

Abstract
A variety of cytokines have been detected in inflamed intestinal mucosal tissues, including the pro-inflammatory cytokine, interleukin-1 (IL-1), along with growth factors involved in wound healing processes such as proliferation and cell migration. However, little is known about how IL-1 and growth factors interact with intestinal epithelial cells to regulate the production of inflammatory cytokines such as interleukin-8 (IL-8). Previously, we have shown that hepatocyte growth factor (HGF) could significantly enhance IL-1-stimulated IL-8 secretion by the Caco-2 colonic epithelial cell line, yet HGF, by itself, did not stimulate IL-8 secretion. In this report, a second growth factor, keratinocyte growth factor (KGF), was also found to significantly enhance IL-1-induced IL-8 secretion by Caco-2 cells, yet KGF, by itself, also had no effect. Simultaneous addition of both IL-1 and KGF was also required for the enhancing effect. Treatment of the Caco-2 cells with wortmannin or triciribine suppressed the enhancing effect of HGF, suggesting that the effect was mediated by signaling through phosphatidylinositol-3-kinase (PI3K) and the kinase AKT. The enhancing effect of KGF was not affected by wortmannin, but was suppressed by triciribine, suggesting that the effect of KGF was through a PI3K-independent activation of AKT. These results suggest that the growth factors HGF and KGF may play a role in enhancing IL-1-stimulated production of IL-8 by epithelial cells during mucosal inflammations. However, the mechanism by which the growth factors enhance the IL-1 response may be through different initial signaling pathways.
Keywords: AKT, Colon, HGF, IL-1, IL-8, Inflammation, Intestine, KGF, PI3K
Introduction
The intestinal epithelium represents a complex and incompletely understood environment. Multiple levels of interaction between the epithelium, immunological cells, and signaling factors form a network responsible for regulating the inflammatory and wound healing processes. During an inflammatory response, activation of resident and infiltrating inflammatory cells leads to the production of several cytokines and chemokines. One of the main cytokines produced during this response is interleukin-1 (IL-1; Dinarello 2009). IL-1 stimulation of intestinal epithelial cells (IEC) results in the production of several cytokines, including the chemokine interleukin-8 (IL-8; Eckmann et al. 1993; Gross et al. 1995). As a pro-inflammatory cytokine, IL-8 provides a chemotaxic gradient for neutrophil and macrophage migration and activation (Zimmerman et al. 2008). In patients with inflammatory bowel disease (IBD), levels of IL-1 (Ligumsky et al 1990), IL-8, and other chemokines (Zimmerman et al. 2008) are significantly up-regulated. Furthermore, research implicates this increased expression of cytokines and chemokines as one of the key factors responsible for the widespread inflammation and resulting damage that is a hallmark of IBD (Laukoetter et al. 2008).
Reports also indicate that there is a significant increase in the levels of certain growth factors in many IBD patients, including keratinocyte growth factor (KGF; Brauchle et al. 1996; Finch and Cheng 1999) and hepatocyte growth factor (HGF) (Kitamura et al. 2000). In fact, studies have shown that IL-1 may act on fibroblasts to enhance the production of both HGF (Tamura et al. 1993) and KGF (Finch and Cheng 1999). However, little is known of how HGF or KGF may affect the expression of inflammatory cytokines by IECs.
Hepatocyte growth factor (also known as scatter factor) is a heparin-binding glycoprotein of the plasminogen-related growth factor family (Ido et al. 2005). Expressed primarily in an inactive pro-HGF monomeric form, cleavage of pro-HGF by proteolytic digestion yields the mature heterodimer, which is a potent mitogen for epithelial cells, including those of the gastrointestinal tract (Ido et al. 2005). It has also been well-established that subepithelial myofibroblasts and stromal cells of the intestinal basal lamina can provide a significant source of HGF (Goke et al. 1998).
Keratinocyte growth factor (also known as fibroblast growth factor 7) is a member of the heparin-binding fibroblast growth factor family (Rubin et al. 1995). This factor is known to be a potent mitogen (Housley et al. 1994) and has been implicated as an important component of wound healing as in IBD (Zeeh et al. 1996; Greenwood-Van Meerveld et al. 2003). In the intestine of IBD patients, KGF production by mesenchymal cells in the mucosa was significantly up-regulated over that in undamaged tissue (Brauchle et al. 1996). These mesenchymal cells are located directly beneath the intestinal epithelial cells and could provide a potentially direct source for KGF to IEC in the event of wounds or IBD damage. This is especially noteworthy as most epithelial cell types, including those of the intestinal epithelium, are known to express the KGF receptor, FGFR2-IIIb (Werner 1998). Unfortunately, little is known about how KGF may affect the production of pro-inflammatory cytokines by IEC.
Previously, we have shown that HGF alone had no effect on IL-8 cytokine secretion by the Caco-2 colon carcinoma cell line, yet co-stimulation of the cells with both HGF and IL-1 caused a significant increase in pro-inflammatory IL-8 secretion above the levels generated by IL-1 alone (Grygas et al. 2007). This suggests a link between pro-inflammatory and wound healing events in IEC. Therefore, the focus of the present study was twofold. Our first goal was to determine if KGF could induce a similar increase in IL-1-induced IL-8 secretion by intestinal epithelial cells. Secondly, we sought to determine how HGF and/or KGF may exert an enhancing effect on IL-1-induced IL-8 secretion. As with HGF, we found that KGF could also enhance IL1-induced IL-8 secretion. However, using chemical inhibitors, we provide evidence that the HGF-induced enhancement of IL-1-stimulated IL-8 secretion was mediated by phosphatidylinositol-3-kinase (PI3K) and AKT (also known as protein kinase B) signaling. Yet, the KGF-enhanced IL-1-stimulated IL-8 secretion was not affected by inhibiting PI3K, though the effect was sensitive to the inhibition of AKT. This suggests a differential mechanism of regulation between these two growth factors with IL-1 stimulation, which may play an important role in mucosal inflammation and wound healing.
Materials and Methods
Cell culture
The human colonic adenocarcinoma cell line Caco-2 (HTB-37) was purchased from American Type Culture Collection (Manassas, VA) in March of 2007, and serially passaged and stored in liquid nitrogen. The cells were cultured in RPMI-1640 (Cellgro, Washington, DC) with 10% fetal bovine serum (FBS; Hyclone, Logan, UT), 2 g/L sodium bicarbonate (J.T. Baker, Phillipsburg, NJ), 25 U/ml penicillin (Cellgro), 25 μg/ml streptomycin (Cellgro), 2 mM L-glutamine (Cellgro), and 10 mM non-essential amino acids (Sigma, St. Louis, MO), referred to as 10% FBS-RPMI. The cells were maintained at 37°C in a 90% air–10% CO2 humid environment.
The cells were removed from flasks by brief trypsin–ethylenediaminetetraacetic acid (EDTA; Sigma) treatment and were cultured at 5 × 104 cells/well in 24-well tissue culture plates for 24 h. The supernatants were then removed and replaced with fresh medium containing recombinant human (rh) KGF (1, 10, or 100 ng/ml; R&D Systems, Minneapolis, MN), rhHGF (Sigma) alone or in combination with rhIL-1β (0.5 or 1.0 ng.ml; R&D Systems). These levels of HGF and KGF were appropriate for the suggested ED50 values of 20–40 ng/ml and 15–25 ng/ml, respectively, (R&D Systems) and IL-1 published optimal levels of IL-1β for stimulating Caco-2 IL-6 production (Vitkus et al. 1998). For some experiments, KGF was added 24 h prior to treatment with IL-1 at various concentrations. At 24 h, the culture supernatants were collected and stored at −80°C.
Inhibitors
The Caco-2 cells were pre-treated with the irreversible PI3K inhibitor, wortmannin (Sigma) at a concentration of 10 or 100 nM 6 h prior to adding the KGF, HGF, and/or IL-1. Then, 24 h later, the supernatants were collected for determining the IL-8 content. The AKT Inhibitor V, triciribine, (Calbiochem/EMD Chemicals, Gibbstown, NJ) was added at a final concentration of 0.1 or 1.0 μM 30 min prior to adding KGF, HGF, and/or IL-1 and collected as above (Yang et al. 2004).
ELISA for determination of secreted IL-8 levels
The IL-8 content in the Caco-2 cell culture supernatants was determined using the DuoSet enzyme-linked immunosorbent assay (ELISA) development systems for human IL-8 (R&D Systems). The absorbances of the samples were measured using a Bio-Tek EL×808 microplate reader (Winooski, VT) and compared with a standard curve using the DeltaSoft3 software for Macintosh (BioMetallics, Inc., Princeton, NJ).
Statistics
The results from three separate experiments were analyzed for significant differences by ANOVA and Fisher's protected least significant difference test using the Statview program (SAS Institute, Cary, NC).
Results
The effect of KGF on IL-1-induced IL-8 secretion in Caco-2 cells
While there are many papers describing the increased presence of HGF during inflammatory responses (Beck and Podolsky 1999; Ido et al. 2005), it is not the only growth factor-type cytokine known to be up-regulated in IBD conditions. KGF is a potent mitogenic growth factor which is also known to be present during both wound healing (Werner et al. 1992) and in the tissue of patients with IBD (Brauchle et al. 1996; Finch and Cheng 1999). While IECs do not produce KGF, it is known that these cells do express the specific receptor for KGF, FGFR2-IIIb, and that IL-1 can induce KGF production by nearby stromal cell types (Rubin et al. 1995). However, no information is available on how the combination of KGF and IL-1 may affect the IL-1-induced production of pro-inflammatory cytokines within the intestinal epithelium. Since previous work (Grygas et al. 2007), has indicated that HGF in addition to IL-1 can induce a significant increase of pro-inflammatory IL-8 or MCP-1 secretion by IEC cell lines, our next study was to determine whether or not KGF could also synergize with IL-1 to enhance IL-8 secretion by Caco-2 cells.
For this experiment, Caco-2 cells were prepared as specified in the Methods section. After 24 h, the media was removed and replaced with media containing 10% FBS and 0.5 or 1.0 ng/ml IL-1, KGF at various concentrations or both. After 24 h, culture supernatants were collected and an IL-8 ELISA was performed. As shown in Fig. 1, the introduction of KGF by itself at concentrations up to 100 ng/ml induced no noticeable change in the levels of IL-8 secretion in Caco-2 cells. However, KGF, in combination with IL-1, led to a dose-dependent increase in IL-8 secretion which was significant with both 0.5 and 1.0 ng/ml IL-1 in combination with 100 ng/ml KGF, as compared with respective levels of IL-1-only-induced IL-8 secretion. In both cases, KGF-enhanced IL-1-induced IL-8 secretion was approximately twofold greater than IL-1-only-induced IL-8 secretion (2.1-fold for 0.5 ng/ml and 2.4-fold for 1.0 ng/ml IL-1). Since 0.5 ng/ml IL-1 induced a similar effect as 1.0 ng/ml IL-1 and prior studies have indicated that 0.5 ng/ml is a suboptimal concentration for IL-1-induced cytokine secretion (Vitkus et al. 1998), 0.5 ng/ml IL-1 was used in all subsequent experiments.
Figure 1.
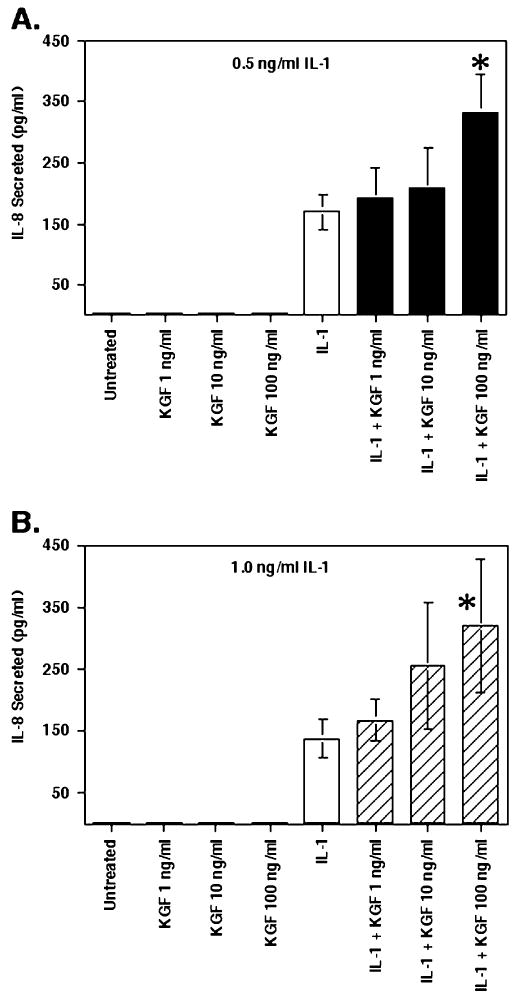
KGF-enhanced IL-1-induced IL-8 secretion in Caco-2 cells. Caco-2 cells were incubated at 5 × 104 cells/well in 24-well plates. After 24 h, the culture supernatants were removed and replaced with 10% FBS-RPMI containing rhKGF at 1, 10, or 100 ng/ml with or without rhIL-1β at either 0.5 (A) or 1.0 ng/ml (B). Twenty-four h later, supernatants were collected for determining IL-8 concentration by specific ELISA. Shown are the mean ± SEM of three separate experiments. Asterisk indicates a significant difference compared with IL-1-only control (p<0.05).
It is well known that KGF is a potent factor capable of inducing proliferation, so the next experiment was to determine if the addition of KGF resulted in an increase in overall cell numbers, which may affect the total levels of IL-8 in the samples. While the brief exposure time (24 h) was unlikely to be sufficient for significant proliferation, viability counts were performed on the cells in the wells used for the experiments in Fig. 1 to determine if the KGF-treated samples exhibited greater total numbers of cells than the untreated or IL-1 only samples. As shown in Fig. 2, there were no significant changes in the total cells per well in any of the samples (P≥0.3 for all conditions compared with the controls), suggesting that the results in Fig. 1 were due to an effect of KGF and IL-1, rather than proliferative effects caused by KGF itself. A similar finding of no effect of IL-1 or HGF on the proliferation of Caco-2 cells after 24 h has been previously shown (Grygas et al. 2007).
Figure 2.
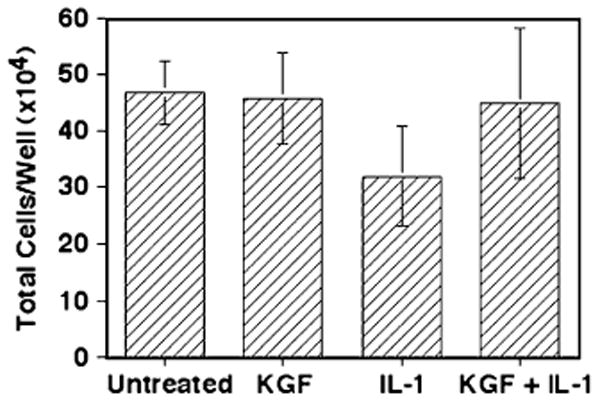
KGF has no effect on Caco-2 proliferation within 24 h. Caco-2 cells were cultured as in Fig. 1, and after the supernatants were removed at 24 h, the cells were removed by trypsin–EDTA treatment and counted using Trypan Blue. The values shown represent the mean ± SEM of three separate experiments.
While we suspect that KGF is exerting an effect on the activated IL-1 pathway, the possibility exists that KGF may somehow be “priming” elements of the IL-1 pathway to enhance IL-1-induced IL-8 secretion. KGF may also be inducing the cells to produce another factor, which may then act in an auto- or paracrine manner to enhance IL-1-stimulated cytokine secretion. To examine this possibility, Caco-2 cells were pre-treated with KGF for 24 h prior to IL-1 stimulation, and then IL-8 secretion was compared with IL-1 alone, or IL-1 and KGF administered simultaneously.
Briefly, cells were prepared and cultured 24 h before pre-treatment with 100 ng/ml KGF or an equivalent amount of ITS-RPMI (see Fig. 3). Then, 24 h later, the culture supernatants were removed and fresh medium with or without IL-1 and KGF was added. The cells were then cultured for 24 h before collecting the supernatants for determination of secreted IL-8 levels. Of importance, the values shown in Fig. 3 are the picograms of IL-8 secreted per 105 cells to account for any potential proliferation of the Caco-2 cells over the extra 24 h (48 h total) for this experiment which could result in a different number of cells in each condition. Figure 3 shows that neither KGF alone or KGF-pre-treated samples resulted in any measurable increase in IL-8 production compared with the unstimulated control samples, further confirming that KGF alone does not induce secretion of IL-8. Prestimulation of the Caco-2 cells with KGF for 24 h before addition of IL-1 led to no increase in IL-1-induced IL-8 secretion as compared with the IL-1-only-stimulated levels (p=0.4). Equally as important were the cultures pre-treated with KGF for 24 h prior to addition of IL-1 that induced a level of IL-8 secretion which was significantly lower than that of cultures which were not pre-treated with KGF, but were stimulated with KGF and IL-1 added simultaneously (P<0.0001), the latter inducing the synergistic increase in IL-8 secretion similar to Fig. 1. This strongly indicates that the presence of KGF does not lead to a sensitization or priming of the Caco-2 cells, rather that KGF and IL-1 are required simultaneously for the enhanced effect to occur.
Figure 3.
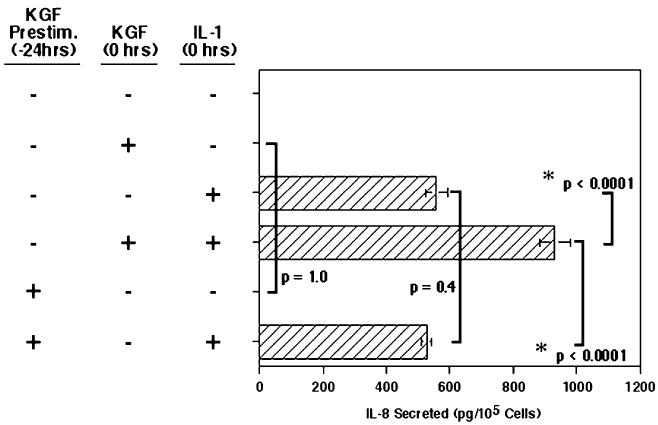
Prestimulation with KGF had no effect on IL-1-induced IL-8 secretion in Caco-2 cells. The Caco-2 cells were cultured for 24 h as in Fig. 1. Then, the supernatants were removed (at −24 h) and 100 ng/ml rhKGF was added to the KGF prestimulation wells, or an equivalent amount of FBS-RPMI was added to all others. After incubation for 24 h, all supernatants were removed and replaced with 10% FBS-RPMI either with or without rhKGF at 100 ng/ml and/or rhIL-1β at 0.5 ng/ml, as indicated (time 0 h). The cells were incubated for an additional 24 h, then, the supernatants were collected for determination of IL-8 levels. Shown are the mean ± SEM of three separate experiments. Bars indicate the significance compared between appropriate conditions.
Role of PI3K in the HGF/KGF enhancement of the IL-1 response
Sources widely implicate PI3K as a key downstream kinase activated by both HGF and KGF (Rubin et al. 1995; Rahimi et al. 1996; Chandrasekher et al. 2001). Hence, inhibition of this kinase could provide information on how HGF or KGF enhances IL-1-induced IL-8 secretion. Therefore, the role of PI3K was examined using wortmannin, an extremely specific, irreversible inhibitor of PI3K, with an IC50 of 1.9 to 5 nM (Powis et al. 1994; EMD Biosciences product data sheet).
For ease in comparison, the data is now presented as the percent of the IL-1 only (no inhibitor) samples. The results presented in Fig. 4 demonstrate that the addition of the PI3K inhibitor wortmannin at 10 nM to Caco-2 cells stimulated with both HGF and IL-1 led to a significant reduction in IL-8 secretion, reducing the IL-8 secretion levels to essentially the IL-1-only-stimulated levels (p=0.38). However, there was no effect of wortmannin at 10 nM on overall IL-1-stimulated IL-8 secretion levels compared with IL-1-only-treated controls (p>0.9). This suggests that the enhancing effect of HGF on IL-1-stimulated IL-8 secretion is mediated through PI3K.
Figure 4.
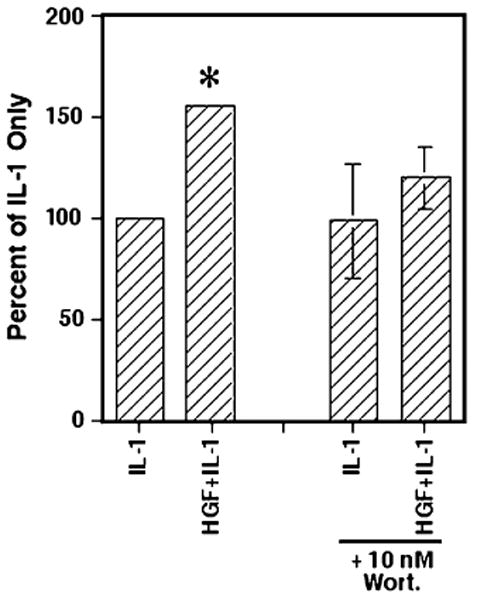
Inhibition of PI3K significantly reduces HGF-enhanced IL-1-induced IL-8 secretion. As in Fig. 1, the Caco-2 cells were cultured at 5 × 104 cells/well in 24-well plates. After 24 h, wortmannin (10 nM) or an equivalent amount of FBS-RPMI was added. Six h later, rhHGF at 100 ng/ml, rhIL-1β at 0.5 ng/ml, or both were then added to wells as indicated. After 24 h, the supernatants were collected for determination of IL-8 levels. The graph represents the IL-8 secreted as a percentage of IL-1-only controls. Shown are the mean ± SEM of three separate experiments. Asterisk indicates significance compared with IL-1-only condition (p<0.05).
In contrast to the results seen with HGF, addition of wortmannin at 10 nM caused no significant change in the enhanced level of IL-8 secreted by IL-1- and KGF-stimulated Caco-2 cells (Fig. 5). This suggests that KGF may be acting in a PI3K-independent manner to enhance IL-1-stimulated IL-8 secretion in Caco-2 cells. Experiments were also conducted using wortmannin at a concentration of 100 nM. However, this high concentration of the inhibitor led to a significant reduction in IL-1-induced IL-8 secretion, suggesting that in Caco-2 cells, this higher concentration of wortmannin may also inhibit IL-1-induced signaling (data not shown). Yet, interestingly, even at this high 100 nM wortmannin concentration, KGF plus IL-1-induced IL-8 levels were still significantly higher than that of the IL-1-only plus wortmannin-treated cells (data not shown). Therefore, HGF and KGF clearly act through different mechanisms to enhance IL-1-stimulated IL-8 secretion.
Figure 5.
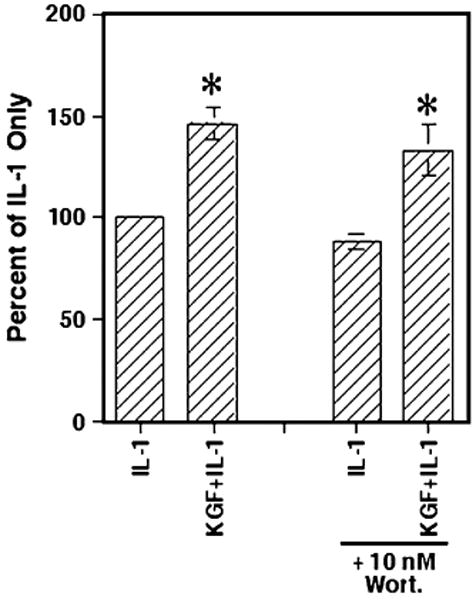
Inhibition of PI3K had no effect on KGF-enhanced IL-1-induced IL-8 secretion. Caco-2 cells were cultured with wortmannin as in Fig. 4, and after 6 h, rhKGF (100 ng/ml), rhIL-1β (0.5 ng/ml), or both were added. After 24 h, the supernatants were collected and the levels of secreted IL-8 were determined. The information shown represents the IL-8 secreted as a percentage of the IL-1-only controls and are the mean ± SEM of three experiments. The asterisks indicate significance compared with IL-1-only samples (p<0.05).
PI3K-independent AKT signaling in KGF-enhanced IL-1-induced IL-8 secretion
Since our studies suggest that HGF-enhanced IL-1-induced IL-8 secretion may be functioning in a PI3K-dependent manner, while KGF-enhanced effects seem to be PI3K-independent, investigation turned to a downstream signaling factor. AKT (also known as protein kinase B) is a known downstream target of PI3K (Chandrasekher et al. 2001; Song et al. 2005). AKT has also been shown to be activated by both PI3K and non-PI3K-dependent signaling pathways as a result of growth factor signaling (Song et al. 2005).
To inhibit AKT, we chose triciribine (also known as AKT inhibitor V and API-2), an inhibitor of all three primary AKT family members (Yang et al. 2004). Because triciribine interacts directly with the pleckstrin homology domain on AKT required for protein–protein interaction (Brazil and Hemmings 2001; Cheng et al. 2005), the inhibitor is highly selective for AKT. Yang et al. (2004) have reported that a concentration of 1 μM was sufficient to significantly reduce AKT phosphorylation levels in trans-fected 3T3 cells, while having no effect on other common kinases. For our experiments, Caco-2 cells were cultured as in Fig. 4, except triciribine (at a final concentration of 0.1 μM or 1.0 μM) was added 30 min prior to stimulation with KGF, IL-1, or both for 24 h before supernatants were collected for determining secreted IL-8 levels. As shown in Fig. 6, treatment of KGF- and IL-1-stimulated cells with 0.1 or 1.0 μM triciribine significantly reduced the level of IL-8 secreted to IL-1-only-stimulated levels. This strongly suggests that KGF enhancement of IL-1-induced IL-8 secretion involves AKT, although this appears to occur in a PI3K-independent method.
Figure 6.
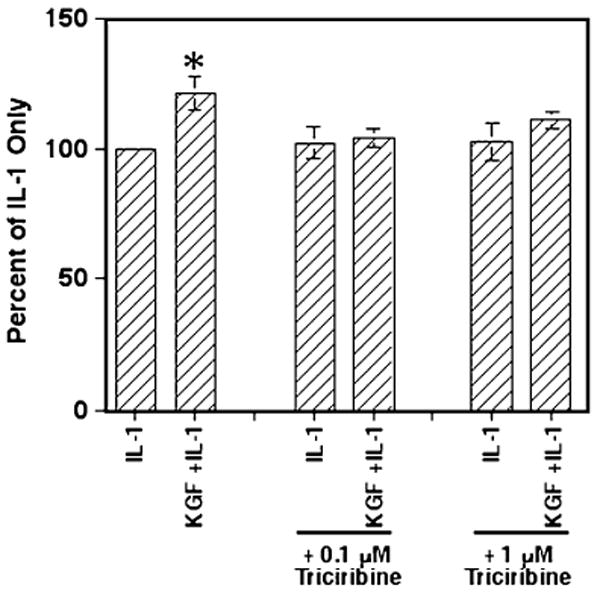
Triciribine abrogates KGF-enhanced IL-1-induced IL-8 secretion in Caco-2 cells. Caco-2 cells (5 × 104 cells/well) were cultured in 24-well plates for 24 h before adding triciribine to a final concentration of 0.1 or 1.0 μM. Then, after 30 min, rhHGF at 100 ng/ml, IL-1 at 0.5 ng/ml, or both were added as indicated. After 24 h, the supernatants were collected and the total IL-8 concentrations determined. The graphs represent the mean ± SEM of three experiments and the asterisk indicates a significance difference from all other values (p<0.03).
However, treatment of the Caco-2 cells with the lower level of triciribine at 0.1 μM had no effect on either IL-1-only-stimulated IL-8 secretion as compared with the control IL-1-stimulated cells (p=0.3) or the HGF-induced enhancement of IL-1-induced IL-8 secretion compared with the IL-1-only-stimulated cultures (p<0.01; Fig. 7). Yet, high levels of triciribine at 1 μM did abolish the HGF enhancement of IL-1-stimulated IL-8 secretion (p>0.2) suggesting that the enhancing effect of HGF may also be through AKT. But, triciribine treatment did affect the IL-1-stimulated IL-8 secretion levels with the high levels of triciribine in this experiment with 1 μM triciribine-treated IL-1-stimulated levels being lower than that of the untreated IL-1-stimulated cells (p=0.04). Others have indicated that IL-1 can also signal through AKT (Parhar et al. 2007; Kenny and O'Neill 2008) and therefore IL-1 responses could be sensitive to triciribine as seen above. Perhaps, the level of 1 μM triciribine is at the threshold of having an effect on IL-1 signaling as Fig. 6 shows no significant effect. Still, these experiments with triciribine treatment suggest that both KGF and HGF enhance IL-1-stimulated IL-8 secretion probably via AKT-based signaling, although a tenfold greater concentration of triciribine was required to abrogate the increased production of IL-8 with HGF as compared with KGF.
Figure 7.
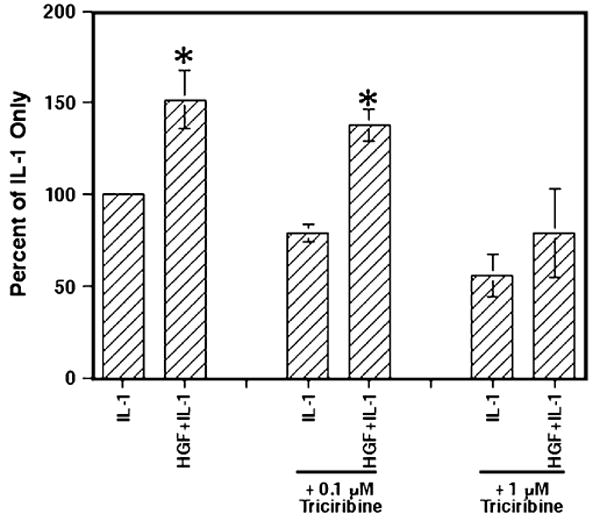
Inhibition of AKT with triciribine significantly decreased HGF-enhanced IL-1-induced IL-8 secretion. The Caco-2 cells were cultured and treated with triciribine as in Fig. 6 and then stimulated with rhHGF (100 ng/ml), IL-1 (0.5 ng/ml), or both. After 24 h, the supernatants were collected and the IL-8 concentration determined in each sample. The information shown represents the mean ± SEM of three experiments. Asterisks indicate significance compared with the appropriate IL-1-only samples (p<0.05).
Discussion
It is clear that growth factors, cytokines, and chemokines interact at the intestinal mucosa to form a regulatory network during inflammation. Patients with IBD show increased levels of pro-inflammatory cytokines, such as IL-1 and IL-8 (Ligumsky et al. 1990; Laukoetter et al. 2008), along with elevated levels of growth factors like HGF and KGF (Brauchle et al. 1996; Finch and Cheng 1999; Kitamura et al. 2000). Reports also indicate that IL-1 and IL-8 can significantly increase the expression of HGF (Tamura et al. 1993) and KGF (Finch and Cheng 1999) by fibroblasts. However, little else is known about the interplay between these inflammatory and wound healing factors on IEC. Previous research by our group has shown that the introduction of HGF to IL-1-stimulated Caco-2 cells resulted in a significant increase in IL-8 secretion over IL-1-only-stimulated IL-8 levels (Grygas et al. 2007). This suggests that the presence of at least one growth factor may lead to an increased production of IL-8. In the present study, we have further examined the role of HGF as well as KGF, and our results indicate that both growth factors may be involved in increasing levels of IL-8 produced during inflammatory situations at the mucosa.
As with HGF (Grygas et al. 2007), KGF (Fig. 1) alone did not induce IL-8 secretion above control levels suggesting that, alone, neither growth factor is involved in IL-8 secretion by IEC. This is in contrast to the findings of others who have shown that HGF alone, via PI3K and AKT, could enhance IL-8 secretion by squamous-cell carcinoma cells (Dong et al. 2001) and KGF alone could induce VEGF and MMP-9 production by pancreatic ductal epithelial cells (Niu et al. 2007). However, KGF, like HGF, could act to enhance IL-1-induced IL-8 secretion. Also, as with HGF (Grygas et al. 2007), pre-treatment of the cells with KGF did not result in an enhancement of IL-1-stimulated IL-8 secretion, suggesting that KGF did not prime the cells, possibly by enhancing IL-1 receptor expression or inducing the secretion of a second factor that actually caused the effect. Therefore, both HGF (Grygas et al. 2007) and KGF were required to be present with IL-1 to produce the enhancing effect, suggesting a potential cross-talk between the growth factor and IL-1 signaling pathways.
In both the KGF- and HGF-induced signaling pathways, PI3K has been identified as a key signaling component (Rubin et al. 1995; Rahimi et al. 1996). Therefore, we used the specific PI3K inhibitor, wortmannin, to examine the role this kinase may be playing in HGF- or KGF-enhanced IL-1-induced IL-8 secretion. Wortmannin treatment of cells co-stimulated with IL-1 and HGF eliminated the HGF-enhanced IL-1-induced IL-8 secretion down to IL-1-only-stimulated IL-8 levels suggesting that the enhancing effect of HGF was highly dependent upon PI3K. However, addition of wortmannin to the KGF- and IL-1-stimulated cells resulted in no significant effect on the KGF-enhanced IL-1-induced IL-8 secretion, even at high concentrations of wortmannin. This suggests that the enhancing effect of KGF may be PI3K-independent. Therefore, HGF and KGF may both work to enhance the effect of IL-1 on IL-8 secretion, yet this effect must occur via different mechanisms.
AKT (protein kinase B) is a known downstream target of PI3K activated phosphoinositides in multiple situations (Cheng et al. 2005). Using the AKT-specific inhibitor, triciribine, we demonstrated that both KGF- and HGF-enhanced IL-1-stimulated IL-8 secretion could be significantly reduced to IL-1-only levels. However, the KGF-enhanced effect was highly sensitive to AKT inhibition requiring tenfold less triciribine than the HGF-enhanced effect. The reason behind this difference in sensitivity remains unclear, but may be dependent upon the PI3K-dependent or PI3K-independent mechanism for activating AKT. Regardless, we have shown that the HGF-induced enhancement of IL-1-stimulated IL-8 secretion is mediated via the classic PI3K/AKT pathway, and this effect on IL-8 secretion by intestinal epithelial cells is novel. However, the enhancing effect of KGF was determined to be due to a PI3K-independent activation of AKT. While the literature suggests PI3K-mediated AKT signaling is a key downstream component of KGF activity (Bao et al. 2005; Chang et al. 2005), Song et al. (2005) have shown that AKT can be activated in a PI3K-independent manner involving protein kinase A in some contexts.
AKT has been shown to result in activation of many signaling components including IκBα, which can lead to NF–κB transcription factor activity (Kane et al. 1999; Song et al. 2005). Since IL-1 is also known to activate NF–κB, this may suggest a possible common mechanism through AKT for the enhancement with both KGF and HGF. KGF can also activate the p38 MAP kinase (Mehta et al. 2001) as well as JNK (Cheng et al. 2005), both of which can enhance IL-1-induced responses (Dinarello 2009). In addition, IL-1 has been shown in some contexts to also signal via AKT (Parhar et al. 2007; Kenny and O'Neill 2008), and the HGF- and KGF-enhanced AKT activation in combination with IL-1 could allow for a more rapid and prolonged activation of the NF–κB transcription factor via IκBα, leading to increased downstream cytokine production.
Clearly, the role of growth factors and immunological factors in the intestine remains incompletely elucidated. However, research is ongoing into the use of factors such as HGF (Arthur et al. 2004; Ido et al. 2005) in therapies designed to alleviate symptoms and intestinal damage as a result of the inflammation, as in IBD. While the data suggests that introduction of these growth factors can lead to a significant reduction in IBD symptoms, little research has been done to suggest how the addition of significant levels of HGF or KGF may be affected by pro-inflammatory cytokines already present during inflammation. This study suggests that the rise in HGF and KGF in wound healing may work synergistically with IL-1 to increase the levels of secreted IL-8. IL-8 is a potent a pro-inflammatory cytokine, yet it may also play a role in wound healing by inducing angiogenesis (Danese et al. 2006) and IEC migration (Wilson et al. 1999; Sturm et al. 2005). Indeed, the angiogenic functions of increased IL-8 levels in IBD may be a key factor of increased neovascularization in those patients with the disorder (Danese et al. 2006). Therefore, HGF- or KGF-enhanced IL-1-induced IL-8 secretion may function as a mediator to induce angiogenesis via IL-8 and help with the transition to wound healing at the end of the inflammatory response.
Acknowledgments
The authors wish to gratefully acknowledge Jill Grygas for her assistance with the technical aspects of this paper. This work was supported by US PHS Grant DK54049.
References
- Arthur LG, Schwartz MZ, Kuenzler KA, Birbe R. Hepatocyte growth factor treatment ameliorates diarrhea and bowel inflammation in a rat model of inflammatory bowel disease. J Pediatr Surg. 2004;39:139–143. doi: 10.1016/j.jpedsurg.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Bao S, Wang Y, Sweeney P, Chaudhuri A, Doseff AI, Marsh CB, Knoell DL. Keratinocyte growth factor induces Akt kinase activity and inhibits Fas-mediated apoptosis in A549 lung epithelial cells. Lung Cellular Molecular Physiol. 2005;288:36–42. doi: 10.1152/ajplung.00309.2003. [DOI] [PubMed] [Google Scholar]
- Beck PL, Podolsky DK. Growth factors in inflammatory bowel disease. Inflamm Bowel Dis. 1999;5:44–60. doi: 10.1097/00054725-199902000-00007. [DOI] [PubMed] [Google Scholar]
- Brauchle M, Madlener M, Wagner AD, Angermeyer K, Lauer U, Hofschneider PH, Gregor M, Werner S. Keratinocyte growth factor is highly overexpressed in inflammatory bowel disease. Am J Pathol. 1996;149:521–529. [PMC free article] [PubMed] [Google Scholar]
- Brazil DP, Hemmings BA. Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci. 2001;26:657–664. doi: 10.1016/s0968-0004(01)01958-2. [DOI] [PubMed] [Google Scholar]
- Chandrasekher G, Kakazu AH, Bazan HE. HGF- and KGF-induced activation of PI-3 K/p70 s6 kinase pathway in corneal epithelial cells: its relevance in wound healing. Exp Eye Res. 2001;73:191–202. doi: 10.1006/exer.2001.1026. [DOI] [PubMed] [Google Scholar]
- Chang Y, Wang J, Lu X, Thewke DP, Mason RJ. KGF induces lipogenic genes through a PI3K and JNK/SREBP-1 pathway in H292 cells. J Lipid Res. 2005;46:2624–2635. doi: 10.1194/jlr.M500154-JLR200. [DOI] [PubMed] [Google Scholar]
- Cheng JQ, Lindsley CW, Cheng GZ, Yang H, Nicosia SV. The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene. 2005;24:7482–7492. doi: 10.1038/sj.onc.1209088. [DOI] [PubMed] [Google Scholar]
- Danese S, Papa A, Scaldaferri F, Graziani C, Bonizzi M, Armuzzi A, Fedeli G, Gasbarrini G, Gasbarrini A. A novel pathogenic role for microvasculature in inflammatory bowel disease. Eur Rev Med Pharmacol Sci. 2006;10:3–5. [PubMed] [Google Scholar]
- Dong G, Chen Z, Li ZY, Yen NT, Bancroft CC, Van Waes C. Hepatocyte growth factor/scatter factor-induced activation of MEK and PI3K signal pathways contributes to expression of proangiogenic cytokines interleukin-8 and vascular endothelial growth factor in head and neck squamous cell carcinoma. Cancer Res. 2001;61:5911–5918. [PubMed] [Google Scholar]
- Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- Eckmann L, Hung HC, Schurer-Maly C, Panja A, Morzycka-Wroblewska E, Kagnoff MF. Differential cytokine expression by human intestinal epithelial cell lines: regulated expression of interleukin-8. Gastroenterology. 1993;105:1689–1697. doi: 10.1016/0016-5085(93)91064-o. [DOI] [PubMed] [Google Scholar]
- Finch PW, Cheng AL. Analysis of the cellular basis of keratinocyte growth factor overexpression in inflammatory bowel disease. Gut. 1999;45:848–855. doi: 10.1136/gut.45.6.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goke M, Kanai M, Podolsky DK. Intestinal fibroblasts regulate intestinal epithelial cell proliferation via hepatocyte growth factor. Am J Physiol. 1998;274:G809–G818. doi: 10.1152/ajpgi.1998.274.5.G809. [DOI] [PubMed] [Google Scholar]
- Greenwood-Van Meerveld B, Venkova K, Connolly K. Efficacy of repifermin (keratinocyte growth factor-2) against abnormalities in gastrointestinal mucosal transport in a murine model of colitis. J Pharm Pharmacol. 2003;55:67–75. doi: 10.1111/j.2042-7158.2003.tb02435.x. [DOI] [PubMed] [Google Scholar]
- Gross V, Andus T, Daig R, Aschenbrenner E, Scholmerich J, Falk W. Regulation of interleukin-8 production in a human colon epithelial cell line (HT 29) Gastroenterology. 1995;108:653–661. doi: 10.1016/0016-5085(95)90436-0. [DOI] [PubMed] [Google Scholar]
- Grygas J, Steiger N, LeSeur C, Unger B, McGee D. Hepatocyte growth factor enhances IL-1β stimulated IL-8 secretion by Caco-2 epithelial cells. In Vitro Cell Dev Biol. 2007;43:147–152. doi: 10.1007/s11626-007-9018-4. [DOI] [PubMed] [Google Scholar]
- Housley RM, Morris CF, Boyle W, Ring B, Biltz R, Tarpley JE, Aukerman SL, Devine PL, Whitehead RH, Pierce GF. Keratinocyte growth factor induces proliferation of hepatocytes and epithelial cells throughout the rat gastrointestinal tract. J Clin Invest. 1994;94:1764–1777. doi: 10.1172/JCI117524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ido A, Numata M, Kodama M, Tsubouchi H. Mucosal repair and growth factors: recombinant human hepatocyte growth factor as an innovative therapy for inflammatory bowel disease. J Gastroenterol. 2005;40:925–931. doi: 10.1007/s00535-005-1705-x. [DOI] [PubMed] [Google Scholar]
- Kenny EF, O'Neill LAJ. Signaling adaptors used by Toll-like receptors: an update. Cytokine. 2008;43:342–349. doi: 10.1016/j.cyto.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF–κB by the Akt/PKB kinase. Curr Biol. 1999;9:601–604. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- Kitamura S, Kindo S, Shinomura Y, Isozaki K, Kanavama S, Higashimoto Y, Minami T, Kivohara T, Yasunaga Y, Ishikawa H, Ohtani T, Ishiguro S, Matsuzawa Y. Expression of hepatocyte growth factor and c-met in ulcerative colitis. Inflamm Res. 2000;49:320–324. doi: 10.1007/PL00000212. [DOI] [PubMed] [Google Scholar]
- Laukoetter M, Nava M, Nusrat A. Role of the intestinal barrier in inflammatory bowel disease. World J Gastroenterol. 2008;14:401–407. doi: 10.3748/wjg.14.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligumsky M, Simon PL, Karmeli F, Rachmilewitz D. Role of interleukin 1 in inflammatory bowel disease-enhanced production during active disease. Gut. 1990;31:686–689. doi: 10.1136/gut.31.6.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta PB, Robson CN, Neal DE, Leung HY. Keratinocyte growth factor activates p38 MAPK to induce stress fibre formation in human prostate DU145 cells. Oncogene. 2001;20:5359–5365. doi: 10.1038/sj.onc.1204688. [DOI] [PubMed] [Google Scholar]
- Niu H, Chang Z, Peng B, Xia Q, Lu W, Huang P, Tsao MS, Chiao PJ. Keratinocyte growth factor/fibroblast growth factor-7-regulated cell migration and invasion through activation of NF–κB transcription factors. J Biol Chem. 2007;282:6001–6011. doi: 10.1074/jbc.M606878200. [DOI] [PubMed] [Google Scholar]
- Parhar K, Eivemark S, Assi K, Gomez-Munoz A, Yee A, Salh B. Investigation of interleukin 1beta-mediated regulation of NF–kappaB activation in colonic cells reveals divergence between PKB and PDK-transduced events. Mol Cell Biochem. 2007;300:113–127. doi: 10.1007/s11010-006-9375-4. [DOI] [PubMed] [Google Scholar]
- Powis G, Bonjouklian R, Berggren MM, Gallegos A, Abraham R, Ashendel C, Zalkow L, Matter WF, Dodge J, Grindey G. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 1994;54:2419–2423. [PubMed] [Google Scholar]
- Rahimi N, Tremblay E, Elliott B. Phosphatidylinositol 3-kinase activity is required for hepatocyte growth factor-induced mitogenic signals in epithelial cells. J Biol Chem. 1996;271:24850–24855. doi: 10.1074/jbc.271.40.24850. [DOI] [PubMed] [Google Scholar]
- Rubin JS, Bottaro DP, Chedid M, Miki T, Ron D, Cheon G, Taylor WG, Fortney E, Sakata H, Finch PW. Keratinocyte growth factor. Cell Biol Int. 1995;19:399–411. doi: 10.1006/cbir.1995.1085. [DOI] [PubMed] [Google Scholar]
- Sturm A, Baumgart DC, d'Heureuse JH, Hotz A, Wiedenmann B, Dignass AU. CXCL8 modulates human intestinal epithelial cells through a CXCR1 dependent pathway. Cytokine. 2005;29:42–48. doi: 10.1016/j.cyto.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura M, Arakaki N, Tsubouchi H, Takada H, Daikuhara Y. Enhancement of human hepatocyte growth factor production by interleukin-1 alpha and -1 beta and tumor necrosis factor-alpha by fibroblasts in culture. J Biol Chem. 1993;268:8140–8145. [PubMed] [Google Scholar]
- Vitkus SJ, Hanifin SA, McGee DW. Factors affecting Caco-2 intestinal epithelial cell interleukin-6 secretion. In Vitro Cell Dev Biol. 1998;34:660–664. doi: 10.1007/s11626-996-0017-7. [DOI] [PubMed] [Google Scholar]
- Werner S. Keratinocyte growth factor: a unique player in epithelial repair processes. Cytokine Growth Factor Rev. 1998;9:153–165. doi: 10.1016/s1359-6101(98)00010-0. [DOI] [PubMed] [Google Scholar]
- Werner S, Peters KG, Longaker MT, Fuller-Pace F, Banda MJ, Williams LT. Large induction of keratinocyte growth factor expression in the dermis during wound healing. Proc Nat Acad Sci USA. 1992;89:6896–6900. doi: 10.1073/pnas.89.15.6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AJ, Byron K, Gibson PR. Interleukin-8 stimulates the migration of human colonic epithelial cells in vitro. Clin Sci Lond. 1999;97:385–390. [PubMed] [Google Scholar]
- Yang L, Dan HC, Sun M, Liu Q, Sun XM, Feldman RI, Hamilton AD, Polokoff M, Nicosia SV, Herlyn M, Sebti SM, Cheng JQ. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res. 2004;64:4394–399. doi: 10.1158/0008-5472.CAN-04-0343. [DOI] [PubMed] [Google Scholar]
- Zeeh JM, Procaccino F, Hoffmann P, Aukerman SL, McRoberts JA, Soltani S, Pierce GF, Lakshmanan J, Lacey D, Eysselein VE. Keratinocyte growth factor ameliorates mucosal injury in an experimental model of colitis in rats. Gastroenterology. 1996;110:1077–1083. doi: 10.1053/gast.1996.v110.pm8612996. [DOI] [PubMed] [Google Scholar]
- Zimmerman NP, Vongsa RA, Wendt MK, Dwinell MB. Chemokines and chemokine receptors in mucosal homeostasis at the intestinal epithelial barrier in inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:1000–1011. doi: 10.1002/ibd.20480. [DOI] [PMC free article] [PubMed] [Google Scholar]