

Abstract
During the first week after sexual exposure to HIV, HIV infection does not appear to trigger a strong innate immune response. Here we describe some recent studies that show that HIV may avoid triggering antiviral innate immune responses by not replicating efficiently in dendritic cells and by avoiding detection in infected CD4 T cells and macrophages by harnessing a host cytoplasmic DNase TREX1 to digest nonproductive HIV reverse transcripts.
The innate immune response alerts the host to infection before an adaptive immune response has a chance to develop. Innate immunity is triggered by recognition of pathogen-associated molecular patterns (PAMP) that distinguish foreign components from host molecules by structure or intracellular location. Innate immunity protects the host by inducing expression and secretion of anti-pathogenic mediators, such as type I interferons (IFN); recognizing and eliminating infected cells; recruiting leukocytes to sites of pathogen invasion; enhancing antigen-presenting cell function; and enhancing the adaptive response to foreign antigens. Antiviral type I IFNs are not usually activated during acute HIV infection, which suggests that the virus avoids triggering innate immunity. Here we review some recent studies that begin to describe how HIV interacts with the innate immune system.
A central question of HIV pathogenesis is what regulates the host’s ability to recognize and respond effectively to acute infection. The patient response to HIV is heterogenous. A small number of highly exposed individuals resists infection. Biallelic mutations of CCR5, the HIV coreceptor used in HIV transmission, block transmission, but additional mechanisms for resisting infection are likely. Another small group of “elite controllers” become infected but control the infection and avoid T cell depletion and immunodeficiency without antiviral drugs. Some of these less susceptible individuals may be able to mount an effective innate immune response to HIV.
HIV Transmission
HIV sexual transmission, which potentially involves multiple CD4+ cell types in genital tissue (Langerhans cells (LC), dendritic cells (DC), macrophages, T cells) [1–6], is inefficient. It takes on average hundreds of unprotected encounters to become infected [7]. Of the swarms of virus in the semen, only a single virion usually establishes a foothold in a new host [5,8–13]. HIV is localized to the genital tissue for about a week and is clinically silent until the virus disseminates. This “eclipse phase of infection”, before adaptive immunity develops, provides a window of opportunity for intervening to prevent transmission [6]. The first week not only determines whether transmission occurs, but likely also sets the equilibrium between the virus and host immunity, which could have a lifelong effect on disease course.
Because the initial phase of infection is asymptomatic, studies of human acute infection usually do not begin until after viral dissemination. Therefore our models of what happens locally during transmission, when innate immunity plays a critical role, are largely based on experiments in Rhesus macaques infected intravaginally with Simian Immunodeficiency Virus (SIV) [10,14–24]. During the first week of macaque infection, a small focus of infection begins and amplifies in CD4 T cells in the subepithelial cervicovaginal mucosa [22,24]. Although initial studies suggested that the first infected cell might be a LC or intraepithelial DC, HIV-infected T cells are detected within a day, but infected DCs are only detected beginning about 4 days after exposure. The current consensus is that DCs, in which viral replication is very inefficient, mostly transmit the infection to T cells by a process termed “trans-infection” that occurs via an “infectious or virological synapse” that resembles the immunological synapse between a T cell and a DC [25–28]. DCs may be more important in viral dissemination to draining lymph nodes than in viral amplification within genital tissue. In fact, rather than being the first cell to replicate HIV, LCs may actually serve as sentinels to protect against infection by degrading virions [29]. However, this protective role is compromised during coinfection or inflammation, because of increased HIV replication and trans-infection by activated LCs [30–32].
In the critical first days, Type I IFNs are not induced [23]. HIV infection does not trigger a cell-autonomous interferon or inflammatory response in CD4 T cells and macrophages, its primary target cells [33]. Plasmacytoid dendritic cells (pDC), the main source of Type I IFNs in chronic infection, are not present in uninflamed genital tissue. Trafficking of immune cells to nonlymphoid tissues is tissue-specific, depending on selective expression of subtypes of integrins and chemokine receptors [34–36]. Although homing to the gut and skin has been well studied [36–38], not much is known about the molecules that direct leukocytes into and out of the genital mucosa. Proinflammatory cytokines, such as GM-CSF, IL-1, IL-6 and IL-8, produced by genital epithelial cells, recruit a broad array of leukocytes (including lymphocytes, DCs and macrophages) to sites of local inflammation. Inflamed genital epithelia, also produce chemokines, such as MIP-3α, that recruit immature DC [39–41]. MIP-3α expression is strongly up-regulated by IL-1. MIP-3α and IL-8 is rapidly upregulated in the vaginal epithelium after SIV infection. The inflammatory infiltrate includes subepithelial pDCs that produce IFNs and MIP-1α and MIP-1β which can recruit additional CD4 T cells [24]. In one study, a small molecule inhibitor of the IL-8 receptor CXCR2 decreased HIV replication in cervical explants [42].
SIV infection is confined to genital tissue for at least a week, while the virus amplifies mostly in CD4 T cells to generate virus to seed secondary lymphoid organs. It is not known whether dissemination occurs primarily by migrating cells or by release of infectious virions into lymphatics or blood. Within 2 weeks, the infection explodes in LN, gut-associated lymphoid tissue and other secondary lymphoid tissues, leading to T cell depletion. At this time, SIV/HIV virions are easily detected in extracellular fluids, and the adaptive immune response begins to kick in. Although the expansion of HIV-specific T cells coincides with a dramatic reduction of systemic viral burden, the adaptive immune response is unable to prevent the establishment of viral reservoirs and eliminate the infection.
The HIV life cycle
HIV enters T cells and macrophages by its envelope gp120 protein binding to CD4, which enables the membrane proximal portion of the envelope gp41 subunit to bind to the CCR5 or CXCR4 co-receptor on the target cell membrane, triggering viral envelope fusion with the plasma membrane (Fig. 1). HIV can also bind to cell surface lectins and enter cells by endocytosis. Endocytosed virus is released into the cytosol by viral membrane fusion with the endosomal membrane or destroyed when lysosomes fuse with endosomes. Endocytosis is the predominant mode of entry into DCs and also occurs in macrophages. Once the viral core is released into the cytosol, HIV reverse transcriptase converts RNA into DNA within the reverse transcription complex (RTC). The RTC matures into the preintegration complex (PIC), where the viral integrase prepares DNA ends for integration by removing a GT dinucleotide in a step called 3’-processing. However, reverse transcriptase is a sloppy enzyme that produces many incomplete reverse transcripts that can’t bind integrase or continue the viral life cycle. 3’-processing makes the viral DNA vulnerable to autointegration in which the reactive ends attack sites within the viral DNA [43,44]. Autointegration is mechanistically analogous to chromosomal integration, but is a suicidal side pathway [44–47]. The PIC delivers reverse-transcribed HIV DNA to the nucleus for chromosomal integration. Few copies of HIV DNA integrate, leaving behind HIV DNA in the cytosol to be cleared by host enzymes. Once the viral genomic DNA is integrated into a host chromosome, viral transcription is activated by the same transcription factors that activate the host cell with the assistance of HIV tat. HIV produces three major transcripts, 2 kb (spliced), 4.3 kb (partially spliced) and 9.2 kb (unspliced), all capped and polyadenylated, like host RNAs. Alternately spliced transcripts are translated into both structural and regulatory viral proteins. The unspliced RNA is both translated to generate Gag-Pol gene products and incorporated as genomic RNA into nascent virions at cell membrane sites where the envelope and capsid proteins assemble before budding.
Figure 1. Innate immune detection of HIV PAMPs.
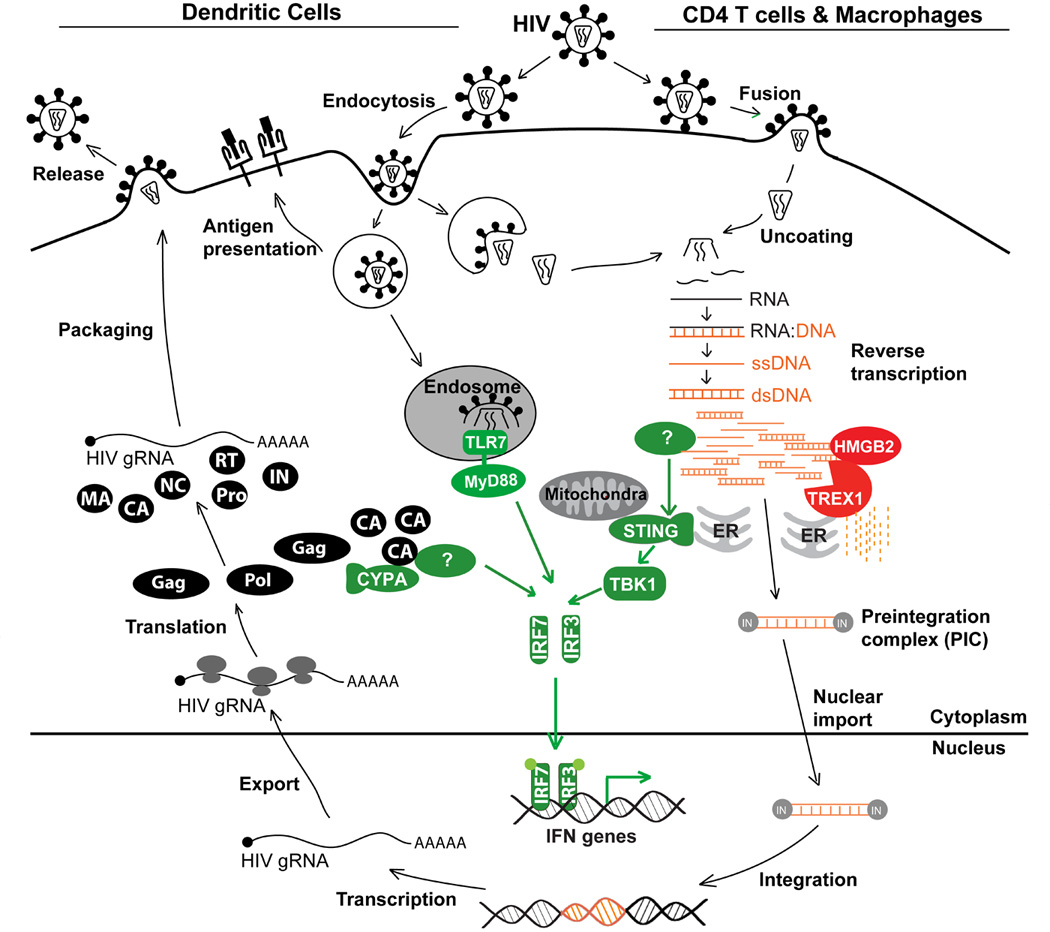
Model of the HIV life cycle and known interactions between HIV and innate immunity. HIV RNA and nascent capsid protein (CA) can be detected in dendritic cells (left) by TLR7 and an unknown sensor, respectively. HIV DNA can be detected in CD4 T cells and macrophages (right) by an unknown cytsolic DNA sensor that signals through STING and TBK1. The host factor TREX1 inhibits innate immune detection of HIV DNA by metabolizing non-productive RT products. Detection of HIV by any of these three innate immune pathways activates IFN genes.
Immune detection of HIV RNA and DNA
HIV infection of pDCs leads to IFN production, even though in these cells infection is inefficient [48] and viral replication is limited (1–2 logs lower than in T cells [49]). IFN stimulation in pDCs is triggered mostly by TLR7 recognition of endocytosed viral genomic RNA within endosomes [50]. Productive HIV infection of macrophages and T cells, however, involves viral membrane fusion with the cell membrane and direct uncoating of the viral capsid into the cytosol, bypassing the endosomal compartment and TLR signaling. It is unknown whether genomic RNA within the capsid is accessible to host RNA sensors, but reverse-transcribed HIV DNA is accessible to exogenous and endogeous nucleases [51–54]. In recent years, a handful of intracellular innate immune DNA sensors have been described, including endosomal TLR9 and cytosolic DAI, POL III/RIG-I, LRRFIP1, IFI16, and HMGB proteins [55–60]. The cytosolic SET complex, which contains three nucleases (Ape1, NM23-H1 and TREX1) and the HMGB protein HMGB2, binds to HIV DNA, but not RNA, in the cytosol and protects HIV DNA from autointegration [51,52]. TREX1, the most abundant cellular exonuclease, also inhibits the innate immune response to HIV DNA in T cells and macrophages by digesting excess HIV DNA [51]. Although ubiquitous, TREX1 is especially highly expressed in lymphocytes. In Trex1−/− mouse cells and human CD4+ T cells and macrophages in which TREX1 expression was suppressed by RNA interference, cytosolic HIV DNA accumulates, and HIV infection induces type I IFNs that inhibit HIV replication and spreading. IL-6, but not IL-1, expression is also activated. Whether other proinflammatory cytokines are stimulated by the accumulation of cytosolic HIV DNA has not been examined. HIV DNA activates IFN through a TLR- and NLR-independent cytosolic pathway that involves a yet to be identified DNA sensor, the adaptor STING, protein kinase TBK1 and IRF3. The known DNA sensors DAI, POL III, or LRRFIP1 are not involved. HMGB2 facilitates TREX1’s digestion of HIV DNA and also inhibits activation of the IFNβ promoter. The Type I IFNs expressed and secreted in TREX1-deficient cells inhibit multiple steps of HIV replication [51]. Therefore HIV protects itself from the antiviral effects of IFNs by harnessing the TREX1 DNase to evade recognition by an unknown DNA sensor of innate immunity. It remains to be seen whether HIV DNA triggers the same signaling in pDCs when TREX1 is inhibited. It is uncertain whether a host RNase in T cells and macrophages inhibits innate immune detection of HIV genomic RNA or whether HIV genomic RNA is shielded within the RTC. Digestion of HIV genomic RNA by the RNase H activity of the viral reverse transcriptase may circumvent detection by cytosolic RNA sensors.
TREX1 – a link between HIV infection, innate immunity and autoimmunity
TREX1, isolated nearly a decade ago as a 3’– 5’ exonuclease from mammalian cell extracts [61,62], is expressed in all tissues. Its exonuclease motifs are closely homologous to E. coli DNA polymerase (DnaQ/MutD). Recombinant TREX1 metabolizes a variety of DNA substrates and acts as a proofreading nuclease for repairing oxidative DNA damage via base excision repair in vitro. TREX1 more efficiently digests single-strand DNA (ssDNA) and DNA containing mismatched 3’ termini [61,62]. However, Trex1−/− mice do not exhibit increased cancer incidence or DNA mutations [63]. Instead, Trex1−/− mice develop inflammatory myocarditis and die of heart failure, perhaps before they would have a chance to show a predilection to cancer [63]. Trex1−/− cells accumulate cytosolic ssDNA enriched for sequences of endogenous retroelements [64,65]. Therefore TREX1 likely digests DNA generated by reverse transcription of endogenous retroviruses as well as by pathogenic lentiviruses. Accumulation of endogenous retroelement DNA may trigger IFN induction in TREX1-deficient mice and humans. TREX1 might also protect against innate immune triggering by other RNA or DNA viruses whose lifecycle involves cytosolic DNA, but this has not been investigated. TREX1 mutations in humans are associated with autoimmune and inflammatory diseases, including Aicardi-Goutieres syndrome (AGS, a severe neurological brain disease that mimics congenital viral infection [66,67]), systemic lupus erythematosus (SLE) and familial chilbain lupus (FCL) [68,69]. Some TREX1 mutations associated with these diseases interfere with nuclease activity or result in protein mislocalization from the endoplasmic reticulum (ER). Excess IFN is the hallmark of many of these diseases, suggesting that inappropriate innate immune activation because of inadequate DNA digestion is an important contributor to the pathogenesis of lupus-like autoimmune disease. Of note, there are hints that lupus patients may be underrepresented in HIV infection cohorts [70]. It will be worthwhile to investigate whether polymorphisms in TREX1 or other genes linked to autoimmunity or clinical autoimmune syndromes are overrepresented in groups of highly exposed, but uninfected, patients or elite controllers.
TREX1 is also known as DNase III. DNase I and II clear extracellular DNA and engulfed DNA from dying cells in macrophage lysosomes, respectively. Mutations in DNASEI are associated with SLE, and Dnase1−/− mice develop lupus-like disease [71,72]. Knockout of Dnase2−/− is embryonically lethal due to excessive IFN expression and can be rescued by Ifn αr1 deficiency [73]. Mortality of Trex1−/− mice can also be rescued by Irf3, Ifnαr1 or Rag2 deficiency, confirming that excessive IFNs underlie the pathogenesis of TREX1 deficiency or mutation [64].
TREX1 is a component of the SET complex, which associates with the ER, but translocates to the nucleus during oxidative stress [74]. In addition to its 3 DNases, the SET complex contains the chromatin modifying proteins, SET, HMGB2 and pp32 [75]. SET inhibits TREX1’s nuclease activity. TREX1 is thought to anchor the SET complex to the ER by its transmembrane domain. The SET complex is responsible for DNA damage activated by granzyme A in killer cells [74]. Some TREX1 mutations render cells resistant to granzyme A-mediated cell death, which might affect the cytolytic function of innate immune NK cells. The SET complex may also activate DNA damage in some forms of caspase-independent neuronal cell death [76]. This other function of TREX1 might affect AGS neurological symptoms. TREX1 functions in multiple cellular processes besides digestion of cytosolic DNA, including DNA damage repair and DNA degradation during caspase-independent programmed cell death. These other functions likely also play roles in autoimmunity and the innate immune response to viral infection.
Innate immune detection of HIV proteins
The nascent HIV capsid (CA) protein interacts with host cyclophilin A (CYPA) to trigger IFN via an IRF3-dependent pathway in monocyte-derived DCs (MDDC) [77]. For these experiments, IFN induction required productive infection of MDDCs, which do not normally efficiently replicate HIV. This was artificially achieved by co-infection with VSV-G pseudotyped HIV-GFP and SIV viral-like particles to enhance HIV replication in DCs [78]. CYPA was previously found to act as a pro-viral host factor that binds a proline-rich loop on the surface of HIV CA [79]. Studies of the CYPA-CA interaction led to the co-discovery of TRIM5 [80,81], an intrinsic antiretroviral host factor that regulates uncoating of the incoming virion core [82]. The CYPA interaction with newly synthesized CA, which occurs late in the viral life cycle when new viral proteins are synthesized, inhibits HIV replication by increasing DC activation and inducing type I IFN expression [77]. Although innate immune signaling in response to CA is IRF3-dependent, it is unclear how the CA is recognized and what other innate immune factors are involved. This innate immune detection of nascent CA appears to be DC-specific and does not occur in CD4+ T cells. Therefore, another way that HIV manages to avoid triggering innate immunity is by not replicating efficiently in DCs. HIV infection of DCs may become more efficient during chronic infection, when proinflammatory cytokines are elevated. More studies are needed to elucidate the innate immune pathway that detects HIV capsid and to understand how CYPA could have opposing pro- and anti-viral roles during early and late stages of the HIV lifecycle.
HIV infection down-regulates IRF3 expression
HIV also inhibits the innate immune response by suppressing IRF3 expression [83]. HIVLAI infection reduces IRF3 protein levels by 92% in a human T cell line and by 26% in peripheral blood mononuclear cells after 2 days. IRF3 down-regulation in HIV-infected cells correlates with increased viral replication. Because of the delay in IRF3 decline (compared to reverse transcription which happens within hours of infection), this phenomenon is unlikely to be the main mechanism of viral evasion of innate immunity, particularly in T cells, which die about 2 days after infection. Because IRF3 is essential for many innate immune signaling pathways, inhibiting IRF3 expression could render infected cells vulnerable to co-infection with other pathogens. However, since the frequency of HIV-infected cells is extremely low, even in end-stage AIDS patients, it remains to be seen whether reduced IRF3 expression in HIV-infected cells has a significant effect on susceptibility to other pathogens at the organismal level.
Concluding remarks
Here we describe several ways by which HIV may gain a foothold during transmission by avoiding triggering the innate immune alarm system. The published studies are just the beginnings of research in this arena. Not much is known about the role of NK cells, the cytotoxic arm of innate immunity, during HIV transmission. Polymorphisms in killer-cell immunoglobulin-like receptor (KIR) genes that regulate NK cell activity have been linked to favorable disease outcome, suggesting that NK cell recognition of HIV-infected cells affects transmission [84,85]. The interaction of HIV with innate immunity will undoubtedly be complex. During chronic infection circulating blood cells show the signature of increased expression of Type I IFN-regulated genes [86,87]. This could be due to circulating IFNs produced by activated pDCs or to chronic activation of overlapping proinflammatory pathways. The effect of systemic, chronic IFN induction on the immune system likely has pleiotropic effects on the ability of the host to handle HIV. Although IFNs and innate immune responses inhibit viral replication and promote antigen-specific immunity, they also lower the threshold for CD4 T cell activation, thereby enhancing the pool of susceptible cells. Viral RNA triggering of TLR8 in conjunction with another signal from gp120 binding to DC-SIGN on DCs activates NF-κB and enhances viral transcription in DCs [88]. Nonetheless, in a recently published clinical study, IFNαtreatment, given with or without AZT during the pre-HAART era, significantly reduced viral load, suggesting that even systemic Type I IFNs might be beneficial overall [89].
Some important questions that remain are: which HIV nucleic acids generated in the cytoplasm during infection can be sensed? What are the sensors for HIV DNA and capsid? Are there additional mechanisms HIV uses to avoid innate immunity? Could sexual transmission be blocked by inducing IFN during acute infection? Are individuals who have chronic innate immune activation less susceptible to transmission? We look forward to seeing these questions answered.
Acknowledgment
This work was supported by NIH AI045587 (JL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
* of special interest
** of outstanding interest
- 1.Shattock RJ, Moore JP. Inhibiting sexual transmission of HIV-1 infection. Nat Rev Microbiol. 2003;1:25–34. doi: 10.1038/nrmicro729. [DOI] [PubMed] [Google Scholar]
- 2.Haase AT. Perils at mucosal front lines for HIV and SIV and their hosts. Nat Rev Immunol. 2005;5:783–792. doi: 10.1038/nri1706. [DOI] [PubMed] [Google Scholar]
- 3.Margolis L, Shattock R. Selective transmission of CCR5-utilizing HIV-1: the 'gatekeeper' problem resolved? Nat Rev Microbiol. 2006;4:312–317. doi: 10.1038/nrmicro1387. [DOI] [PubMed] [Google Scholar]
- 4.Hladik F, McElrath MJ. Setting the stage: host invasion by HIV. Nat Rev Immunol. 2008;8:447–457. doi: 10.1038/nri2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keele BF, Derdeyn CA. Genetic and antigenic features of the transmitted virus. Curr Opin HIV AIDS. 2009;4:352–357. doi: 10.1097/COH.0b013e32832d9fef. [DOI] [PubMed] [Google Scholar]
- 6. Haase AT. Targeting early infection to prevent HIV-1 mucosal transmission. Nature. 2010;464:217–223. doi: 10.1038/nature08757. This review provides a comprehensive overview of early HIV transmission events.
- 7.Pope M, Haase AT. Transmission, acute HIV-1 infection and the quest for strategies to prevent infection. Nat Med. 2003;9:847–852. doi: 10.1038/nm0703-847. [DOI] [PubMed] [Google Scholar]
- 8.Zhang LQ, MacKenzie P, Cleland A, Holmes EC, Brown AJ, Simmonds P. Selection for specific sequences in the external envelope protein of human immunodeficiency virus type 1 upon primary infection. J Virol. 1993;67:3345–3356. doi: 10.1128/jvi.67.6.3345-3356.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu T, Wang N, Carr A, Nam DS, Moor-Jankowski R, Cooper DA, Ho DD. Genetic characterization of human immunodeficiency virus type 1 in blood and genital secretions: evidence for viral compartmentalization and selection during sexual transmission. J Virol. 1996;70:3098–3107. doi: 10.1128/jvi.70.5.3098-3107.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Z, Schuler T, Zupancic M, Wietgrefe S, Staskus KA, Reimann KA, Reinhart TA, Rogan M, Cavert W, Miller CJ, et al. Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science. 1999;286:1353–1357. doi: 10.1126/science.286.5443.1353. [DOI] [PubMed] [Google Scholar]
- 11.Derdeyn CA, Decker JM, Bibollet-Ruche F, Mokili JL, Muldoon M, Denham SA, Heil ML, Kasolo F, Musonda R, Hahn BH, et al. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science. 2004;303:2019–2022. doi: 10.1126/science.1093137. [DOI] [PubMed] [Google Scholar]
- 12.Abrahams MR, Anderson JA, Giorgi EE, Seoighe C, Mlisana K, Ping LH, Athreya GS, Treurnicht FK, Keele BF, Wood N, et al. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-poisson distribution of transmitted variants. J Virol. 2009;83:3556–3567. doi: 10.1128/JVI.02132-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A. 2008;105:7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller CJ, Alexander NJ, Vogel P, Anderson J, Marx PA. Mechanism of genital transmission of SIV: a hypothesis based on transmission studies and the location of SIV in the genital tract of chronically infected female rhesus macaques. J Med Primatol. 1992;21:64–68. [PubMed] [Google Scholar]
- 15.Lu Y, Brosio P, Lafaile M, Li J, Collman RG, Sodroski J, Miller CJ. Vaginal transmission of chimeric simian/human immunodeficiency viruses in rhesus macaques. J Virol. 1996;70:3045–3050. doi: 10.1128/jvi.70.5.3045-3050.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marx PA, Spira AI, Gettie A, Dailey PJ, Veazey RS, Lackner AA, Mahoney CJ, Miller CJ, Claypool LE, Ho DD, et al. Progesterone implants enhance SIV vaginal transmission and early virus load. Nat Med. 1996;2:1084–1089. doi: 10.1038/nm1096-1084. [DOI] [PubMed] [Google Scholar]
- 17.Spira AI, Marx PA, Patterson BK, Mahoney J, Koup RA, Wolinsky SM, Ho DD. Cellular targets of infection and route of viral dissemination after an intravaginal inoculation of simian immunodeficiency virus into rhesus macaques. J Exp Med. 1996;183:215–225. doi: 10.1084/jem.183.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trichel AM, Roberts ED, Wilson LA, Martin LN, Ruprecht RM, Murphey-Corb M. SIV/DeltaB670 transmission across oral, colonic, and vaginal mucosae in the macaque. J Med Primatol. 1997;26:3–10. doi: 10.1111/j.1600-0684.1997.tb00313.x. [DOI] [PubMed] [Google Scholar]
- 19.Harouse JM, Tan RC, Gettie A, Dailey P, Marx PA, Luciw PA, Cheng-Mayer C. Mucosal transmission of pathogenic CXCR4-utilizing SHIVSF33A variants in rhesus macaques. Virology. 1998;248:95–107. doi: 10.1006/viro.1998.9236. [DOI] [PubMed] [Google Scholar]
- 20.Sodora DL, Gettie A, Miller CJ, Marx PA. Vaginal transmission of SIV: assessing infectivity and hormonal influences in macaques inoculated with cell-free and cell-associated viral stocks. AIDS Res Hum Retroviruses. 1998;14 Suppl 1:S119–S123. [PubMed] [Google Scholar]
- 21.Harouse JM, Gettie A, Eshetu T, Tan RC, Bohm R, Blanchard J, Baskin G, Cheng-Mayer C. Mucosal transmission and induction of simian AIDS by CCR5-specific simian/human immunodeficiency virus SHIV(SF162P3) J Virol. 2001;75:1990–1995. doi: 10.1128/JVI.75.4.1990-1995.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller CJ, Li Q, Abel K, Kim EY, Ma ZM, Wietgrefe S, La Franco-Scheuch L, Compton L, Duan L, Shore MD, et al. Propagation and dissemination of infection after vaginal transmission of simian immunodeficiency virus. J Virol. 2005;79:9217–9227. doi: 10.1128/JVI.79.14.9217-9227.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abel K, Rocke DM, Chohan B, Fritts L, Miller CJ. Temporal and anatomic relationship between virus replication and cytokine gene expression after vaginal simian immunodeficiency virus infection. J Virol. 2005;79:12164–12172. doi: 10.1128/JVI.79.19.12164-12172.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Q, Estes JD, Schlievert PM, Duan L, Brosnahan AJ, Southern PJ, Reilly CS, Peterson ML, Schultz-Darken N, Brunner KG, et al. Glycerol monolaurate prevents mucosal SIV transmission. Nature. 2009;458:1034–1038. doi: 10.1038/nature07831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Romano G, Guan M, Long WK, Henderson EE. Efficient virus transmission from dendritic cells to CD4+ T cells in response to antigen depends on close contact through adhesion molecules. Virology. 1997;239:259–268. doi: 10.1006/viro.1997.8895. [DOI] [PubMed] [Google Scholar]
- 26.Geijtenbeek TB, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GC, Middel J, Cornelissen IL, Nottet HS, KewalRamani VN, Littman DR, et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100:587–597. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- 27.Nobile C, Petit C, Moris A, Skrabal K, Abastado JP, Mammano F, Schwartz O. Covert human immunodeficiency virus replication in dendritic cells and in DC-SIGN-expressing cells promotes long-term transmission to lymphocytes. J Virol. 2005;79:5386–5399. doi: 10.1128/JVI.79.9.5386-5399.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arrighi JF, Pion M, Garcia E, Escola JM, van Kooyk Y, Geijtenbeek TB, Piguet V. DC-SIGN-mediated infectious synapse formation enhances X4 HIV-1 transmission from dendritic cells to T cells. J Exp Med. 2004;200:1279–1288. doi: 10.1084/jem.20041356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Witte L, Nabatov A, Pion M, Fluitsma D, de Jong MA, de Gruijl T, Piguet V, van Kooyk Y, Geijtenbeek TB. Langerin is a natural barrier to HIV-1 transmission by Langerhans cells. Nat Med. 2007;13:367–371. doi: 10.1038/nm1541. [DOI] [PubMed] [Google Scholar]
- 30.Fahrbach KM, Barry SM, Ayehunie S, Lamore S, Klausner M, Hope TJ. Activated CD34-derived Langerhans cells mediate transinfection with human immunodeficiency virus. J Virol. 2007;81:6858–6868. doi: 10.1128/JVI.02472-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Jong MA, de Witte L, Oudhoff MJ, Gringhuis SI, Gallay P, Geijtenbeek TB. TNF-alpha and TLR agonists increase susceptibility to HIV-1 transmission by human Langerhans cells ex vivo. J Clin Invest. 2008;118:3440–3452. doi: 10.1172/JCI34721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Jong MA, Geijtenbeek TB. Human immunodeficiency virus-1 acquisition in genital mucosa: Langerhans cells as key-players. J Intern Med. 2009;265:18–28. doi: 10.1111/j.1365-2796.2008.02046.x. [DOI] [PubMed] [Google Scholar]
- 33.Goldfeld AE, Birch-Limberger K, Schooley RT, Walker BD. HIV-1 infection does not induce tumor necrosis factor-alpha or interferon-beta gene transcription. J AcquirImmune Defic Syndr. 1991;4:41–47. [PubMed] [Google Scholar]
- 34.Rodrigo Mora J, Von Andrian UH. Specificity and plasticity of memory lymphocyte migration. Curr Top Microbiol Immunol. 2006;308:83–116. [PubMed] [Google Scholar]
- 35.Mora JR, von Andrian UH. T-cell homing specificity and plasticity: new concepts and future challenges. Trends Immunol. 2006;27:235–243. doi: 10.1016/j.it.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 36.Sigmundsdottir H, Butcher EC. Environmental cues, dendritic cells and the programming of tissue-selective lymphocyte trafficking. Nat Immunol. 2008;9:981–987. doi: 10.1038/ni.f.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campbell DJ, Butcher EC. Rapid acquisition of tissue-specific homing phenotypes by CD4(+) T cells activated in cutaneous or mucosal lymphoid tissues. J Exp Med. 2002;195:135–141. doi: 10.1084/jem.20011502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, Von Andrian UH. Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature. 2003;424:88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- 39.Dieu-Nosjean MC, Massacrier C, Homey B, Vanbervliet B, Pin JJ, Vicari A, Lebecque S, Dezutter-Dambuyant C, Schmitt D, Zlotnik A, et al. Macrophage inflammatory protein 3alpha is expressed at inflamed epithelial surfaces and is the most potent chemokine known in attracting Langerhans cell precursors. J Exp Med. 2000;192:705–718. doi: 10.1084/jem.192.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dieu-Nosjean MC, Vicari A, Lebecque S, Caux C. Regulation of dendritic cell trafficking: a process that involves the participation of selective chemokines. J Leukoc Biol. 1999;66:252–262. doi: 10.1002/jlb.66.2.252. [DOI] [PubMed] [Google Scholar]
- 41.Caux C, Ait-Yahia S, Chemin K, de Bouteiller O, Dieu-Nosjean MC, Homey B, Massacrier C, Vanbervliet B, Zlotnik A, Vicari A. Dendritic cell biology and regulation of dendritic cell trafficking by chemokines. Springer Semin Immunopathol. 2000;22:345–369. doi: 10.1007/s002810000053. [DOI] [PubMed] [Google Scholar]
- 42.Narimatsu R, Wolday D, Patterson BK. IL-8 increases transmission of HIV type 1 in cervical explant tissue. AIDS Res Hum Retroviruses. 2005;21:228–233. doi: 10.1089/aid.2005.21.228. [DOI] [PubMed] [Google Scholar]
- 43.Shoemaker C, Hoffman J, Goff S, Baltimore D. Intramolecular integration within Moloney murine leukemia virus DNA. J Virol. 1981;40:164–172. doi: 10.1128/jvi.40.1.164-172.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, Kappes J, Conway J, Price R, Shaw G, Hahn B. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: identification of replication-competent and -defective viral genomes. J Virol. 1991;65:3973–3985. doi: 10.1128/jvi.65.8.3973-3985.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garfinkel D, Stefanisko K, Nyswaner K, Moore S, Oh J, Hughes S. Retrotransposon suicide: formation of Ty1 circles and autointegration via a central DNA flap. J Virol. 2006;80:11920–11934. doi: 10.1128/JVI.01483-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li L, Farnet C, Anderson W, Bushman F. Modulation of activity of Moloney murine leukemia virus preintegration complexes by host factors in vitro. J Virol. 1998;72:2125–2131. doi: 10.1128/jvi.72.3.2125-2131.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee M, Craigie R. A previously unidentified host protein protects retroviral DNA from autointegration. Proc Natl Acad Sci USA. 1998;95:1528–1533. doi: 10.1073/pnas.95.4.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smed-Sörensen A, Loré K, Vasudevan J, Louder MK, Andersson J, Mascola JR, Spetz A-L, Koup RA. Differential susceptibility to human immunodeficiency virus type 1 infection of myeloid and plasmacytoid dendritic cells. J Virol. 2005;79:8861–8869. doi: 10.1128/JVI.79.14.8861-8869.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McIlroy D, Autran B, Cheynier R, Wain-Hobson S, Clauvel JP, Oksenhendler E, Debré P, Hosmalin A. Infection frequency of dendritic cells and CD4+ T lymphocytes in spleens of human immunodeficiency virus-positive patients. J Virol. 1995;69:4737–4745. doi: 10.1128/jvi.69.8.4737-4745.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Beignon A-S, McKenna K, Skoberne M, Manches O, DaSilva I, Kavanagh DG, Larsson M, Gorelick RJ, Lifson JD, Bhardwaj N. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J Clin Invest. 2005;115:3265–3275. doi: 10.1172/JCI26032. The first study that shows TLR7 detects HIV RNA and induces IFN-α, using a replication competent HIV stain to infect human pDCs.
- 51. Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol. 2010;11:1005–1013. doi: 10.1038/ni.1941. This study shows that HIV evades innate immune detection in CD4 T cells and macrophages by harnessing a host cytoplasmic DNase TREX1 to digest nonproductive HIV reverse transcripts. This is the first study that shows HIV DNA can be detected by a cytosolic DNA sensing pathway that involves an unknown DNA sensor, the adaptor STING, protein kinase TBK1 and IRF3 to activate IFN gene expression.
- 52.Yan N, Cherepanov P, Daigle JE, Engelman A, Lieberman J. The SET Complex Acts as a Barrier to Autointegration of HIV-1. PLoS Pathog. 2009;5:e1000327. doi: 10.1371/journal.ppat.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bowerman B, Brown N, Bishop K, Varmus H. A nucleoprotein complex mediates the integration of retroviral DNA. Genes Dev. 1989;3:469–478. doi: 10.1101/gad.3.4.469. [DOI] [PubMed] [Google Scholar]
- 54.Miller M, Farnet C, Bushman F. Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. J Virol. 1997;71:5382–5390. doi: 10.1128/jvi.71.7.5382-5390.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, Nakasato M, Lu Y, Hangai S, Koshiba R, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. 2009;462:99–103. doi: 10.1038/nature08512. [DOI] [PubMed] [Google Scholar]
- 57.Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol. 2009;10:1065–1072. doi: 10.1038/ni.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chiu Y-H, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138:576–591. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 60.Hornung V, Latz E. Intracellular DNA recognition. Nat Rev Immunol. 2010;10:123–130. doi: 10.1038/nri2690. [DOI] [PubMed] [Google Scholar]
- 61.Hoss M, Robins P, Naven T, Pappin D, Sgouros J, Lindahl T. A human DNA editing enzyme homologous to the Escherichia coli DnaQ/MutD protein. Embo J. 1999;18:3868–3875. doi: 10.1093/emboj/18.13.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mazur D, Perrino F. Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3'-->5' exonucleases. J Biol Chem. 1999;274:19655–19660. doi: 10.1074/jbc.274.28.19655. [DOI] [PubMed] [Google Scholar]
- 63.Morita M, Stamp G, Robins P, Dulic A, Rosewell I, Hrivnak G, Daly G, Lindahl T, Barnes D. Gene-targeted mice lacking the Trex1 (DNase III) 3'-->5' DNA exonuclease develop inflammatory myocarditis. Mol Cell Biol. 2004;24:6719–6727. doi: 10.1128/MCB.24.15.6719-6727.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yang Y, Lindahl T, Barnes D. Trex1 Exonuclease Degrades ssDNA to Prevent Chronic Checkpoint Activation and Autoimmune Disease. Cell. 2007;131:873–886. doi: 10.1016/j.cell.2007.10.017. Reference 64 and 65 showed that Trex1 deficiency allows ssDNA to build up in the cytosol. Stetson et al further demonstrated that those excess ssDNAs are derived from endogenous retroelements. They also provided genetic evidence that mortality of Trex1−/− mice can be rescued by deficiency of Irf3 or Ifnar1 or Rag2, confirming that excessive IFN activation underlies the pathogenesis of diseases linked to TREX1 mutation.
- 66.Crow Y, Hayward B, Parmar R, Robins P, Leitch A, Ali M, Black D, van Bokhoven H, Brunner H, Hamel B, et al. Mutations in the gene encoding the 3'–5' DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006;38:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 67.Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum Mol Genet. 2009;18:R130–R136. doi: 10.1093/hmg/ddp293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee Y-A, de Silva U, Bailey SL, Witte T, Vyse TJ, et al. Mutations in the gene encoding the 3'–5' DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39:1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 69. Lee-Kirsch MA, Chowdhury D, Harvey S, Gong M, Senenko L, Engel K, Pfeiffer C, Hollis T, Gahr M, Perrino FW, et al. A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J Mol Med. 2007;85:531–537. doi: 10.1007/s00109-007-0199-9. Reference 66, 68, 69 described association of TREX1 mutations in humans with autoimmune diseases, such as AGS, SLE and FCL.
- 70.Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329–337. doi: 10.1016/s1568-9972(02)00086-1. [DOI] [PubMed] [Google Scholar]
- 71.Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, Kuroda Y. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet. 2001;28:313–314. doi: 10.1038/91070. [DOI] [PubMed] [Google Scholar]
- 72.Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Möröy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. 2000;25:177–181. doi: 10.1038/76032. [DOI] [PubMed] [Google Scholar]
- 73.Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol. 2005;6:49–56. doi: 10.1038/ni1146. [DOI] [PubMed] [Google Scholar]
- 74.Chowdhury D, Beresford P, Zhu P, Zhang D, Sung J, Demple B, Perrino F, Lieberman J. The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol Cell. 2006;23:133–142. doi: 10.1016/j.molcel.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 75.Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol. 2008;26:389–420. doi: 10.1146/annurev.immunol.26.021607.090404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu Z, Jang SW, Liu X, Cheng D, Peng J, Yepes M, Li XJ, Matthews S, Watts C, Asano M, et al. Neuroprotective actions of PIKE-L by inhibition of SET proteolytic degradation by asparagine endopeptidase. Mol Cell. 2008;29:665–678. doi: 10.1016/j.molcel.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Manel N, Hogstad B, Wang Y, Levy DE, Unutmaz D, Littman DR. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature. 2010;467:214–217. doi: 10.1038/nature09337. This study shows that newly synthesized HIV capsid can by detected by an unknown sensor to trigger an IRF-3 dependent signaling pathway. The authors used monocyte derived dendritic cells (MDDC) in the study and achieved productive infection by co-infecting with VSV-G pseudotyped HIV and SIVmac239 virus-like particles. The infection induces DC maturation, IFN expression and T cell activation. This innate immune response is also dependent on host factor cyclophilin A, which is known to interact with HIV capsid.
- 78.Goujon C, Jarrosson-Wuillème L, Bernaud J, Rigal D, Darlix J-L, Cimarelli A. With a little help from a friend: increasing HIV transduction of monocyte-derived dendritic cells with virion-like particles of SIV(MAC) Gene Ther. 2006;13:991–994. doi: 10.1038/sj.gt.3302753. [DOI] [PubMed] [Google Scholar]
- 79.Luban J, Bossolt K, Franke E, Kalpana G, Goff S. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell. 1993;73:1067–1078. doi: 10.1016/0092-8674(93)90637-6. [DOI] [PubMed] [Google Scholar]
- 80.Sayah DM, Sokolskaja E, Berthoux L, Luban J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature. 2004;430:569–573. doi: 10.1038/nature02777. [DOI] [PubMed] [Google Scholar]
- 81.Stremlau M, Owens C, Perron M, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 82.Song B. TRIM5alpha. Curr Top Microbiol Immunol. 2009;339:47–66. doi: 10.1007/978-3-642-02175-6_3. [DOI] [PubMed] [Google Scholar]
- 83. Doehle B, Hladik F, McNevin J, McElrath M, Gale M. HIV-1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J Virol. 2009 doi: 10.1128/JVI.00849-09. This study showed that infection of human T cells with replication competent HIV reduces IRF3 protein and increases chances of co-infection with other viruses or pathogens.
- 84.Carrington M, Martin MP, van Bergen J. KIR-HLA intercourse in HIV disease. Trends Microbiol. 2008;16:620–627. doi: 10.1016/j.tim.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Martin MP, Qi Y, Gao X, Yamada E, Martin JN, Pereyra F, Colombo S, Brown EE, Shupert WL, Phair J, et al. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat Genet. 2007;39:733–740. doi: 10.1038/ng2035. This study analyzed 1,500 HIV patients and found a strong correlation between NK cell receptor/ligand genes (KIR3DL1/HLA-B loci) and AIDS progression. It suggests a critical role of NK cell activity in HIV infection and progression to AIDS.
- 86.Lehmann C, Harper JM, Taubert D, Hartmann P, Fatkenheuer G, Jung N, van Lunzen J, Stellbrink HJ, Gallo RC, Romerio F. Increased interferon alpha expression in circulating plasmacytoid dendritic cells of HIV-1-infected patients. J Acquir Immune Defic Syndr. 2008;48:522–530. doi: 10.1097/QAI.0b013e31817f97cf. [DOI] [PubMed] [Google Scholar]
- 87.Trinchieri G. Type I interferon: friend or foe? J Exp Med. 207:2053–2063. doi: 10.1084/jem.20101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gringhuis SI, van der Vlist M, van den Berg LM, den Dunnen J, Litjens M, Geijtenbeek TB. HIV-1 exploits innate signaling by TLR8 and DC-SIGN for productive infection of dendritic cells. Nat Immunol. 2010;11:419–426. doi: 10.1038/ni.1858. This study showed that productive HIV infection in DCs requires two innate immune signaling events: first, TLR8 recognizes HIV genomic RNA and activates NF-κB, which activates proviral transcription; second, DC-SIGN recognizes HIV gp120 and activates the kinase Raf-1 which leads to transcription elongation.
- 89.Tavel JA, Huang CY, Shen J, Metcalf JA, Dewar R, Shah A, Vasudevachari MB, Follmann DA, Herpin B, Davey RT, et al. Interferon-alpha produces significant decreases in HIV load. J Interferon Cytokine Res. 30:461–464. doi: 10.1089/jir.2009.0090. [DOI] [PMC free article] [PubMed] [Google Scholar]