

Abstract
We report the 1.7 Å resolution crystal structure of the Lip2 lipase from Yarrowia lipolytica in its closed conformation. The Lip2 structure is highly homologous to known structures of the fungal lipase family (Thermomyces lanuginosa, Rhizopus niveus, and Rhizomucor miehei lipases). However, it also presents some unique features that are described and discussed here in detail. Structural differences, in particular in the conformation adopted by the so-called lid subdomain, suggest that the opening mechanism of Lip2 may differ from that of other fungal lipases. Because the catalytic activity of lipases is strongly dependent on structural rearrangement of this mobile subdomain, we focused on elucidating the molecular mechanism of lid motion. Using the x-ray structure of Lip2, we carried out extensive molecular-dynamics simulations in explicit solvent environments (water and water/octane interface) to characterize the major structural rearrangements that the lid undergoes under the influence of solvent or upon substrate binding. Overall, our results suggest a two-step opening mechanism that gives rise first to a semi-open conformation upon adsorption of the protein at the water/organic solvent interface, followed by a further opening of the lid upon substrate binding.
Introduction
Interest in lipases (triacylglycerol acyl hydrolase EC 3.1.1.3) has greatly increased over the past few years, mainly due to the high degree of versatility displayed by this class of enzymes. Their natural function is to hydrolyze the ester moiety of triglycerides. However, they are also able to perform a variety of synthesis reactions on a broad range of unnatural substrates. These reactions are carried out in organic solvent with high chemo-, regio-, and enantioselectivity. The versatility of these enzymes makes them biocatalysts of choice for numerous biotechnological applications (1).
In a continuation of our efforts to develop efficient biocatalysts with required selectivity and specificity for industrial applications, we recently focused on improving the properties of a very promising lipase, Lip2, the main extracellular lipase secreted by the yeast Yarrowia lipolytica (2), by molecular engineering. This enzyme was recently characterized by several different groups (2–4) and is currently being used for various environmental and nutraceutical (e.g., Artechno SA, Gembloux, Belgium) applications. Thanks to its high resistance to acidic pH and detergents (4), Lip2 is a promising therapeutic replacement lipase for pancreatic enzyme insufficiency, a disease that is often related to cystic fibrosis. Moreover, Lip2 has been shown to be a very efficient catalyst for the resolution of 2-bromo-arylacetic esters, an important class of intermediates in the pharmaceutical industry as analgesics and semisynthetic penicillin precursors (5).
To be able to design enzymes for specific applications, it is clearly essential to understand the structural determinants involved in their selectivity. A particular structural feature of many lipases is the presence of a mobile subdomain, called lid, whose conformational changes control the access of substrate molecules to the catalytic center. Indeed, in available x-ray structures of lipases, the lid has been observed in two distinct conformations: 1), a closed form in which the subdomain shields the active site rendering the enzyme inactive; and 2), an open active form with an accessible active site. Such conformational rearrangements of the lid have been reported to be closely related to the interfacial activation of lipases (6). These conformational rearrangements are generally assumed to be induced either by the adsorption of the enzyme to a hydrophobic interface or by substrate binding. However, these phenomena are not mutually restrictive, as they can intervene together. Lid opening is usually accompanied by the exposure of hydrophobic residues, creating a lipid substrate anchoring area around the active site.
The activation mechanism by which the lid motion operates is still poorly understood, and the characterization of such large conformational transitions at the atomic level and in real time by experimental biophysical methods remains a challenge (7). X-ray techniques provide key information on static conformational states but offer poor dynamic insight. Thus, molecular modeling techniques are used in an attempt to predict and understand the conformational changes of macromolecular systems. To date, a number of molecular-dynamics (MD) studies have been carried out to gain some insight into the conformational changes of the lid and the role played by the surrounding environment (solvent, pH, and temperature) on the dynamic properties of several lipases. However, only a few studies have investigated the entire conformational transition between the closed and open conformations (8–10).
Herein, we report an investigation based on a combination of crystallographic, biochemical, and MD studies that allowed us to obtain a detailed description of the active site at the atomic level, as well as a better understanding of the factors that trigger interfacial activation of the Lip2 protein. This structural knowledge is of central importance for the design of lipase variants with improved properties for industrial applications.
Materials and Methods
Experimental procedures
Chemicals
Tryptone was purchased from Difco (Difco, Paris, France). Other chemicals, unless otherwise stated, were purchased from Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO).
Strains, plasmids, and culture conditions
For solid media, 20 g/L of agar were added. Escherichia coli strain DH5α, used for transformation and for amplification of recombinant plasmid DNA, was grown at 37°C in Luria-Bertani medium supplemented with kanamycin (40 μg/L). The yeasts were grown on YPD. Selection of Ura+ yeast transformants was performed on YNBcasaD plates (YNB with 2 g of casamino acids/L and glucose at 10 g/L). The plates were kept at 28°C for growth. The pH of all media was kept steady with 50 mM phosphate buffer (pH 6.8).
Plasmid JMP61-pTEF-LIP2 construction
The JMP61-LIP2 vector (strain JME719) was constructed by cloning the LIP2 gene as an HindIII-EcoRI 1079 bp fragment from plasmid JMP6 (11) into JMP61 (12) at the corresponding sites. The JMP61-pTEF-LIP2 derivative (strain JME870) was constructed by exchange of the ClaI-HindIII of JMP61-pPOX2-LIP2 by a 389 bp ClaI-HindIII fragment carrying the pTEF promoter. The pTEF was amplified with primer pair oligo (olg23 cccatcgatagagaccgggttggcggcg and olg24 cccaagcttcattgtgggatccttcgggtgtgagttgacaagg, containing a ClaI and HindIII/BamHI sites, respectively).
General genetics techniques
Standard molecular genetics techniques were used (13). Restriction enzymes were obtained from Invitrogen (Cergy Pontoise, France) or New England Biolabs (Beverly, MA). Yeast cells were transformed by the lithium acetate method (14).
LIP2 expression cassette
The LIP2 gene was carried by the expression cassette (EC) contained in plasmid JMP61. The EC is flanked by ζ regions and is composed of URA3 marker (ura3d1), TEF promoter, and LIP2 gene. The EC liberated from the plasmid upon NotI digestion was used directly for yeast transformation.
Construction of Lip2 mutants
The derivative plasmids JMP61-pTEF-LIP2-R86A and JMP61-pTEF-LIP2-R86K carrying single amino acid changes in the LIP2 gene were constructed by site-directed mutagenesis using the QuikChange site-directed mutagenesis kit (Stratagene, Santa Clara, CA). The technique used the double-stranded DNA vector JMP61-pTEF-LIP2 and two complementary reversed synthetic oligonucleotide primers containing the desired mutation (Eurogentec, Liège, Belgium).
The following forward primers and their complementary reverse counterparts were used to construct the variant enzymes:
R86A: 5′-ccttgttattgcaggaacccactctctgg-3′
R86K: 5′-ccttgttattaaaggaacccactctctgg-3′
T88 mutants were constructed as described in a previous work (15).
Production of Lip2
A culture of strain JMY1212 – JMP61-pTEF-LIP2 in YPD was inoculated at 10% in YT2D5 medium (yeast extract 10 g/L, tryptone 20 g/L, glucose 50 g/L) and cultivated at 28°C for 48 h.
Purification of Lip2
The culture supernatant collected after centrifugation (10,000 g, 30 min) was concentrated by ultrafiltration (10 kDa cutoff) and dialyzed against Tris 20 mM, pH 7.8. It was loaded at 8 mL/min on a Q sepharose fast-flow gel (Amersham, Massy, France). The column was washed at 13 mL/min with Tris 20 mM, NaCl 50 mM, pH 7.8, and the target protein was eluted with Tris 20 mM, NaCl 150 mM, pH 7.8. Active fractions were identified by monitoring p-nitrophenol butyrate hydrolysis at 405 nm (16) and were concentrated and dialyzed against Tris 20 mM, (NH4)2SO4 1 M, pH 7.8, on Amicon apparatus with 10 kDa cutoff (Millipore, Billerica, MA). It was then loaded at 5 mL/min on a butyl sepharose gel (Amersham). Lip2 was eluted with a linear salt gradient ranging from 1 M to 0 M in 20 mM, Tris pH 7.8, on 125 mL. The purity was checked by silver-stained sodium dodecyl sulfate polyacrylamide gel electrophoresis (data not shown).
Deglycosylation of Lip2
Enzymatic deglycosylation was performed at 30°C for 36 h using 5 mg of concentrated purified Lip2 with 5000 units of endoglycosydase Endo Hf (New England Biolabs). Endo Hf was then removed using Amylose resin (New England Biolabs).
Crystallization of Lip2
Initial crystallization conditions were determined for the glycosylated and deglycosylated enzymes using a nanodrop ExtY robot (Innovadyne, Santa Rosa, CA). All crystallization optimization trials were carried out at 12°C using the hanging drop vapor diffusion method in 24 well plates. The glycosylated enzyme crystallized as multilayer hexagonal crystals (data not shown).
Subsequent crystallization assays were therefore undertaken using the deglycosylated lipase. The best crystals (200 × 80 × 50 μm3) were obtained with a 1:1 (v/v) ratio of protein (15 g/L in Tris 20 mM pH 7.8, NaCl 500 mM) to precipitant solution (30% 2-methyl-1,3-pentanediol (MPD), 100 mM MES pH 5.5, 20 mM CaCl2, 200 mM NaCl).
Data collection
X-ray experiments were carried out at 100 K on crystals of the deglycosylated enzyme. Crystals were directly flash-cooled in a stream of nitrogen gas. Native data to 1.7 Å were collected at a wavelength of 0.9395 Å on the ID14-4 beamline at the European Synchrotron Radiation Facility (ESRF, Grenoble, France). Diffracted intensities were integrated using MOSFLM (17) and scaled with SCALA (18) from the CCP4 software suite (19).
The crystal parameters and data collection statistics are given in Table 1. The structure was solved by the molecular replacement method using MOLREP (20) and the mutated structure of the chain A of the lipase II from Rhizopus niveus (PDB ID: 1LGY) (21) as a search model. The best result was obtained with a search of six copies in the asymmetric unit. The R- and Rfree-values of the refined molecular replacement solution were 0.37 and 0.47, respectively.
Table 1.
Data collection and refinement statistics
| Data collection | |
|---|---|
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 115.35, 132.14, 137.26 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| No. of molecules/asymmetric unit | 7 |
| % of solvent | 46 |
| Matthews coefficient (A3/Da) | 2.3 |
| Resolution (Å) | 20.00–1.70 (1.79–1.70) |
| Rsym | 0.073 (0.343) |
| I / σI | 6.7 (2.2) |
| Completeness (%) | 99.5 (99.9) |
| Redundancy | 4.1 (4.2) |
| Refinement | |
| Resolution (Å) | 1.70 |
| No. of reflections | 216,938 |
| Rwork / Rfree | 0.171 / 0.211 |
| No. of atoms | |
| Protein | 16698 |
| Ligand/ion | 210 (NAG∗); 536 (MPD∗); 7 (K+) |
| Water | 1648 |
| B-factors (Å2) | 23 |
| Protein | 22 |
| Ligand/ion | 33 (NAG∗); 35 (MPD∗); 25(K+) |
| Water | 29 |
| RMSD | |
| Bond lengths (Å) | 0.027 |
| Bond angles (°) | 2.109 |
NAG = N-acetyl glucosamine; MPD = 2-methyl-1,3-pentanediol
Refinement
Structure refinement was performed with refmac5 from the CCP4 GUI (22) and models were manually reconstructed in SigmaA weighted electron density maps using coot (23). After several refinement cycles, a seventh, partially occupied molecule appeared in the electron density map. Water molecules were automatically assigned in residual maps. The final model contained 2095 amino acids among the 2107 theoretically present from the sequence of the Lip2 construct used, 1670 solvent molecules comprising 1646 water molecules, 17 MPD molecules, and 7 potassium ions. Each polypeptide chain displayed two N-acetyl-glucosamine residues covalently bound to N113 and N134, respectively. The crystallographic R- and Rfree-values of the final model were 0.171 and 0.211, respectively, for a crystallographic data set between 20 and 1.7 Å. The refinement statistics are summarized in Table 1. The final structure has 96.3% of the residues in the most favored region of the Ramachandran plot and 1.2% in the disallowed region, as defined by PROCHECK (24).
Computational procedures
Structural alignment
Multiple structure alignment was performed using the Mammoth server (25) with the A chain of the Lip2 structure and the closed structures of lipases from Thermomyces lanuginosa (PDB ID: 1DT3), R. niveus (PDB ID: 1LGY), and Rhizomucor miehei (PDB ID: 3TGL). The alignment was represented using ESPript (26). The root mean-square deviations (RMSDs) among three-dimensional structures were calculated using superpose (27) from the CCP4 suite (19).
Construction of Lip2 tetrahedral intermediate model
Coordinates of the Lip2 model used to create tetrahedral intermediate were extracted from the semi-open conformation obtained after 8 ns of MD simulation in octane/water environment starting from the closed x-ray structure. Tetrahedral intermediate was constructed by covalently bonding the catalytic Ser (S162) to the (S) ethyl 2-bromophenylacetate molecule as previously described (15). The covalent intermediate model was then refined using a standard energy minimization procedure within the AMBER 9 suite of programs (28).
MD simulations
All MD simulations were carried out using the AMBER 9 suite of programs and the all-atom ff03 force field (28). The starting model was derived from the high-resolution crystal structure of Lip2 in its closed conformation. Eleven Na+ cations were added to neutralize the molecular system. MD simulations were performed under different conditions to investigate the effect of solvent on the dynamics of Lip2. Solvent molecules were taken into account explicitly. For all simulations, the protein or the protein/substrate complex together with the counterions was embedded in a rectangular parallelepiped solvent box that left a space of 1 nm around the solute. To generate the octane/water interface, a periodic octane/water box was built according to a previously described procedure (9). For explicit water simulations, TIP3P water molecules were added using the LEaP module integrated in the AMBER9 package.
For each environment under study, the preparation and production phases of the MD simulations were performed as described previously (9). The simulations were carried out for a total of 20 ns at constant temperature (303 K) and pressure (1 bar) conditions, and some trajectories were extended up to 60 ns. To avoid artifacts, several MD simulations were run twice with different starting atomic velocities from the Maxwellian distribution. The resulting trajectories were analyzed using the Ptraj module of the AMBER 9 package. The RMSD was calculated for the protein backbone atoms using least-squares fitting. Distances between given residues were calculated with respect to their center of mass.
Computing of enantiomer trajectories
Starting from the docked position, enantiomer trajectories were computed using the Biomove3D integrated path planner (29), going from the bottom toward the entrance of the active site. The substrate trajectories were computed according to a previously described procedure (30).
Results and Discussion
Overall structure and crystal packing
The crystal structure of Lip2 (PDB code: 3O0D) adopts the typical α/β fold observed for lipases from the fungal lipase family. The central β-sheet is formed of nine β-strands (β2 being the only one antiparallel) and five α-helices that are packed on the two sides of the sheet (Fig. 1 a).
Figure 1.
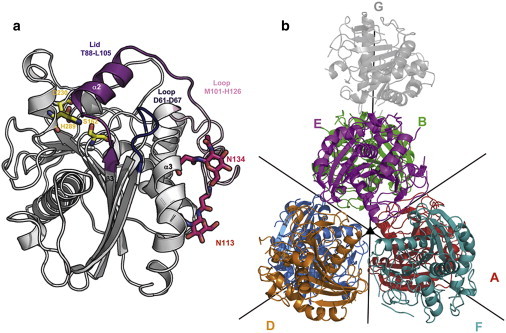
(a) Ribbon representation of the structure of Lip2 from Y. lipolytica. The lid (T88–L105), the loops D61–D67, and M101–H126 have been pointed out. Residues forming the catalytic triad (S162, D230, and H289) and the remaining N-acetyl glucosamine residues from the two glycosylation sites N113 and N134 are represented in sticks. (b) Packing of the seven molecules of the asymmetric unit (chains D–F in front, chains A–C at the rear, and chain G on top). Noncrystallographic pure rotational symmetry operators applicable to chains A–F are represented (triangle: threefold symmetry axis; lines: twofold symmetry axis).
Seven molecules of Lip2 (molecules A–G) were found in each asymmetric unit. Six of these seven molecules are organized in two layers of three polypeptide chains related by a threefold symmetry axis. A twofold symmetry occurs between the two layers (Fig. 1 b). Chain G, located on the top of the pyramid formed by chains A–F (in close contact with chains B and E), has a particular status in the crystal lattice. Not only is it not involved in any noncrystallographic pure rotational symmetry operator, its electron density is also less accurately defined (see Fig. S1 in the Supporting Material), as revealed by an average B-factor of 45 Å2, which is twice as high as the average B-factors of the other chains (16–22 Å2). We considered lowering the crystal symmetry by generating the three possible monoclinic space groups. However, the quality of the resulting electron density maps for the G molecules observed in the monoclinic asymmetric unit was neither improved nor deteriorated (data not shown).
Lip2 is expressed as a 301 amino acid glycosylated protein of 38 kDa. The glycosylation of this protein was recently characterized (31). An endoglycosidase (Endo Hf) was used to remove the sugar moiety of the protein, except for the first N-acetyl glucosamine residue. Deglycosylation of Lip2 did not significantly affect enzyme activity. Two glycosylation sites were found in the structure of Lip2 at positions N113 (loop M101–H126) and N134 (helix α3; Fig. 1 a). The two remaining N-acetyl glucosamines found on the asparagine residues N113 and N134 of both glycosylation sites are well defined in all seven lipase molecules of the asymmetric unit, and were found positioned in the same manner. However, these sugar moieties are involved in different interaction patterns within the protein. The N-acetyl glucosamine unit linked to N113 is tightly involved in at least five polar interactions with residues D145 (helix α3), D174 (helix α4), N178 (helix α4), and H180 (loop V177–L183). Conversely, the N-acetyl glucosamine linked to N134 is less constrained because it is only involved in one or two polar interactions with L110, depending on the considered chain of Lip2.
A comparison of the Lip2 structure with the closed structures of lipases from T. lanuginosa (TLL, PDB ID: 1DT3), R. niveus (RNL, PDB ID: 1LGY), and R. miehei (RML, PDB ID: 3TGL) shows a conservation of the three-dimensional fold among all four enzymes. The RMSD values obtained after superimposition based on secondary structures using superpose from the CCPS4 suite of program of Lip2 with RML, RNL, and TLL were 1.9 Å (on 245 of 265 Cα atoms), 1.8 Å (on 238 of 265 Cα atoms) and 1.7 Å (on 248 of 269 Cα atoms), respectively.
Catalytic center
The catalytic triad of lipases is invariably composed of a serine, a histidine, and an aspartic/glutamic acid. The nucleophile serine is comprised in a consensus pentapeptide G-X-S-X-G in a γ-like turn between the C-terminal end of the β5-strand (referring to the canonical α/β hydrolase fold nomenclature) and the helix α4. Another important component of the catalytic machinery is the oxyanion hole, which is composed of two hydrogen-bond donor amino acid residues that stabilize the tetrahedral intermediate. On the basis of a multiple sequence alignment, the catalytic machinery residues of Lip2 have been identified as S162, H289, and D230 for the catalytic triad, whereas T88 and L163 are putatively assigned as the residues forming the oxyanion hole. A structural comparison of Lip2 with homologous lipases shows a perfect superimposition of the catalytic triad and one of the two residues of the oxyanion hole (L163 located at the N-extremity of the helix α4 (Fig. 2 a)). However, the location of the second putative residue forming the oxyanion hole (T88 located in the G87–S90 loop) differs considerably from the corresponding amino acid in other homologous lipases (S82, S83, and T83 in RML, TLL, and RNL, respectively (Fig. 2)). The relevance of such differences is discussed in the “Lid region” section below.
Figure 2.
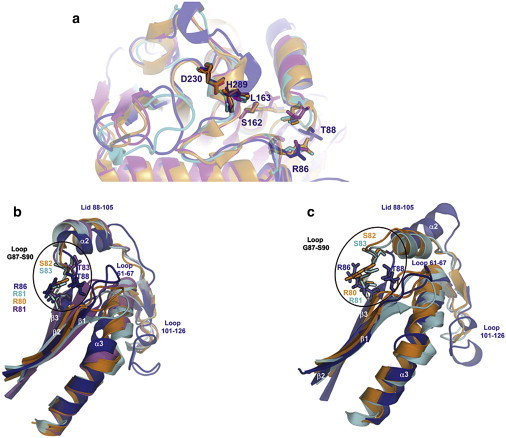
(a) Superposition of closed lipases from the fungal family. Structures of Lip2 (blue), TLL (cyan), RNL (magenta), and RML (orange) are represented in ribbon mode. Amino acids from the catalytic triad and the putative oxyanion hole are represented as sticks. (b) Representation in ribbon mode of the lid: strands β1, β2, and β3; helix α3; and loops D61–D67, G87–S90, and M101–H126 of the closed form of Lip2 (blue), and equivalent elements belonging to TLL (1DT3) (cyan), RNL (1LGY) (magenta), and RML (3TGL) (orange). The amino acids involved in the formation of the oxyanion hole (putative for Lip2) are represented as sticks. (c) Representation of the same elements shown in b for the semi-open form obtained from MD experiments performed on Lip2 (blue) and open forms of TLL (1GT6) (cyan) and RML (4TGL) (orange). For clarity, the G87–S90 loop is encircled in black.
Disulfide bridges
Four disulfide bridges were found in Lip2, instead of the three that were identified in known structures of other fungal lipases. Among these disulfide bridges, two (C30–C299 and C43–C47; bridges 1 and 2 in Fig. 3) are conserved within all of these lipases. The third disulfide bridge (C120–C123) is also found in TLL, whereas the disulfide bridge C265–C273 is conserved among RML (32), RNL (21), and Lip2. Of interest, C123 located in the M101–H126 loop was found in an alternative conformation in chains A, B, C, E, and G (in both reduced and oxidized forms), whereas only the oxidized form was observed in chains D and F.
Figure 3.
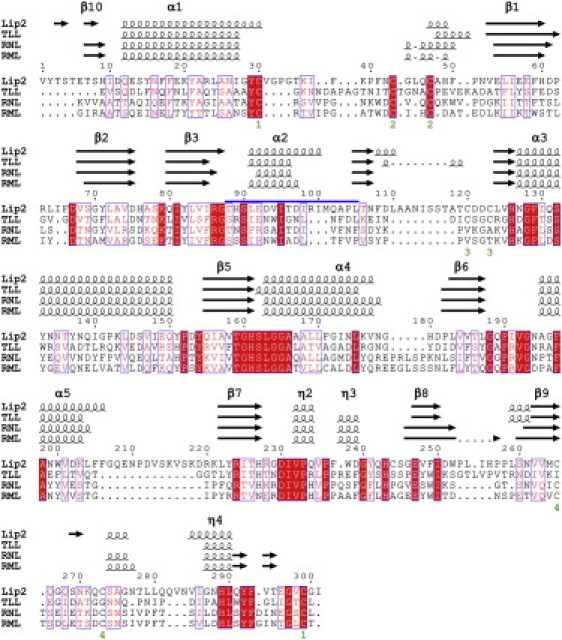
Multiple sequence alignment of lipases from Lip2, TLL, RNL, and RML with their secondary structure elements shown on top. The lid region of Lip2 is highlighted with a thick line on the top of the alignment. β-sheets are numbered according to the nomenclature of Ollis et al. (44) .
Likelihood of a potassium ion
Close inspection of electron density maps highlighted a strongly positive residual density around some automatically assigned water molecules with a very low B-factor value. They are located near the thiol group of the free C244. Distances between this positive density and atoms C244-SE1, C244-N, H243-ND1, Q189-NE2, and O of an adjacent water molecule were measured as 3.8, 3.4, 3.1, 3.1, and 3.0 Å, respectively. These large interatomic distances in conjunction with the exclusive electrostatic interaction type led us to assume the presence of a potassium ion at this position. Potassium ion is well known to form predominantly electrostatic interactions with protein atoms. The coordination geometry of this ion was shown to be a distorted square pyramidal geometry with a coordination number of 5. This geometry is highly conserved among all seven molecules in the asymmetric unit.
Lid region
The lid region, a key element in the activity and specificity of lipases (33–35), is composed of an α-helix (α2) and two hinge segments in fungal lipases (Fig. 3). A comparison of the open and closed structures of RML and TLL enabled the identification of the lid region of these enzymes as being composed of residues S82–P96 and S83–L97, respectively. By analogy, we could infer that the Lip2 lid is formed by residues T88–L105. It is noteworthy that the helix α2 forming the lid in Lip2 is three residues longer than in homologs. Thus, the additional amino acid residues I98-R99-I100 form a supplementary α-helical turn at the C-terminus of the lid that is not observed in any of the homologous fungal lipases (Fig. 3). Additionally, because the strand β3 is two residues longer in Lip2, the interconnecting loop G87–S90, located between strand β3 and helix α2, is two residues shorter. Consequently, the conformation of this loop is drastically different compared to other lipases in their closed conformation (Fig. 2 b). This loop is packed against the D61–D67 loop, which also adopts an unusual conformation compared to other closed structures. Although the D61–D67 loop does not appear to be constrained by crystal packing, it is located closer to the lid than in other homologous lipases where it is stabilized by a network of polar and hydrophobic interactions. As a result, both sides of the lid interact with counterparts in the closed conformation of Lip2, whereas only residues from the inside of the lid are involved in interactions in the closed structures of homologous lipases. No interaction could be seen between the lid and the corresponding D61–D67 loop in Lip2 homologs. Only during the opening of the lid are these two elements brought together, allowing the formation of the numerous stabilizing interactions observed in the open conformation of lipases. Because these two structural elements are already found in close contact in the closed conformation of Lip2, one could assume that Lip2 might adopt a different lid-opening mechanism compared to homologous fungal lipases. In its closed conformation, interactions occurring between the lid and the D61–D67 loop involve hydrophobic residues such as I65, F66, V94, and I98, as well as polar interactions between the NE2 atom of H89 and the main-chain oxygen atoms of P62 and L64. Residue T88, the putative residue of the oxyanion hole, is also involved in polar interactions with the D61–D67 loop (with distances T88-OG1/S69-O and T88-N/D67-O of 2.9 Å). This hydrogen-bonding pattern is completely different from what is found in homologous lipases. Indeed, the hydroxyl groups of S82 (RML) and S83 (TLL), equivalent to T88 of Lip2, are respectively coordinated to S84 (RML) and S85 (TLL), which are both located at the beginning of the lid helix (with distances S82-OG1/S84-N and S83-OG1/S85-N of 3.2 Å, respectively). During the activation process, the side chains of these residues undergo a reorientation and become available for interaction bonding with the substrate while their backbone stays in a similar conformation in both the open and closed conformations. This is clearly reflected by the distances measured in the closed structures between the Cα atoms of the residue of the oxyanion hole and the catalytic serine residue (7.4 Å for TLL, and 7.6 Å for RML) which are close to those measured in the open conformations (7.5 Å for TLL and 7.4 Å for RML). Strikingly, the formation of the oxyanion hole in Lip2 may differ significantly from homologous lipases, as both the side chain and the backbone amide of T88 are not conveniently oriented toward the catalytic center due to the unusual conformation of the G87–S90 loop (Fig. 2 b). Therefore, the G87–S90 loop should undergo a larger conformational rearrangement (not observed in homologous lipases) to bring T88 into a proper position and allow the stabilization of the tetrahedral intermediate of Lip2.
The M101–H126 loop, located at the C-terminal end of the lid, also differs from the loop usually found in other lipases because it is 11–12 residues longer (Fig. 3). This loop and helix α3 show a larger surface of interactions (involving in particular the N-acetyl glucosamine moieties borne by N113 and N134; Figs. 1 a and 2 b). The other side of helix α3 interacts with the D61–D67 loop (also in close contact with the lid) while the N-terminal extremity of helix α3 stabilizes the C-terminus of the lid. Such a network of interactions in the lid region is not observed in closed conformations of homologous lipases. Therefore, one can assume that the activation mechanism of Lip2 could involve drastic conformational rearrangements that may be also accompanied by changes in the interaction network involving the D61–D67 and M101–H126 loops.
The interfacial mechanism followed by lipases can vary significantly depending on their structure and origin. Some lipases have been described to involve conformational changes upon adsorption at the lipid interface (36) or substrate binding (37). Other lipases have also been described to follow a two-step mechanism initiated by the adsorption of the enzyme at the lipid interface, followed by the formation of the enzyme/substrate complex. To further investigate the mechanism by which the lid of Lip2 might switch from a closed to an open state, we carried out a series of large-scale MD simulations using different solvent conditions (water and octane/water interface) to investigate the effects of substrate binding.
Chain G
Chain G differs significantly from the six other molecules in the asymmetric unit, with a mean RMSD value on Cα atoms of 0.8 Å compared to 0.3 Å found when superimposing chains A–F. Of note, the main differences are observed around the lid region (L91–L105) and in the D61–D67 loop (Fig. S2). Because these differences are inferred by crystal packing constraints, they are not considered in the following discussion; instead, they are described in the Supporting Material section.
Dynamic insights into lid opening—effect of solvent
The starting model of Lip2 was built from the x-ray structure of the closed conformation described above. The model was solvated in explicit water and an octane/water interface, and a 20 ns MD simulation was performed for each of the two enzyme-solvent systems. The MD simulations were run twice using different starting velocities to avoid artifacts. Because they showed essentially the same fluctuations, the analysis of only one MD simulation is described below.
As expected, no significant conformational change was observed during the MD simulation carried out in water, and the structure remained in a closed conformation. The core of the enzyme was stable and slight deviations were only observed for loops located at the surface. Of note, the structural features observed at the water/octane interface (and described below) were not observed. The backbone RMSD between the conformation after 20 ns and the initial structure was 1.5 Å for the global structure (Fig. 4 a) and 1.3 Å for the lid.
Figure 4.
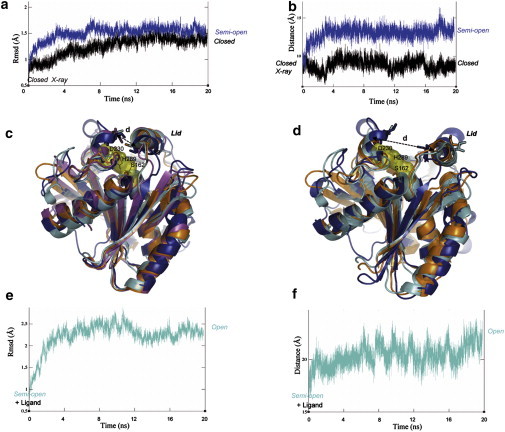
(a) RMSD of backbone atoms of Lip2 and (b) variation of the distance between the amino acid residues I95 and I286 as a function of time during 20 ns MD simulations in water (black) or at the octane/water interface (blue and cyan) starting from the x-ray structure of the closed form. (c and d) Superposition based on backbone atoms of the structures of Lip2, TLL, RNL and RML, with the lid in a closed (c) or open (d) conformation. Residues forming the catalytic triad (S162, D230, and H289 in Lip2) are colored in yellow. (c) Closed x-ray structures of Lip2 (blue), TLL (cyan), RNL (magenta), and RML (orange) are represented in cartoon mode. For analysis of these closed conformations, distances d(I95-Cα/I286-Cα) = 9.0 Å, d(I90-Cα/I255-Cα) = 9.3 Å, d(I90-Cα/I254-Cα) = 9.1 Å, and d(I89-Cα/V254-Cα) = 8.6 Å were respectively measured for Lip2, TLL, RNL, and RML, and are represented as dotted lines. (d) Open x-ray structures of TLL (cyan) and RML (orange) are represented in cartoon mode. The semi-open model of Lip2 obtained after 20 ns of MD simulation at the water/octane interface is also shown (blue). For analysis of these open conformations, distances d(I95-Cα/I286-Cα) = 13.4 Å, d(I90-Cα/I255-Cα) = 19.0 Å, and d(I89-Cα/V254-Cα) = 18.3 Å were respectively measured for Lip2, TLL, and RML. (e) RMSD of backbone atoms of Lip2 and (f) variation of the distance between the amino acid residues I95 and I286 as a function of time during 20 ns MD simulations in water (black) or at the octane/water interface (blue and cyan) starting from the semi-open conformation of Lip2 (obtained after 8 ns of MD simulation in octane/water interface) with the catalytic serine residue covalently bound to the (S) ethyl 2-bromophenylacetate substrate (cyan).
At the octane/water interface, slightly faster (<4 ns) and larger-amplitude changes were observed. The backbone RMSD of the conformation after 20 ns compared to the initial structure was 1.6 Å for the global structure (Fig. 4 a) and 2.0 Å for the lid. A more detailed analysis of this simulation showed that conformational changes resulted mainly from motions of the lid helix α2. Analysis of the distance between the Cα atoms of I95 and I286 during the MD simulation revealed a significant displacement of the lid subdomain by ∼4–5 Å (Fig. 4 b). Despite the structural differences observed in the Lip2 lid region, the distance d(I95-Cα/ I286-Cα) in Lip2 (∼8.5 Å) was nearly comparable to the corresponding distances measured in x-ray structures of the closed conformation of TLL, RNL, and RML (Fig. 4 c). A comparison of this distance in Lip2 after 20 ns of MD simulation at the octane/water interface (d(I95-Cα/I286-Cα) ∼13.4 Å) with that between corresponding conserved residues in the x-ray structures of open TLL and RML revealed differences in the degree of opening of the structures (Fig. 4 d), suggesting that the open conformation reached by Lip2 during the MD simulation is in fact a partially open conformation. Such a partially open conformation was previously described for TLL in an MD study (38). This semi-open conformation of Lip2 could not be further opened even when the MD simulation was pursued up to 60 ns. Striking differences were observed in the semiopen conformation adopted by the lid, particularly in the orientation of helix α2, which was tilted by ∼45° compared to other homologous lipases. The hinge-type motion of the lid helix α2 is accompanied by concomitant conformational changes within surrounding loops (G87–S90, D61–D67, and M101–H126) and to a lesser extent of helix α3, which is embedded between the D61–D67 and M101–H126 loops. During such a conformational rearrangement, in particular the G87–S90 loop movement, the putative residue of the oxyanion hole (T88) is moved farther away from the catalytic center, as indicated by the increase of the distance d-(T88-Cα/S162-Cα) from 10.9 Å to 12.3 Å (Fig. 2 c). Thus, T88 is involved in polar interactions with D67 and S69 from the D61–D67 loop (d(T88-N/D67-O) = 2.6 Å, d-(T88-O-D67-O) = 2.5 Å, and d(T88-O-S69-O) = 2.8 Å), rather than being engaged in stabilizing the tetrahedral intermediate, as seen in x-ray structures of TLL and RML in their open conformation. Analysis of the MD simulation at the octane/water interface thus revealed that Lip2 is trapped in a semi-open conformation.
Because MD simulations carried out at the octane/water interface did not show the complete transition from the closed to a fully open conformation of Lip2, we suspected the presence of a high-energy barrier associated with the transition. A partial lid opening was also previously observed for RML (39), TLL (38), and CRL (40) during MD simulations in explicit solvents. Lid opening of TLL was then suggested to occur through a multistep process involving several conformational transitions before a fully activated conformation is attained. For Lip2, one can assume that either the transition from the closed to the fully open conformation cannot be attained when starting from the closed crystallographic structure described herein and within the time frame of our MD simulation (60 ns), or the sole adsorption of the protein at the lipid/water interface cannot allow a complete lid opening, and substrate binding might be needed for full activation of the enzyme.
Dynamic insight into lid opening—effect of ligand binding
To explore the potential role of the substrate in the interfacial activation mechanism, we used a structure snapshot obtained after 8 ns of MD simulation carried out at the octane/water interface where a substrate was covalently bound to the catalytic residue S162 using a previously described procedure (15). (S) Ethyl 2-bromophenylacetate, a molecule of pharmaceutical importance that is well recognized by Lip2, was chosen as a substrate (5). Next, we ensured that this substrate could access the catalytic center of the semiopen conformation of Lip2 by computing the substrate trajectory from the catalytic site to the protein surface using a robotics-based approach (29,30). Our results clearly show that the partially open conformation of the lid does not impede the access of the substrate to the active site from the external environment. The tetrahedral intermediate was then further subjected to 20 ns of MD simulation at the octane/water interface. The overall RMSD quickly increased (i.e., in <3 ns) by ∼1.5 Å (Fig. 4 e). Upon ligand binding, the conformation of the lid in Lip2 was moved farther away. The main conformational changes that occurred under solvent effect and upon ligand binding are shown in Fig. S3. In particular, the distance between the Cα atoms of I95 and I286 went from 13.4 Å in the semi-open unliganded state up to 22 Å in the final open ligand-bound conformation of Lip2 (Fig. 4 f). A detailed analysis of the tetrahedral intermediate conformation obtained after 20 ns of MD simulation at the octane/water interface showed the presence of the commonly found hydrogen-bonding interactions between the substrate and the catalytic residues (S162, H289, and D230). Strikingly, no interaction was observed during the entire 20 ns simulation between the substrate and T88, the putative residue forming the oxyanion hole. However, T88 is found to be involved in polar interactions with the D61–D67 loop, helping to stabilize the open conformation of the lid.
Questioning the stabilizing role of T88 and R86
The MD simulations described above reveal that the putative oxyanion hole T88 is oriented in a catalytically unproductive conformation during the entire simulations, which brings into question its real role in catalysis. On the other hand, another residue, R86 from the conserved RG motif, reorients its side chain toward the catalytic center during the MD simulation at octane/water interface, bringing the guanidinium group into a proper positioning for the stabilization of the negative charge of the tetrahedral intermediate in the MD simulation with ligand. The conformational rearrangement observed for R86 residue in Lip2 is similar to that observed for the residue known to form the oxyanion hole in other fungal lipases (S82 in RML, S83 in TLL). These side-chain switches are often observed during the activation process of homologous lipases.
Although features of the oxyanion hole are conserved in most filamentous fungal lipases, closely related lipases, such as the M37 lipase from Photobacterium lipolyticum, have been reported to exhibit distinct oxyanion hole features, in particular a larger cavity due mostly to the RG sequence (41). Of interest, a novel family of bacterial lipases very similar to fungal lipases was recently described to use an RG sequence as an oxyanion hole (42). It is also worth mentioning that a conserved arginine residue was found to play an important stabilizing role during opening of the lid in the bacterial BTL2 lipase from Geobacillus thermocatenulatus (43). On the basis of these data, we propose that R86 may participate in stabilizing the transition state during Lip2 catalysis, and thus together with L163 may form an unusual oxyanion hole within the fungal lipase family. Although MD simulations seem to indicate that T88 may not be involved in stabilizing the transition state, T88 reorientation toward the catalytic center may require some conformational rearrangements associated with a high-energy barrier that could not be observed within the MD simulation timescale. To discriminate between these two candidates for the stabilization of the tetrahedral intermediate, we performed mutagenesis experiments. A detrimental loss of activity was observed when R86 was mutated into an alanine or a lysine amino acid (data not shown), as well as when T88 was substituted into a leucine, a valine, or an alanine amino acid (15). Thus, these results did not allow us to draw any conclusions regarding the role of these residues. Of note, when T88 was mutated into a serine residue, a significant part of the activity was conserved (∼30% residual activity). Thus, the nature of the side chain at position 88 may be instrumental in the activity of the enzyme. Replacement of the polar T88 side chain by a serine could keep some of the hydrogen-bonding interactions with the D61–D67 loop observed during the MD simulation. Overall, our results show that both R86 and T88 residues have a crucial role in Lip2 activity.
Conclusions
By combining x-ray crystallography and MD simulations, we were able to show for the first time the three-dimensional structure of the Lip2 lipase from Y. lipolytica in its closed conformation and the extensive structural rearrangements required for full activation of the enzyme. The 1.7 Å structure of Lip2 revealed a typical α/β-hydrolase fold, although some unique features compared to homologous fungal lipases were also observed. The unusual conformation of a central loop containing the putative T88 residue that comprises the oxyanion hole led us to suggest the alternative participation of R86 in the stabilization of the transition state during catalysis. However, mutagenesis experiments did not permit us to clearly determine which one of these residues was involved in the oxyanion hole formation, although their role in Lip2 activity was shown to be crucial.
From the crystallographic structure, we performed large-scale MD simulations that revealed that the adsorption of Lip2 at the water/octane interface resulted in a partially open conformation that could still allow access of the substrate to the catalytic site. Only upon subsequent binding of the substrate did the lipase adopt a fully open form. Altogether, our data suggest a two-step activation mechanism driven by adsorption of the enzyme at the water/octane interface followed by enzyme-substrate binding. These results provide a comprehensive view of the molecular determinants that trigger the conformational transition undergone by the enzyme during its activation. This information will be used in our laboratory to engineer Lip2 mutants with improved properties for biotechnological purposes.
Acknowledgments
We thank the ESRF for giving us access to the synchrotron radiation facilities; Dr. A.-M. Goncalves for assistance in using beamline ID14-4; V. Guillet, F. Bergeret, L. Maveyraud, and J.-D. Pédelacq for their help with data collection at the ESRF; and the Computing Center of Region Midi-Pyrénées (Toulouse, France) and the Center for Computing Resources of INSA Toulouse for providing calculation resources and support.
Contributor Information
Isabelle André, Email: isabelle.andre@insa-toulouse.fr.
Alain Marty, Email: marty@insa-toulouse.fr.
Samuel Tranier, Email: tranier@ipbs.fr.
Supporting Material
References
- 1.Pandey A., Benjamin S., Soccol V.T. The realm of microbial lipases in biotechnology. Biotechnol. Appl. Biochem. 1999;29:119–131. [PubMed] [Google Scholar]
- 2.Pignède G., Wang H., Nicaud J.M. Characterization of an extracellular lipase encoded by LIP2 in Yarrowia lipolytica. J. Bacteriol. 2000;182:2802–2810. doi: 10.1128/jb.182.10.2802-2810.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu M., Lange S., Schmid R.D. High-level expression of extracellular lipase Lip2 from Yarrowia lipolytica in Pichia pastoris and its purification and characterization. Protein Expr. Purif. 2007;53:255–263. doi: 10.1016/j.pep.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 4.Aloulou A., Puccinelli D., Carrière F. A comparative study on two fungal lipases from Thermomyces lanuginosus and Yarrowia lipolytica shows the combined effects of detergents and pH on lipase adsorption and activity. Biochim. Biophys. Acta. 2007;1771:1446–1456. doi: 10.1016/j.bbalip.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 5.Cancino M., Bauchart P., Marty A. A variant of Y. lipolytica lipase with improved activity and enantioselectivity for resolution of 2-bromo-arylacetic esters. Tetrahedron Asymmetry. 2008;19:1608–1612. [Google Scholar]
- 6.Ferrato F., Carriere F., Verger R. A critical reevaluation of the phenomenon of interfacial activation. Methods Enzymol. 1997;286:327–347. doi: 10.1016/s0076-6879(97)86018-1. [DOI] [PubMed] [Google Scholar]
- 7.Henzler-Wildman K., Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 8.Cherukuvada S.L., Seshasayee A.S., Pennathur G. Evidence of a double-lid movement in Pseudomonas aeruginosa lipase: insights from molecular dynamics simulations. PLOS Comput. Biol. 2005;1:e28. doi: 10.1371/journal.pcbi.0010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbe S., Lafaquière V., André I. Insights into lid movements of Burkholderia cepacia lipase inferred from molecular dynamics simulations. Proteins. 2009;77:509–523. doi: 10.1002/prot.22462. [DOI] [PubMed] [Google Scholar]
- 10.Trodler P., Schmid R.D., Pleiss J. Modeling of solvent-dependent conformational transitions in Burkholderia cepacia lipase. BMC Struct. Biol. 2009;9:38. doi: 10.1186/1472-6807-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pignède G., Wang H.J., Nicaud J.M. Autocloning and amplification of LIP2 in Yarrowia lipolytica. Appl. Environ. Microbiol. 2000;66:3283–3289. doi: 10.1128/aem.66.8.3283-3289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicaud J.M., Madzak C., Gaillardin C. Protein expression and secretion in the yeast Yarrowia lipolytica. FEM. Yeast Res. 2002;2:371–379. doi: 10.1016/S1567-1356(02)00082-X. [DOI] [PubMed] [Google Scholar]
- 13.Sambrook J., Fritsh E.F., Maniatis T. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- 14.Le Dall M.T., Nicaud J.M., Gaillardin C. Multiple-copy integration in the yeast Yarrowia lipolytica. Curr. Genet. 1994;26:38–44. doi: 10.1007/BF00326302. [DOI] [PubMed] [Google Scholar]
- 15.Bordes F., Cambon E., Marty A. Improvement of Yarrowia lipolytica lipase enantioselectivity by using mutagenesis targeted to the substrate binding site. Chembiochem. 2009;10:1705–1713. doi: 10.1002/cbic.200900215. [DOI] [PubMed] [Google Scholar]
- 16.Quinn D.M., Shirai K., Harmony J.A. Lipoprotein lipase catalyzed hydrolysis of water-soluble p-nitrophenyl esters. Inhibition by apolipoprotein C-II. Biochemistry. 1982;21:6872–6879. doi: 10.1021/bi00269a038. [DOI] [PubMed] [Google Scholar]
- 17.Leslie A.G.W. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography. 1992 No. 26. [Google Scholar]
- 18.Evans P.R. SCALA. Joint CCP4 & ESF-EACBM Newslett. 1997;33:22–24. [Google Scholar]
- 19.Collaborative Computational Project, Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 20.Vagin A., Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Cryst. 1997;30:1022–1025. [Google Scholar]
- 21.Kohno M., Funatsu J., Morita Y. The crystal structure of lipase II from Rhizopus niveus at 2.2 A resolution. J. Biochem. 1996;120:505–510. doi: 10.1093/oxfordjournals.jbchem.a021442. [DOI] [PubMed] [Google Scholar]
- 22.Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 23.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 24.Laskowski R.A., MacArthur M.W., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 25.Lupyan D., Leo-Macias A., Ortiz A.R. A new progressive-iterative algorithm for multiple structure alignment. Bioinformatics. 2005;21:3255–3263. doi: 10.1093/bioinformatics/bti527. [DOI] [PubMed] [Google Scholar]
- 26.Gouet P., Courcelle E., Métoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 27.Krissinel E., Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 28.Case D.A., Darden T.E., Kollman P.A. University of California; San Francisco: 2006. AMBER 9. [Google Scholar]
- 29.Cortés J., Siméon T., Tran V. A path planning approach for computing large-amplitude motions of flexible molecules. Bioinformatics. 2005;21(Suppl 1):i116–i125. doi: 10.1093/bioinformatics/bti1017. [DOI] [PubMed] [Google Scholar]
- 30.Lafaquière V., Barbe S., Remaud-Simeon M. Control of lipase enantioselectivity by engineering the substrate binding site and access channel. Chembiochem. 2009;10:2760–2771. doi: 10.1002/cbic.200900439. [DOI] [PubMed] [Google Scholar]
- 31.Jolivet P., Bordes F., Nicaud J.M. Analysis of Yarrowia lipolytica extracellular lipase Lip2p glycosylation. FEM. Yeast Res. 2007;7:1317–1327. doi: 10.1111/j.1567-1364.2007.00293.x. [DOI] [PubMed] [Google Scholar]
- 32.Brady L., Brzozowski A.M., Norskov L. A serine protease triad forms the catalytic centre of a triacylglycerol lipase. Nature. 1990;343:767–770. doi: 10.1038/343767a0. [DOI] [PubMed] [Google Scholar]
- 33.Brocca S., Secundo F., Lotti M. Sequence of the lid affects activity and specificity of Candida rugosa lipase isoenzymes. Protein Sci. 2003;12:2312–2319. doi: 10.1110/ps.0304003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Santarossa G., Lafranconi P.G., Lotti M. Mutations in the “lid” region affect chain length specificity and thermostability of a Pseudomonas fragi lipase. FEBS Lett. 2005;579:2383–2386. doi: 10.1016/j.febslet.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 35.Secundo F., Carrea G., Lotti M. Activity and enantioselectivity of wildtype and lid mutated Candida rugosa lipase isoform 1 in organic solvents. Biotechnol. Bioeng. 2004;86:236–240. doi: 10.1002/bit.20034. [DOI] [PubMed] [Google Scholar]
- 36.Derewenda Z.S. Structure and function of lipases. Adv. Protein Chem. 1994;45:1–52. doi: 10.1016/s0065-3233(08)60637-3. [DOI] [PubMed] [Google Scholar]
- 37.Thuren T. A model for the molecular mechanism of interfacial activation of phospholipase A2 supporting the substrate theory. FEBS Lett. 1988;229:95–99. doi: 10.1016/0014-5793(88)80805-6. [DOI] [PubMed] [Google Scholar]
- 38.Jensen M.Ø., Jensen T.R., Peters G.H. Orientation and conformation of a lipase at an interface studied by molecular dynamics simulations. Biophys. J. 2002;83:98–111. doi: 10.1016/S0006-3495(02)75152-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peters G.H., Bywater R.P. Influence of a lipid interface on protein dynamics in a fungal lipase. Biophys. J. 2001;81:3052–3065. doi: 10.1016/S0006-3495(01)75944-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.James J.J., Lakshmi B.S., Gautam P. Activation of Candida rugosa lipase at alkane-aqueous interfaces: a molecular dynamics study. FEBS Lett. 2007;581:4377–4383. doi: 10.1016/j.febslet.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 41.Jung S.K., Jeong D.G., Kim S.J. Structural basis for the cold adaptation of psychrophilic M37 lipase from Photobacterium lipolyticum. Proteins. 2008;71:476–484. doi: 10.1002/prot.21884. [DOI] [PubMed] [Google Scholar]
- 42.Lee M.-H., Lee C.-H., Yoon J.H. Isolation and characterization of a novel lipase from a metagenomic library of tidal flat sediments: evidence for a new family of bacterial lipases. Appl. Environ. Microbiol. 2006;72:7406–7409. doi: 10.1128/AEM.01157-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carrasco-López C., Godoy C., Hermoso J.A. Activation of bacterial thermoalkalophilic lipases is spurred by dramatic structural rearrangements. J. Biol. Chem. 2009;284:4365–4372. doi: 10.1074/jbc.M808268200. [DOI] [PubMed] [Google Scholar]
- 44.Ollis D.L., Cheah E., Goldman A. The α/β hydrolase fold. Protein Eng. 1992;5:197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.