

Abstract
A recently determined atomic structure of an H+-coupled ATP-synthase membrane rotor has revived the long-standing question of whether protons may be bound to these structures in the form of a hydronium ion. Using both classical and quantum-mechanical simulations, we show that this notion is implausible. Ab initio molecular dynamics simulations of the binding site demonstrate that the putative H3O+ deprotonates within femtoseconds. The bound proton is thus transferred irreversibly to the carboxylate side chain found in the ion-binding sites of all ATP-synthase rotors. This result is consistent with classical simulations of the rotor in a phospholipid membrane, on the 100-nanosecond timescale. These simulations show that the hydrogen-bond network seen in the crystal structure is incompatible with a bound hydronium. The observed coordination geometry is shown to correspond instead to a protonated carboxylate and a bound water molecule. In conclusion, this study underscores the notion that binding and transient storage of protons in the membrane rotors of ATP synthases occur through a common chemical mechanism, namely carboxylate protonation.
The membrane domain of ATP synthases (and related ATPases) couples their catalytic activity to electrochemical gradients. This coupling is made possible by the rotor subunit, an oligomeric assembly of transmembrane helices. This ring-like structure binds H+ or Na+ and mediates their membrane translocation, either down or against the gradient, through a rotary mechanism that is linked to ATP synthesis or hydrolysis, respectively (1,2).
X-ray crystallography and computer simulations have revealed the atomic structure of the ion-binding sites in several rotors (3–8). These sites are located on the outer face of the rings, half-way across the membrane. Different rotors use different amino acids for ion coordination, thus modulating their specificity and affinity (9). A conserved carboxylate side chain is the most prominent among these, countering most of the energy cost of ion dehydration.
A long-standing question with regard to ion binding is whether H+ binds to the rotor rings through protonation of the conserved carboxylate, or as a hydronium ion (10). This question, raised by Boyer (10) more than 20 years ago, followed the observation that some transport systems are alternatively driven by H+ or Na+ gradients, including ATP synthases. The then recent discovery of the structure of a cation-binding crown ether, with a bound H3O+ (11), proved that a given coordination chemistry can, in principle, suit both Na+ and hydronium. More recently, the possibility of proton binding as H3O+ has been invoked to provide a mechanistic underpinning to the mystifying pH dependence of DCCD-based inhibition assays (12); the concept has since caught on, and for other transport systems as well (13–15).
The subsequent determination of the atomic structures of two H+-coupled rotors, however, appeared to establish that binding of H+ to ATP-synthase rotors occurs through side-chain protonation (4,5,8). In these rings (both of which show anomalous pH profiles), the conserved carboxylate side chain is clearly engaged in an intersubunit hydrogen bond, acting as a donor. This notwithstanding, the more recent structure of the c13 ring from Bacillus pseudofirmus has reopened the controversy (16). The suggested structural similarity of its binding sites to the crown ether mentioned above, and the presence of what appears to be a water molecule, appear to revive the notion of H3O+ binding.
In this work, we use classical and quantum-mechanical computer simulations to assess whether or not protons bind to the Bacillus pseudofirmus rotor ring in the form of a hydronium species. This theoretical analysis unequivocally demonstrates that they do not. Instead, like the c15 and c14 rotors previously described (4,5), the Bacillus c13 ring binds and shuttles H+ in the membrane through protonation of the carboxylate side chain in the binding site.
Our first line of evidence derives from inspection of the structure itself. Like the crown ether, the proton-binding site in the Bacillus ring includes three potential hydrogen-bond acceptors of the putative H3O+ ion; these are the side-chain carboxylate of E54, and the backbone carbonyls of L52 and A53 (Fig. 1 A). However, the first coordination shell also includes a hydrogen-bond donor, namely the backbone amide of V56. This is an important distinction, as the likelihood that H3O+ acts as an acceptor is, in fact, negligible. The electrostatics of such interaction is simply not favorable, as shown by detailed theoretical studies of the structure and dynamics of hydrated protons (17).
Figure 1.
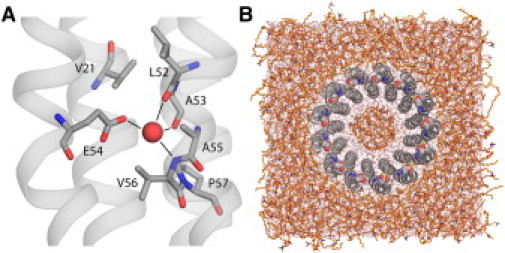
(A) Crystal structure of the proton-binding site in the c13 rotor from Bacillus pseudofirmus OF4. (Red sphere) Putative H3O+. (B) View of the simulation system, with the rotor embedded in a model lipid membrane.
The logical hypothesis that follows this argument is that the coordination geometry in the crystal structure is incompatible with a bound H3O+. To assess this hypothesis, we carried out a series of classical molecular dynamics simulations of the c13 rotor embedded in a lipid membrane. The simulations were prepared either with a bound H3O+ ion and E54 in the ionized state, or with protonated E54 and a bound water molecule. Classical simulations do not allow for dissociation or formation of covalent bonds; thus, these configurations are chemically stable by construction. Three slightly different H3O+ models were used, to account for the inexactness of the classical approximation.
As shown in Fig. 2 and Fig. S1 in the Supporting Material, the simulations of the rotor with a bound H3O+ consistently showed clear deviations from the geometry observed in the crystal structure. The electrostatic repulsion with the amide of V56 appreciably shifts the ion position deeper in the binding site, thereby altering the hydrogen-bonding network. For example, the carbonyl of A53 is no longer engaged, while those of G17 and A18 alternate as transient new acceptors. Also, the coordination by E54 becomes bidentate, with both H2O/O-Oɛ distances outside the range measured. By contrast, in simulations in which E54 is protonated, the geometry of the binding site remains in good agreement with the crystal structure, i.e., the four hydrogen-bond network in Fig. 1 A corresponds to a plain water molecule. Incidentally, the conclusions from these classical simulations are consistent with density-functional-theory structural optimizations and molecular-orbital analysis (see the Supporting Material).
Figure 2.
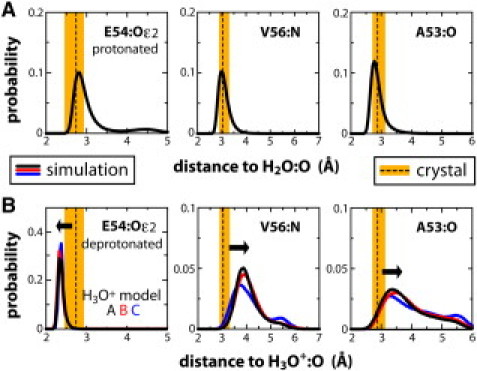
Representative coordination distances in the Bacillus c13 rotor ring. Average and spread crystal-structure values (dashed line, orange band) are compared with calculated ensembles from classical simulations of the rotor in a membrane, at ∼300 K (solid lines). (A) Simulations in which E54 is protonated, and a water is bound to the site; (B) simulations in which H+ is bound as a H3O+ species, and E54 is deprotonated.
The second line of evidence pertains to the differential proton affinity that can be expected of a water molecule and a carboxylate. Clearly, the presence of a competing protonatable group in the coordination shell is another crucial distinction between the aforementioned crown ether and the actual binding site in the Bacillus rotor ring. An estimate of the relative proton affinities of water and carboxylate can be derived from the solution pKa of their conjugate acids, i.e., ∼−1.7 and 4.0. That is, the affinity of a water molecule for an additional proton is 1,000,000-times weaker than that of a carboxylate group.
For the Bacillus rotor to favor a hydronium, then, it must provide an environment that could reverse this inherent bias. We assessed whether this is the case through ab initio molecular dynamics simulations of the binding site, which by design allow for bond-breaking and formation, and thus enable us to study the proton dynamics.
The results from these simulations consistently show that, when H+ is initially bound to the water molecule, transfer to E54 occurs within ∼100 fs (Fig. 3 and Fig. S2). Proton transfer is effectively irreversible; no indication of a back-reaction was detected in simulations with H+ initially bound to E54. These observations demonstrate that the proton affinity of the carboxylate groups is also much stronger than that of water, in the context of the rotor ring.
Figure 3.
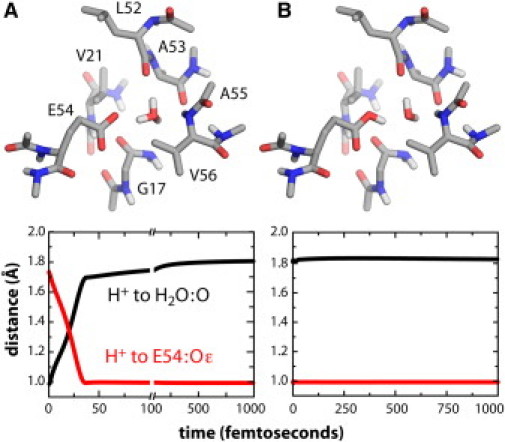
Ab initio simulations of proton dynamics within a reduced model of the ion-binding site of the Bacillus rotor. Configurations at t = 0 are depicted for (A) the putative H3O+ state, and (B) the site with E54 protonated plus a bound H2O. The plots describe the proton trajectories as the distance to the water or carboxylate oxygen atoms.
In conclusion, in the Bacillus c13 ring as well as in other H+-coupled rotors, an incoming proton will ultimately bind covalently to the conserved carboxylate side chain, even if it has reached the site via a hydrated pathway (18). It is in this state that the bound protons are shuttled by the lipid-facing c-subunits as the ring rotates within the membrane, before being released at the a-subunit interface.
The amino-acid composition of the ion-binding sites in ATP-synthase rotors is significantly variable, even among those known or expected to be H+-coupled (Fig. 4). Their stoichiometry, and the arrangement of their constituent subunits, also varies greatly. Nonetheless, all available structural and theoretical evidence points to a common mode of H+ binding, namely protonation of the strictly conserved carboxylate side chain. Intramolecular interactions formed by this protonated E/D stabilize the closed conformation of the site, and thus enable the rotary mechanism to proceed. These interactions may be direct, such as with a backbone carbonyl in the Spirulina c15 rotor, or with a side chain, e.g., a serine hydroxyl, as we anticipate for the Gloeobacter c15; but they may be water-mediated, as in the Bacillus c13 ring, in which the site is mostly hydrophobic and its flanking helices are further apart. To date, however, the notion of H3O+ binding to ATP-synthase rotors remains without a basis in reason or in fact.
Figure 4.

Representative proton-binding sites of ATP-synthase membrane rotors. The Gloeobacter c-ring, whose stoichiometry is most likely 15 (19), is a model based on molecular dynamics simulations of an A63S mutant of the Spirulina c15 rotor (9).
Acknowledgments
We thank Thomas Meier for providing the atomic structure of the Bacillus pseudofirmus OF4 c13 ring before publication, and Qiang Cui for helpful discussions.
This work was supported in part by the Deutsche Forschungsgemeinschaft project No. EXC115, and by the Jülich and Leibniz Supercomputing Centers.
Supporting Material
References and Footnotes
- 1.Junge W., Sielaff H., Engelbrecht S. Torque generation and elastic power transmission in the rotary F0F1-ATPase. Nature. 2009;459:364–370. doi: 10.1038/nature08145. [DOI] [PubMed] [Google Scholar]
- 2.Nakamoto R.K., Baylis Scanlon J.A., Al-Shawi M.K. The rotary mechanism of the ATP synthase. Arch. Biochem. Biophys. 2008;476:43–50. doi: 10.1016/j.abb.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meier T., Krah A., Faraldo-Gómez J.D. Complete ion-coordination structure in the rotor ring of Na+-dependent F-ATP synthases. J. Mol. Biol. 2009;391:498–507. doi: 10.1016/j.jmb.2009.05.082. [DOI] [PubMed] [Google Scholar]
- 4.Pogoryelov D., Yildiz O., Meier T. High-resolution structure of the rotor ring of a proton-dependent ATP synthase. Nat. Struct. Mol. Biol. 2009;16:1068–1073. doi: 10.1038/nsmb.1678. [DOI] [PubMed] [Google Scholar]
- 5.Krah A., Pogoryelov D., Faraldo-Gómez J.D. On the structure of the proton-binding site in the F0 rotor of chloroplast ATP synthases. J. Mol. Biol. 2010;395:20–27. doi: 10.1016/j.jmb.2009.10.059. [DOI] [PubMed] [Google Scholar]
- 6.Meier T., Polzer P., Dimroth P. Structure of the rotor ring of F-type Na+-ATPase from Ilyobacter tartaricus. Science. 2005;308:659–662. doi: 10.1126/science.1111199. [DOI] [PubMed] [Google Scholar]
- 7.Murata T., Yamato I., Walker J.E. Structure of the rotor of the V-type Na+-ATPase from Enterococcus hirae. Science. 2005;308:654–659. doi: 10.1126/science.1110064. [DOI] [PubMed] [Google Scholar]
- 8.Vollmar M., Schlieper D., Groth G. Structure of the c14 rotor ring of the proton translocating chloroplast ATP synthase. J. Biol. Chem. 2009;284:18228–18235. doi: 10.1074/jbc.M109.006916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krah A., Pogoryelov D., Faraldo-Gómez J.D. Structural and energetic basis for H+ versus Na+ binding selectivity in ATP synthase F0 rotors. Biochim. Biophys. Acta. 2010;1797:763–772. doi: 10.1016/j.bbabio.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 10.Boyer P.D. Bioenergetic coupling to protonmotive force: should we be considering hydronium ion coordination and not group protonation? Trends Biochem. Sci. 1988;13:5–7. doi: 10.1016/0968-0004(88)90005-9. [DOI] [PubMed] [Google Scholar]
- 11.Behr J.P., Dumas P., Moras D. The H3O+ cation: molecular structure of an oxonium macrocyclic polyether complex. J. Am. Chem. Soc. 1982;104:4540–4543. [Google Scholar]
- 12.von Ballmoos C., Dimroth P. Two distinct proton binding sites in the ATP synthase family. Biochemistry. 2007;46:11800–11809. doi: 10.1021/bi701083v. [DOI] [PubMed] [Google Scholar]
- 13.Bae L., Vik S.B. A more robust version of the Arginine 210-switched mutant in subunit a of the Escherichia coli ATP synthase. Biochim. Biophys. Acta. 2009;1787:1129–1134. doi: 10.1016/j.bbabio.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smirnova I.N., Kasho V., Kaback H.R. Protonation and sugar binding to LacY. Proc. Natl. Acad. Sci. USA. 2008;105:8896–8901. doi: 10.1073/pnas.0803577105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buch-Pedersen M.J., Palmgren M.G. Mechanism of proton transport by plant plasma membrane proton ATPases. J. Plant Res. 2003;116:507–515. doi: 10.1007/s10265-003-0111-9. [DOI] [PubMed] [Google Scholar]
- 16.Preiss L., Yildiz Ö., Meier T. A new type of proton coordination in an F1F0-ATP synthase rotor ring. PLoS Biol. 2010;8:e1000443. doi: 10.1371/journal.pbio.1000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Markovitch O., Agmon N. Structure and energetics of the hydronium hydration shells. J. Phys. Chem. A. 2007;111:2253–2256. doi: 10.1021/jp068960g. [DOI] [PubMed] [Google Scholar]
- 18.Steed P.R., Fillingame R.H. Aqueous accessibility to the transmembrane regions of subunit c of the Escherichia coli F1F0 ATP synthase. J. Biol. Chem. 2009;284:23243–23250. doi: 10.1074/jbc.M109.002501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pogoryelov D., Reichen C., Meier T. The oligomeric state of c rings from cyanobacterial F-ATP synthases varies from 13 to 15. J. Bacteriol. 2007;189:5895–5902. doi: 10.1128/JB.00581-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.