the BK B1 receptor. This finding is supported by a report demonstrating that treatment of cultured rat DRG neurons with BK for 3 hours significantly up-regulated COX-2 expression [22]. This effect was blocked by the BK B2 antagonist Hoe 140 but not by a BK B1 receptor antagonist. This report also indicates that the long-term BK-induced up-regulation of prostaglandin generation is mediated by both COX-1 and COX-2 although this question was not assessed in our study.
The findings presented herein are generally consistent with a number of studies that examined the BK and/or prostanoid-mediated acute release (usually 5-10 minutes) of CGRP (and SP) in various sensory nerve preparations. For example, using spinal cord slices with attached dorsal roots, Andreeva and Rang [10], demonstrated that BK (0.1-10 μM) enhanced 2-3-fold the acute CGRP release evoked by dorsal root stimulation. This effect was blocked by the BK B2 receptor antagonist Hoe 140 and by the cyclooxygenase inhibitor indomethacin. In addition, the adenylate cyclase activator forskolin mimicked the effect of BK on iCGRP release, as did various prostanoids, having an approximate order of potency of PGE1 = PGD2 > PDE2= PGE2. PGI2 (10 μM) was without effect. It was concluded from these studies that BK activates BK B2 receptors on afferent nerve terminals and other tissues, causing the formation of prostaglandins, which in turn enhance iCGRP release most likely through the stimulation of adenylate cyclase.
Somewhat different results were obtained from experiments using cultured embryonic rat DRG neurons [11]. As expected, exposure of the neuronal cultures to BK (10 nM-1 μM) markedly increased acute (10 minutes) CGRP and SP release in a concentration-dependent manner. In contrast, PGE2 alone did not significantly alter CGRP or SP release, however, this prostanoid augmented the release of these peptides evoked by BK. Pre-exposing the cultures to indomethacin attenuated, but did not abolish the BK-stimulated release of CGRP and SP. It is interesting that the effects of BK on sensory nerve function appear to be similar in a number of different studies, whereas there is more variability in response to PG treatment. While we do not know the mechanisms involved, it may be that the response of sensory nerves to prostanoids may vary depending on the type of sensory nerve preparation used, the method of tissue preparation, and the widely varied experimental conditions employed in these studies. In a study employing primary cultures of adult rat DRG neurons, it was reported that the activation of adenylate cyclase by PGE1, PGE2, and iloprost (a prostaglandin I2 receptor [IP] agonist) were highly dependent on DRG cell density, illustrating the difficulty of directly comparing studies performed under widely varying conditions [3, 11].
In a related study, Southall and Vasko [20] proposed that the biological actions of PGE2 in sensory nerves were mediated by the prostaglandin E receptor (EP) subtypes that have been linked to an increase in cAMP (EP2, EP3C and/or EP4). Experiments performed in cultured rat trigeminal neurons by other investigators indicated that activation of prostaglandin D (DP), EP, and IP receptors, all of which have been shown to be associated with increased cAMP levels, markedly enhanced acute CGRP release [11]. Therefore, it is very likely that the long-term BK/PG-evoked increase in CGRP expression in sensory neurons observed in our studies is mediated by one or more class or subtype of prostanoid receptors that is coupled to adenylate cyclase and requires activation of the protein kinase A signaling pathway.
Although one must be cautious when extrapolating data obtained in vitro to an in vivo setting, these observations provide support for the argument that BK and certain prostanoids may be responsible, at least in part, for the long-term up-regulation of CGRP in affected sensory neurons during certain chronic states of inflammation. While some questions remain concerning the precise role of CGRP in neurogenic inflammation and pain transmission, there is considerable evidence that CGRP is involved in both spinal and peripheral nociceptive signal pathways during both acute and chronic inflammation [3, 4]. In inflammatory states, it is known that many sensory C-fibers that innervate inflamed tissues become spontaneously active and that this peripherally-derived sensory nerve stimulation may play an important role in maintaining certain central sensitization states. Indeed, our results suggest that increased BK and prostaglandin production and release at sites of tissue injury could enhance the synthesis of CGRP (and possibly SP) from sensory neurons. The increase in neuropeptide production and presumably release from central sensory nerve terminals could augment pain signaling and nociceptive behaviors, whereas increased synthesis and release from the peripheral endings could exacerbate the inflammatory response [1-6, 10, 11].
In summary, we have used primary cultures of adult DRG neurons to show, for the first time, that BK and PGE1 can significantly increase CGRP mRNA accumulation and peptide release on a long-term (48 hr) basis. Furthermore, these data indicate that the effects of BK on CGRP expression are mediated by a cyclooxygenase product. Thus, these results are suggestive of a direct relationship between chronic alterations in BK/prostaglandin production that arise from pathophysiological or therapeutic reasons, and a long-term change in sensory nerve CGRP expression. Chronic changes in the synthesis and release of this sensory neuropeptide have important physiological consequences that relate to inflammatory states and sensory neurotransmission.
Acknowledgments
These studies were supported by National Institutes of Health grant HL067472
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Scott C. Supowit, Email: scott.supowit@uscmed.sc.edu, University of South Carolina School of Medicine, Department of Cell Biology and Anatomy, 6439 Garners Ferry Road, Bldg. 1, C46, Columbia, South Carolina 29208, Phone: (803) 733-1511.
Huawei Zhao, Email: Huawei_Zhao@merck.com, Merck, Department of Hypertension, 126 East Lincoln Ave., Rahway, New Jersey 07065, Phone: (732) 594-3056.
Khurshed A. Katki, Email: KKATKI@swmail.sw.org, Texas A&M University College of Medicine, Department of Medicine, Cardiovascular Research Institute, Temple, Texas 76504, Phone: (512) 921-4730.
Prakash Gupta, Email: prakash.gupta@uscmed.sc.edu, University of South Carolina School of Medicine, Department of Cell Biology and Anatomy, 6439 Garners Ferry Road, Bldg. 1, C46, Columbia, South Carolina 29208, Phone: (803) 733-1511.
Donald J. DiPette, Email: Donald.dipette@uscmed.sc.edu, University of South Carolina School of Medicine, Department of Medicine, 3 Medical Park, Suite 220, Columbia, South Carolina 29208, Phone: (803) 545-5215.
References
- 1.Couture R, Harrisson M, Vianna RM, Cloutier F. Kinin receptors in pain and inflammation. Eur J Pharmacol. 2001;429:161–176. doi: 10.1016/s0014-2999(01)01318-8. [DOI] [PubMed] [Google Scholar]
- 2.Dray A, Perkins M. Bradykinin and inflammatory pain. Trends Neurosci. 1993;16:99–104. doi: 10.1016/0166-2236(93)90133-7. [DOI] [PubMed] [Google Scholar]
- 3.Geppetti P. Sensory neuropeptide release by bradykinin: mechanisms and pathophysiological implications. Regul Pept. 1993;47:1–23. doi: 10.1016/0167-0115(93)90268-d. [DOI] [PubMed] [Google Scholar]
- 4.Cuello AC. Peptides as neuromodulators in primary sensory neurons. Neuropharmocology. 1987;26:971–979. doi: 10.1016/0028-3908(87)90075-x. [DOI] [PubMed] [Google Scholar]
- 5.Kumazawa T, Mizumura K, Minagawa M, Tsujii Y. Sensitizing effects of bradykinin on the heat responses of the visceral nociceptor. J Neurophysiol. 1991;66:1819–1824. doi: 10.1152/jn.1991.66.6.1819. [DOI] [PubMed] [Google Scholar]
- 6.Narumiya S, FitzGerald GA. Genetic and pharmacological analysis of prostanoid receptor function. J Clin Invest. 2001;108:25–30. doi: 10.1172/JCI13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moriyama T, Higashi T, Togashi K, Iida T, Segi E, Sugimoto T, Tominga T, Narumiya S, Tomonga M. Sensitization of TRPV1 by EP1 and IP reveals nociceptive mechanism of prostaglandins. Molecular Pain. 2005;1:3, 1–13. doi: 10.1186/1744-8069-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marti E, Gibson SJ, Polak JM, Facer P, Springall DR, Van Aswegan G, Aitchison M, Koltzenberg M. Ontogeny of peptide-and-amine-containing neurons in motor, sensory, and autonomic regions of rat and human spinal cord, dorsal root ganglia, and rat skin. J Comp Neurol. 1987;266:332–359. doi: 10.1002/cne.902660304. [DOI] [PubMed] [Google Scholar]
- 9.Holzer P. Local effector functions of capsaicin-sensitive nerve endings: involvement of tachykinins, calcitonin gene-related peptide and other neuropeptides. Neuroscience. 1988;24:739–768. doi: 10.1016/0306-4522(88)90064-4. [DOI] [PubMed] [Google Scholar]
- 10.Andreeva L, Rang HP. Effect of bradykinin and prostaglandins on the release of calcitonin gene-related peptide-like immunoreactivity from the rat spinal cord in vitro. Br J Pharmacol. 1993;108:185–190. doi: 10.1111/j.1476-5381.1993.tb13460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hingtgen CM, Waite KJ, Vasko MR. Prostaglandins facilitate the peptide release from rat sensory neurons by activating the adenosine 3′, 5′-cyclic monophosphate transduction cascade. J Neurosci. 1995;15:5411–5419. doi: 10.1523/JNEUROSCI.15-07-05411.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grubb BD, Birrell GJ, McQueen DS, Iggo A. The role of PGE2 in the sensitization of mechanoreceptors in normal and inflamed ankle joints of the rat. Exp Brain Res. 1991;84:383–392. doi: 10.1007/BF00231460. [DOI] [PubMed] [Google Scholar]
- 13.Hong D, Byers MR, Oswald RJ. Dexamethasone treatment reduces sensory neuropeptides and nerve sprouting reactions in injured teeth. Pain. 1993;55:171–18. doi: 10.1016/0304-3959(93)90146-G. [DOI] [PubMed] [Google Scholar]
- 14.Supowit SC, Christensen MD, Westlund KN, Hallman DM, DiPette DJ. Dexamethasone and activators of the protein kinase A and C signal transduction pathways regulate neuronal calcitonin gene-related peptide expression and release. Brain Res. 1995;686:77–86. doi: 10.1016/0006-8993(95)00461-x. [DOI] [PubMed] [Google Scholar]
- 15.Lindsay RM, Lockett C, Sternberg J, Winter J. Neuropeptide expression in cultures of adult sensory neurons: modulation of substance P and calcitonin gene-related peptide levels by nerve growth factor. Neuroscience. 1989;33:53–65. doi: 10.1016/0306-4522(89)90310-2. [DOI] [PubMed] [Google Scholar]
- 16.Amara SG, Jonas V, Rosenfeld MG, Ong ES, Evans RM. Alternative RNA processing in calcitonin gene expression generates mRNAs encoding different polypeptide products. Nature. 1982;298:240–244. doi: 10.1038/298240a0. [DOI] [PubMed] [Google Scholar]
- 17.Bowman LH, Rabin B, Schlessinger D. Multiple ribosomal RNA cleavage pathways in mammalian cells. Nucleic Acids Res. 1981;9:4951–4966. doi: 10.1093/nar/9.19.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Narumiya S. Prostanoids and inflammation: a new concept arising from receptor knockout mice. J Mol Med. 2009;87(10):1015–1022. doi: 10.1007/s00109-009-0500-1. [DOI] [PubMed] [Google Scholar]
- 19.Rowlands DK, Kao C, Wise H. Regulation of prostacyclin and prostaglandin E(2) receptor mediated responses in adult rat dorsal root ganglion cells, in vitro. Br J Pharmacol. 2001;133:13–22. doi: 10.1038/sj.bjp.0704028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Southall MD, Vasko MR. Prostaglandin receptor subtypes, EP3C and EP4, mediate the prostaglandin E2-induced cAMP production and sensitization of sensory neurons. J Biol Chem. 2001;276:16083–16091. doi: 10.1074/jbc.M011408200. [DOI] [PubMed] [Google Scholar]
- 21.Akaogi J, Yamada H, Kuroda Y, Nacionales D, Reeves W, Satoh M. Prostaglandin E2 receptors EP2 and EP4 are up-regulated in peritoneal macrophages and joints of pristine-treated mice and modulate TNF-α and IL-6 production. J Leukocyte Biol. 2004;76:227–236. doi: 10.1189/jlb.1203627. [DOI] [PubMed] [Google Scholar]
- 22.Atsuko I, Iwasa M, Nishikura Y, Ogawa S, Naksuka A, Nakata Y. The long-term exposure of rat cultured dorsal root ganglion cells to bradykinin induced the release of prostaglandin E2 by the activation of cyclooxygenase-2. Neurosci Lett. 2006;401:242–247. doi: 10.1016/j.neulet.2006.03.026. [DOI] [PubMed] [Google Scholar]