

Abstract
Patients with type II diabetes exhibit fibrillar deposits of human amylin protein in the pancreas. It has been proposed that amylin oligomers arising along the aggregation or fibril-formation pathways are important in the genesis of the disease. In a step toward understanding these aggregation pathways, in this work we report the conformational preferences of human amylin monomer in solution using molecular simulations and infrared experiments. In particular, we identify a stable conformer that could play a key role in aggregation. We find that amylin adopts three stable conformations: one with an α-helical segment comprising residues 9–17 and a short antiparallel β-sheet comprising residues 24–28 and 31–35; one with an extended antiparallel β-hairpin with the turn region comprising residues 20–23; and one with no particular structure. Using detailed calculations, we determine the relative stability of these various conformations, finding that the β-hairpin conformation is the most stable, followed by the α-helical conformation, and then the unstructured coil. To test our predicted structure, we calculate its infrared spectrum in the amide I stretch regime, which is sensitive to secondary structure through vibrational couplings and linewidths, and compare it to experiment. We find that theoretically predicted spectra are in good agreement with the experimental line shapes presented herein. The implications of the monomer secondary structures on its aggregation pathway and on its interaction with cell membranes are discussed.
Introduction
A broad range of human diseases including Huntington's, Alzheimer's, Creutzfeldt -Jacob's, and type II diabetes are associated with misfolding and aggregation of a specific protein or proteins (1,2). In these diseases, the proteins in question are known to convert from their native functional state into highly organized fibrillar aggregates. Recent investigations have shown that intermediate states that occur along the formation pathway of fibrillar aggregates induce apoptosis of their associated cells, leading to disease symptoms (3–6). Hence, a deeper understanding of the aggregation process would provide useful information for development of therapeutic strategies that could inhibit aggregation and maybe even reverse it.
Its importance notwithstanding, the structure of most of the proteins involved in protein aggregation diseases is not known with atomic-level resolution. This is partly because most of these proteins are membrane-bound and exhibit extremely low solubility. This makes their structural characterization by traditional experimental means, including x-ray crystallography and nuclear magnetic resonance (NMR), particularly demanding. Furthermore, these proteins exhibit very fast aggregation kinetics, thereby complicating the interpretation of experimental data considerably.
In the particular case of type II diabetes, the culprit is amylin, also called human islet amyloid polypeptide (hIAPP), a 37-residue hormone produced by the islet β-cells in the pancreas whose sequence is shown in references 7–9. The aggregation of amylin is widely perceived to be associated with many of the disease's symptoms (10,11). Past efforts have been largely aimed at characterizing the fibrillar aggregates formed by this protein. Using x-ray and electron diffraction techniques, Sumner-Makin and Serpell (12) determined that the fibrils of the human amylin protein are made up of extended β-strands that run perpendicular to the fibril axis. Jayasinghe and Langen (13), using electron paramagnetic resonance spectroscopy, determined that the β-strands adopt a parallel orientation. Similar results were also obtained using two-dimensional infrared measurements (14,15). More recently, using solid-state NMR, it has been proposed that amylin aggregates form a symmetrical structure consisting of striated ribbons containing layers of parallel β-sheet (16). The structure of amylin aggregates has also been studied using molecular models (17,18) and, consistent with experimental observations, past simulations have identified β-sheet structures with interdigitated side chains.
Although some consensus is starting to emerge regarding the structure of the human amylin aggregates, less is known about the structure of the individual hIAPP molecule in solution and the early-stage aggregates that are thought to precede fibril formation. Recent experiments have produced evidence for the occurrence of α-helical intermediates of amylin in the presence of membranes and for associations of oligomers in the lag phase of aggregation (19–22). As shown in this work, our calculations predict that roughly one-third of the ensemble of structures that arises in dilute solution consists of conformations with a partial α-helix, which may be responsible for these experimental observations, or it could be that a structure change occurs upon interactions with membranes or other peptides.
Our approach is to generate an atomic level model of the amylin monomer using advanced atomistic molecular simulations in explicit water, which can then be tested against experiment by calculating infrared spectra from the resulting molecular structures. Infrared spectra by themselves cannot provide atomic level structures, but because the backbone vibrational modes are strongly coupled to one another and because the linewidths are sensitive to solvent exposure (14,15,23,24), the infrared (IR) spectra reflect the secondary structure content of proteins and polypeptides. For instance, β-sheet polypeptides absorb near 1620 cm–1 and have narrow linewidths, whereas random coils absorb near 1645 cm–1 and are broad. Peak fitting is often used to extract secondary structure content from IR spectra, but the accuracy of the conclusions is often suspect because the fits are usually not unique and because the peaks have no rigorous theoretical basis. However, IR spectra can be quantitatively predicted from structures generated by molecular simulations through semiclassical lineshape theories and by including vibrational couplings, which is the approach we use here. This combination of simulation, theory, and experiment offers advantages over more established techniques. For instance, the interpretation of circular dichroism spectra, which is perhaps the most often used spectroscopy for amyloids, is entirely empirical. And NMR and x-ray spectroscopy continue to be difficult to apply to amyloids, particularly in situations where misfolding and aggregation occur over timescales that are faster than the time required for experimental measurements. Our approach is thus unique in that it enables a quantitative cycle of structure prediction and validation for challenging systems such as amyloid aggregation.
Theoretical secondary structure prediction algorithms predict that, in solution, the human amylin polypeptide adopts an α-helical secondary structure that spans residues 8–14 (25). However, in vitro circular dichroism experiments suggest that human amylin is natively unstructured (26–30). Experimentally it is also known that when human amylin is dissolved in hexafluoro-isopropanol (HFIP), it is found in a predominantly monomeric state with a stable α-helical domain spanning residues 5–20 (31). Similarly, in solution, the calcitonin-gene-related peptide, which has a sequence similar to that of human amylin and belongs to the same hormone family, forms a stable α-helical segment spanning residues 8–18 (32). Recently, based on NMR experiments and by using better aggregate removal protocols, Yonemoto et al. (33) and Cort et al. (34) showed that human amylin has a strong preference for an α-helical secondary structure near the N-terminal end of the peptide. In their study, to minimize aggregation the authors use a low temperature of 5°C and pH of 6. Note that other experiments in the presence of membranes also seem to support this secondary structure for the human amylin protein (20,36–38). Very recently, Dupuis et al. (42) studied the hIAPP monomer using a combination of ion mobility mass spectrometry and all-atom replica exchange molecular dynamics simulations with implicit water and found three distinct confomational families, one of which is an extended β-hairpin and one of which is a compact helix-coil.
When studying hIAPP, it is useful to consider the particular sequence of the amylin peptide that arises in rats, called rIAPP. The rat amylin peptide exhibits >80% sequence homology with hIAPP (7,8), and yet its propensity to form aggregates is severely diminished, thereby facilitating studies of the monomer in solution (29). Experiments suggest that rat and human amylin proteins interact similarly with lipid monolayers (39), and it is therefore thought that the human and rat proteins may have several common structural features. High-resolution NMR studies show that rIAPP adopts an α-helical conformation spanning residues 5–19 (40). In the presence of phospholipid micelles, the N-terminus adopts a helical structure (41). Based on available evidence for the rat amylin protein, one could expect the human variant to also exhibit an α-helical structure near its N-terminus. In recent work, using a combination of replica exchange umbrella sampling simulations and IR spectroscopy, we have shown that the folded state of the rat amylin protein exhibits an α-helical secondary structure between residues 7–17 (8). That work also served to establish the accuracy of our molecular simulation advanced sampling algorithms and our protocol for converting the results of simulations into IR spectra; we predicted NMR chemical shifts and IR spectra, and both were found to be in good agreement with experiment. Our results on the structure of rIAPP confirm the recent results of Dupuis et al. (42).
In this work, using all-atom replica exchange molecular dynamics with explicit water, we determine the stable conformations of hIAPP in solution and provide quantitative predictions of their relative stability.
We also compare our findings for human amylin to results for rat amylin, and propose an explanation for the proclivity of human amylin to form aggregates in solution. We do so by resorting to what we believe is a novel simulation technique, developed for this work, that enables extensive sampling and calculation of free energies for different conformers of amylin. We then employ the line shape theory to predict the vibrational absorption spectrum, and compare it to our own Fourier-transform infrared (FTIR) measurements. Our results indicate that the human peptide can adopt α-helical, random coil, and β-hairpin conformations in solution. The relative abundance of these conformers at room temperature is 31%, 29%, and 40%, respectively. The simulated and measured spectra are in quantitative agreement. In contrast, in our previous work we found that the rat peptide can only adopt α-helical and random coil conformations (with relative abundances of 55% and 45%, respectively) (8). Based on these findings, we propose that the ability of human amylin to adopt a stable β-hairpin is a crucial characteristic of the molecule that facilitates its ability to form fibrils, and that such a conformer is a key intermediate.
Materials and Methods
Molecular simulations
The amino-acid sequence of the human amylin protein studied in our work is given in Green et al. (7) and Reddy et al. (8). Consistent with experiments, the C-terminal of the peptide is capped with an NH2 group, and a disulfide bond is present between CYS-2 and CYS-7 residues. The protein is modeled using the GROMOS96 53a6 force field, while the simple point-charge water model is adopted for water (43). The ionization state of the amino-acid side chains was assigned based on their pKa values. The resulting peptide structure was found to have a net positive charge of +3. To make the system charge neutral, three Cl− ions were added to the simulation box. The GROMOS96 53a6 force field has been used extensively in the literature to study the folding of protein molecules, and was found to provide good agreement with experiment in our recent studies of peptide aggregation and of rIAPP (8).
Molecular simulations were performed with the GROMACS molecular simulation package (44,45). Long-range electrostatic interactions were treated with a particle-mesh Ewald sum (46,47). All simulations were performed with rigid bonds (using the linear constraint solver method) and with an integration time step of 2 fs. Once at equilibrium, most simulations were performed at a temperature of 298 K and pressure of 1 bar using Berendsen coupling (48).
Replica-exchange method
The replica exchange molecular dynamics (REMD) or parallel tempering method has been used extensively in the literature to understand the conformational preferences of proteins (49–51). The basic idea of REMD is to simulate copies (replicas) of the same system at different temperatures values. The configurations from adjacent temperature replicas are exchanged at regular intervals using the Metropolis acceptance criteria (Pacc) given below:
| (1) |
In the above equation, Δβ and ΔE represent the difference in the inverse temperature, and the difference in total internal energy of the replicas being considered for an exchange of configurations, respectively.
We used a total of 74 replicas spanning a temperature range of 273–578 K. The simulation length for each replica was 10 ns, with a total simulation length of 740 ns. The exchange moves between adjacent replicas were attempted every 2 ps. The optimal performance of the algorithm requires that the probability distribution curves of the potential energies corresponding to neighboring replicas exhibit a sufficient degree of overlap. Fig. S1 in the Supporting Material shows the potential energy distribution at the different temperatures used in our REMD simulations. As can be seen from the figure, the overlap of the potential energy between adjacent replicas was chosen such that it leads to an average acceptance rate of 20% (51).
Stable conformations of the peptide were extracted from our REMD simulations by resorting to principal component analysis (52–54). As discussed below, our analysis revealed the presence of three major clusters corresponding to the three conformations discussed in this article. Fig. S2 provides the results of that analysis.
Thermodynamic integration
Our REMD simulations indicate the human peptide can adopt α-helical, random coil, and β-hairpin conformations in solution (see Results). The relative free energy (and thermodynamic stability) of these states was determined using thermodynamic integration (TI). The TI scheme used here is similar to that used in the context of simulations of polymeric materials to identify order-disorder transitions (55). For the human peptide, this technique is used to determine the free energy difference between 1), an α-helical conformation and a random coil, and 2), a β-hairpin conformation and a random coil.
In our previous we work we have shown that the rat amylin protein exists in solution in the α-helical and random coil conformations. For comparison, we also used TI to calculate the free energy difference between an α-helical and a random coil conformation of the rat amylin peptide.
The TI pathway used in our problem is illustrated in Fig. S3. As shown in the figure, it consists of four segments. Starting from the folded state, along the segment labeled 1, an external umbrella potential Uf is imposed whose relative strength (λ) is linearly increased from zero to unity, as one travels from vertex a to vertex b. Along the segment labeled 2, denoted by a line connecting vertices b and c, the temperature of the system was gradually raised from 298 K to 498 K. Next, along Segment 3, denoted by the line connecting c and d, the relative strength (λ) of the imposed umbrella potential, Uf, was gradually reduced from unity to zero. Finally, along Segment 4, the temperature of the system was gradually lowered from 498 K back to 298 K.
In the particular case of islet amyloid polypeptide, we found it sufficient to use an umbrella potential, Uf, consisting of a set of harmonic springs imposed between the Cα atoms of the native contacts. The functional form of Uf is simply
| (2) |
where the spring constant was chosen to be equal to k = 500 kJ/mol/nm2. The symbol r0 represents the equilibrium distance between the contacts for an ideally folded structure, the symbol rj represents the actual distance between the Cα atoms of the jth contact, and N represents the total number of contacts. For an α-helical conformation, a native contact is defined as that between two residues which are three residues apart (i and i + 4). The distance r0 that corresponds to an ideal helix was set 0.59 nm. In the case of a β-hairpin, a native contact was defined as that between the two residues that are involved in hydrogen bonding in the β-sheet. The corresponding r0 for β-sheet structure was set to 0.55 nm (based on an ideal β-sheet). Using these criteria, and based on results from exploratory equilibrium MD simulations (see Results), for an α-helical conformation a total of N = 7 umbrellas were used between residue pairs (7,11), (8,12), (9,13), (10,14), (11,15), (12,16), and (13,17). Similarly, for the β-hairpin structure, N = 7 umbrellas were used between residue pairs (13,28), (14,27), (15,26), (16,25), (17,24), (18,23), and (19,24).
IR line shape calculations
We followed the protocol used for rat amylin (8,56) to determine the IR amide I line shape of human amylin. However, herein we use an improved electrostatic amide I backbone map, and a new, to our knowledge, side-chain map for glutamine and asparagine residues, which were developed from experimental spectra of n-methyl-acetamide and acetamide, respectively, in various solvents, and the total simulation time was 10 ns. For backbone chromophores,
| (3) |
with
| (4) |
In Eq. 3, Ei, C is the electric-field component for the ith chromophore in the C=O direction on the C atom and Ei, N is that on the N atom (both in atomic units), and the frequency is in cm–1. The exclusion list for the electric field calculation includes backbone atoms in the (i-1)th, ith, and (i+1)th peptide units. To account for contributions from the nearest-neighbor peptide units, we have adapted the nearest-neighbor frequency shift map developed by la Cour Jansen et al. (57). These contributions depend on the (Φ, Ψ) angles between peptide units and are denoted as Δωi, N and Δωi, C. The side-chain frequencies are calculated as
| (5) |
Couplings between adjacent amide groups have been determined by the nearest-neighbor coupling map of la Cour Jansen et al. (57). All the other coupling constants have been calculated from the transition dipole coupling method (58–61), and we have adopted the parameters optimized by Torii and Tasumi (62).
Experimental peptide preparation and IR spectrum
Peptide preparation
hIAPP were purchased from Bachem (Bubendorf, Switzerland) and dissolved in deuterated hexafluoroisopropanol (d-HFIP) at a concentration of 0.5 mM. An aliquot of the d-HFIP stock solution was made into D2O solution after evaporating d-HFIP. The hIAPP D2O solution was dialyzed in 0.1 mM DCl multiple times and then one time in D2O to remove residual TFA. The sample was then lyophilized and redissolved in d-HFIP. The denaturant HFIP was then removed by evaporation under a stream of nitrogen to generate a film in Eppendorf tubes. Amyloid formation was initiated by redissolving the film in D2O at pH 6. The initial peptide concentration was 0.25 mM.
Fourier-transform infrared
Infrared absorption data were taken by a Mattson Instruments (Thermo Electron, Waltham, MA) Galaxy series Fourier-transform infrared (FTIR) 7000 spectrometer purged with dry air. The sample was placed between two CaF2 plates separated by a 100-μM Teflon spacer. The spectrum was collected immediately after redissolving the film in D2O at pH 6, as mentioned above in Peptide Preparation. It was smoothed by truncating higher frequency noises in time domain.
Results and Discussion
A principal component analysis performed on results from extensive REMD simulations revealed three stable conformations for the peptide:
-
1.
An α-helical conformation where residues 9–17 were folded into an α-helix and a short β-sheet between residues 24–28 and 31–35 (Fig. 1 a).
-
2.
A β-hairpin conformation with a turn at residues between 20 and 23 (Fig. 1 b).
-
3.
A random coil conformation (Fig. 1 c).
The α-helical conformation was found to be similar to that predicted for the rat amylin protein (8) (Fig. 1 d). Our three stable hIAPP conformations are very similar to those found by Dupuis et al. (42). The β-sheet domain in the α-helical conformation includes the region with proline mutations in rat amylin protein. The low amyloidogenicity associated with the proline residues might explain the difference in the structure of human amylin protein from rat amylin protein. Calcitonin-gene-related peptide is known to exist in an α-helical conformation in its active form, and loses its activity upon the loss of α-helicity (63).
Figure 1.
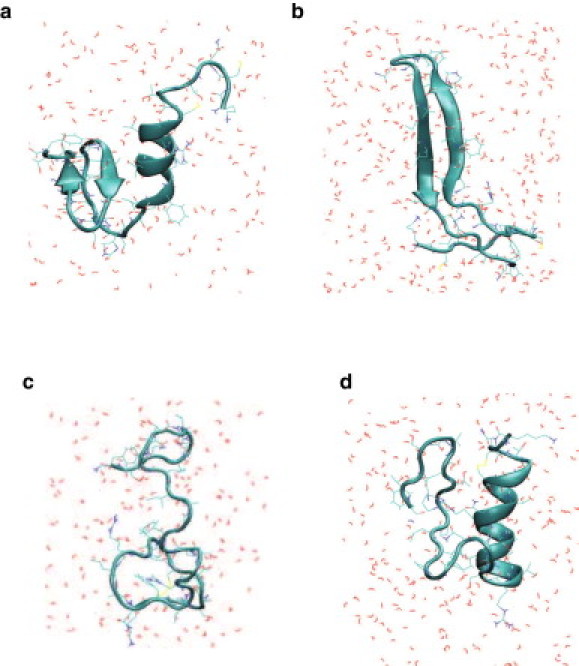
Representative configurations of human amylin peptide in (a) α-helical conformation, (b) β-hairpin conformation, (c) random coil conformation, and (d) rat amylin in α-helical conformation (8).
Long molecular dynamics simulations (100 ns) of human amylin revealed that the α-helical conformation can spontaneously convert into the β-hairpin conformation (observed in two out of four simulations). In contrast, MD simulations started from the β-hairpin conformation were always found to be stable (average RMSD < 3 Å). Several snapshots of the secondary structure of the molecule at different times, along one of the misfolding pathways, are presented in Fig. 2 a. The β-hairpin order parameter, measured as the fraction of native β-hairpin contacts along the misfolding pathway, is presented in Fig. 2 b. As can be seen in Fig. 2 a, the early steps in the misfolding pathway of hIAPP involve loss of contacts between residues 24–28 and 31–35, which in the folded state of the protein correspond to a short β-sheet. This event is followed by residues 24–28 forming new contacts with residues 15–19, leading to the formation of the β-hairpin structure.
Figure 2.
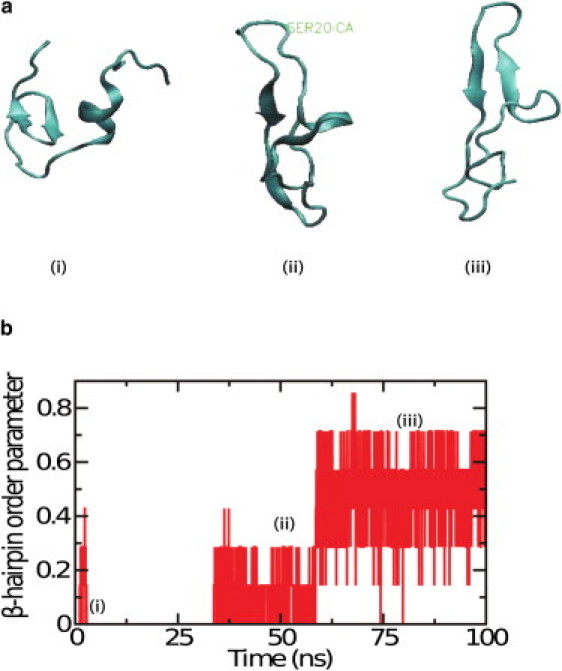
(a) Representative configurations of human amylin peptide along a representative folding trajectory at (i) 0 ns, (ii) 50 ns, and (iii) 80 ns. (b) β-Hairpin order parameter (measured as fraction of native contacts in β-hairpin) along the representative trajectory.
Having identified candidate structures for the α-helical and β-hairpin states of hIAPP in solution, we proceeded to investigate their relative stability as a function of suitable order parameters (see Materials and Methods) by calculating their free energy vis-à-vis that of the random coil state. To that end, we performed extensive TI simulations using a newly proposed algorithm. Table 1 shows the free energy changes of the protein during the α-helix-coil and β-hairpin-coil transformations. Our results show that an α-helix is preferred over a random coil structure. However, our results also show that the β-hairpin is more favorable than both the random coil (−0.77 kJ/mol) and the α-helix (−0.6 kJ/mol). Using this method, we also calculated the free energy during the α-helix-coil transformation of rat amylin protein (−0.47 kJ/mol). This value is consistent with our previously published value, where calculations were performed using a very different simulation technique (8). In Table 2, we report the enthalpy change (ΔH) associated with the transitions studied in the TI. The ΔH is computed as the difference between the average potential energy of the two end states in TI. Our results show that the α-helical and β-hairpin conformations are stabilized enthalpically, and preferred over the random coil conformations. The enthalpy gain for the α-helical conformation is greater than that for the β-hairpin. However, there is an entropic cost associated with folding the protein from a random coil into the α-helical and β-hairpin conformations. These results indicate that the α-helical conformation, which is stabilized by enthalpic contributions to the free energy, would be even more stable and more pronounced at lower temperatures. This observation is consistent with the recent NMR studies by Yonemoto et al. (33), where a larger α-helical content was observed for the peptide at 5°C.
Table 1.
Free energy changes during folding of rat amylin and human amylin proteins calculated using TI, as explained in the text
| Protein | Transition | Free energy (kJ/mol) |
|---|---|---|
| Rat amylin | ΔF (random coil → α-helix) | −0.47 ± 0.08 |
| Human amylin | ΔF (random coil → α-helix) | −0.17 ± 0.09 |
| Human amylin | ΔF (random coil → β-hairpin) | −0.77 ± 0.08 |
Table 2.
Enthalpy, entropy, and number of hydrogen-bond changes for human amylin protein
| Transition | ΔH (kJ/mol) | TΔS (kJ/mol) | ||||
|---|---|---|---|---|---|---|
| Random coil → α-helix | −78.3 | −78.13 | 5.669 | −11.907 | ||
| Random coil → β-hairpin | −70.1 | −69.33 | 4.664 | −11.350 |
For random coil conformation, = 12.032, .
In Table 2, we also report average number of protein-protein and protein-water hydrogen bonds. A hydrogen bond is defined based on a acceptor-donor-hydrogen cutoff angle of 30° and a cutoff distance of 0.35 nm between hydrogen-acceptors (64,65). Our results indicate that the α-helical and β-hairpin states are primarily stabilized by intrapeptide hydrogen bonds. The random coil state on the other hand, is stabilized by peptide-water hydrogen bonds.
The calculated IR spectrum for each conformer (see Materials and Methods) is shown in Fig. 3. The spectra for the α-helical and β-hairpin states peak at ∼1644 and 1643 cm–1, respectively, whereas the spectrum for the random coil peaks at ∼1657 cm–1. We calculated the weighted average of these spectra using the relative populations (listed earlier) that follow from the relative free energies determined above. As shown in Fig. 3, the theoretical line shape is in quantitative agreement with experiment (see Materials and Methods), consistent with our contention that there is a substantial population of the β-hairpin conformer in solution. Note that our experimental line shape showed only minor changes when the amylin concentration was doubled, and that a similar experimental line shape for very dilute (0.01 mM) amylin solution at early times after mixing has been determined by Jha et al. (30), thus providing some confirming evidence that our spectrum is for the amylin monomer. Also, note that the differences in the IR spectra of the three conformations are caused by the vibrational couplings between residues. Standard interpretation of IR spectra based on empirical rules would not assign a peak at 1643 cm–1 to a β-sheet, because β-sheets usually absorb at 1620 cm–1. The β-hairpin absorbs much higher, because the vibrational excitons can only delocalize over two strands rather than 3 or more, which is necessary to obtain a lower β-sheet vibrational frequency.
Figure 3.
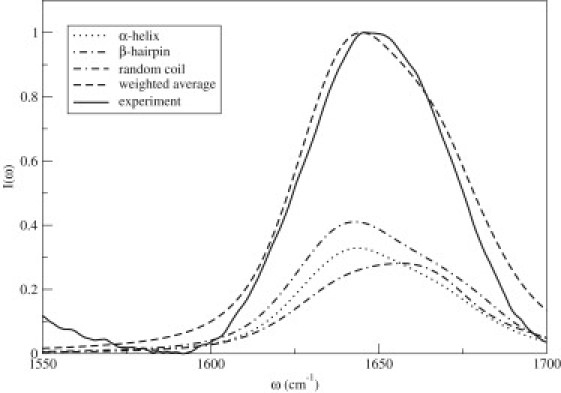
Theoretical (total) and experimental IR line shapes in the amide I stretch region for the human amylin protein at 25°C. Also shown are line shapes for the individual conformations weighted by their relative probabilities. The peptide concentration is 0.25 mM.
The similarity in the sequence between the rat amylin and the human amylin proteins at the N-terminus as well as the similarity of their α-helical structures suggest that the α-helical secondary structure at the N-terminus might be important for the hormonal activity of the protein. We believe that the difference in the aggregation propensity for the rat and human amylin proteins is determined by how the residues near the C-terminus affect the stability of the α-helical segment. In the case of the human amylin protein, the interaction of the residues 24–28 near the C-terminus with the α-helical segment of the protein makes the α-helical structure less stable, causing it to misfold into a β-hairpin. In the rat protein, the presence of proline residues in this domain prevents such unwanted interactions, leading to a more stable native state.
It is of interest to note that the Amyloid β polypeptide is thought to exhibit an aggregation mechanism (and fibril structure) similar to that of the human amylin protein. Recent NMR experiments have shown that the intermediates along the aggregation pathway of the amyloid β peptide include intramolecular β hairpins, similar to those observed in our simulations (66). Hence we believe that, if our predictions are correct, similar experiments should detect β-hairpin structures in hIAPP (see Fig. 4 a and (67)). Also, note that the turn region in the β-hairpin conformation presented here for hIAPP coincides with the turn region proposed by Luca et al. (16) for fully grown amylin aggregates.
Figure 4.
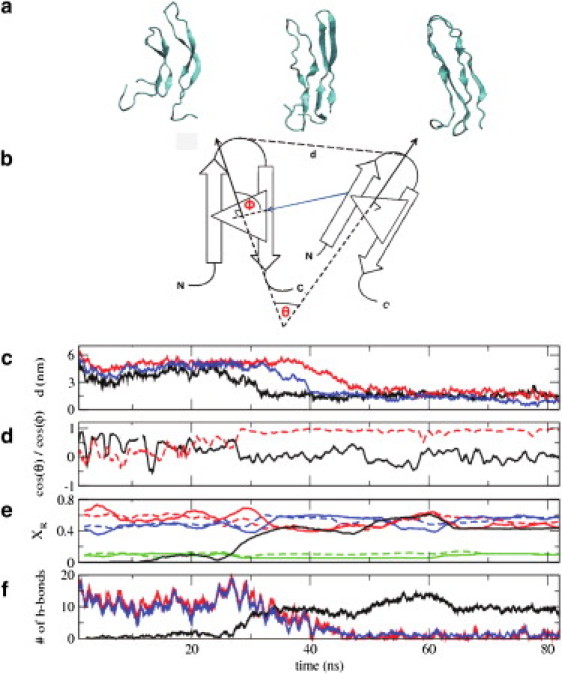
Dimerization of hIAPP monomers in solution. (a) Representative configurations along the dimerization pathway of two human amylin peptides in their β-hairpin conformation. The two β-hairpin structures interact by exchanging their intramolecular hydrogen bonds for intermolecular hydrogen bonds, resulting in a U-shaped β-sheet dimer structure. (b) Schematic representation of the distance and angles between two peptides that are used to characterize their dimerization (see below). (c) Distance (in nm) between residues 22 (the turn residues) of the two peptides as a function of time. Results are shown for three distinct trajectories started from three different initial configurations: (i) molecules at a distance of 5 nm, with a θ angle between the two peptides of 90° (shown in black); (ii) molecules at a distance of 6 nm, with a θ angle between them of 180° (shown in red); and (iii) molecules at a distance of 6.5 nm, with a θ angle between them of 0° (shown in blue). (d) For trajectory (i), the red dotted line shows cos(θ), the angle between the normals of the two molecules. (Black solid line) Cos(Φ), the angle between the line joining two peptides and one normal. (e) For trajectory (i), fraction of residues (XR) in a particular secondary structure. The secondary structure of a residue was assigned using the DSSP (67) approach. (Solid and dotted lines) Results for the two different peptides, respectively. (Red, blue, and green) Residues in β-sheet, random coil, and turn, respectively. (Solid black line) Fraction of residues exhibiting interstrand β-bridge joining two β-sheets. (f) For trajectory (i), (blue and red lines) number of main chain intrastrand hydrogen bonds for the two monomers; (black line) number of main chain interstrand hydrogen bonds.
Having established the existence of β-hairpin intermediates, we proceeded to examine their aggregation propensity. To that end, we conducted 10 independent molecular dynamics simulations starting from different preequilibrated initial conditions (different Cartesian coordinates and momenta). For clarity, representative results from only three of those 100-ns MD trajectories are shown in Fig. 4. The remaining trajectories do not show any qualitative differences. For these calculations, two β-hairpin conformers were placed at a random orientation at distance ranging from 40 to 70 Å between their centers of mass. After an initial equilibration period during which the separation of the peptides was constrained, the aggregation of the two molecules was followed (Fig. 4 a shows representative configurations of the molecule along the aggregation pathway). Fig. 4 b depicts the conformational variables used here to characterize dimerization. Fig. 4 c shows that, regardless of initial orientation, after a period of time in the range of 20–60 ns, the molecules approach each other to form a dimer. Fig. 4 d, which shows the relative angles between the two hairpins as a function of time, shows that in some cases the approach trajectory can be relatively convoluted, with multiple twists and turns, but always ends up in the dimerized state. Fig. 4 e shows the structural evolution of the two hairpins and the dimer as a function of time for one of the 10 independent trajectories considered here (results for all trajectories exhibit the same qualitative features). Previous studies of the aggregation pathway for other amyloidogenic peptides indicate that the molecules aggregate by first unfolding into a random coil (hydrophobic collapse), and then gradually reorganizing into a β-sheet aggregate (68). Our results show that the human amylin peptide exhibits a high secondary structure content throughout the aggregation pathway. Fig. 4 f shows the number of main chain intrastrand and interstrand hydrogen bonds. Our results indicate that, after ∼20–30 ns, the two peptides start interacting through formation of contacts between the two chains. As shown in the figure, intrastrand hydrogen bonds are gradually replaced by interstrand hydrogen bonds, leading to formation of a stable dimer. The entire dimer formation process requires a period of time in the range of tens of nanoseconds. Additional simulations (not shown here) also suggest that the β-hairpin conformer is able to facilitate the transformation of α-helical states into additional β hairpins. The results shown in Fig. 4 provide insights into the mechanism through which dimerization occurs in human amylin, and they also provide initial reactive paths that we are currently pursuing in additional calculations aimed at characterizing the reaction coordinate for amylin dimerization. The results of such calculations will be presented in the future. Note here that recent work by Rivera et al. (69) has also used simulations to examine the dimerization of amylin. However, because those calculations were performed on fragments of the full peptide (residues 20–29), it is difficult to make a comparison to the results presented above.
Recent experiments by Engel et al. (39) for hIAPP with phospholipid monolayers indicate that the protein inserts into such monolayers as a monomer. Furthermore, they suggest that insertion occurs at the N-terminus. More-recent NMR observations by Patil et al. (36) on sodium dodecyl sulfate micelles also indicate that the N-terminus of hIAPP adopts an α-helical state and inserts into the micelles. It is known that the ability of a protein to insert into a monolayers or a bilayer is influenced by the secondary structure in solution state. The α-helical segment on the N-terminus, as predicted in our work, would promote its insertion into monolayers and micelles. Note that when the amylin protein is in an aggregated state, it would lose the α-helical secondary structure, thereby making the insertion of the protein less favorable, as also observed by Engel et al. (39) in their work.
Conclusions
We have presented a first, to our knowledge, atomistic scale description of the full-length human amylin protein in explicit water. We predict that the protein adopts three stable conformations, one of which is a β-hairpin. The respective abundance of each conformer is determined from precise thermodynamic integration simulations. The results of simulations are consistent with FTIR measurements, which exhibit a good level of agreement with theory. Such agreement is worth emphasizing in view of the many levels of theory that have gone into our methods, and suggests that one- and two-dimensional IR spectra can provide detailed structural information that is not accessible in other experimental approaches.
Our results also show that, despite a high degree of sequence homology, there are fundamental differences in the structures of the rat and the human amylin protein in solution. The folded state of the rat amylin protein contains an helical segment spanning residues 7–17. The human amylin protein adopts two stable conformations with significant secondary structure: an α-helical conformation with a short β-sheet near the C-terminus, and a full β-hairpin conformation. The thermodynamic integration simulations indicate that, at room temperature, the β-hairpin conformation is slightly more stable than the α-helical state.
These structural differences are particularly important in that, in contrast to its rat counterpart, the human protein forms amyloid fibrils. We concur with the proposal by Dupuis et al. (42) that a possible pathway for fibril formation involves the formation and aggregation of monomeric β-hairpins, a process in which intramolecular hydrogen bonds are exchanged in favor of intermolecular hydrogen bonds. We note that the turn region in the β-hairpin conformation proposed here coincides with the turn region proposed by Luca et al. (16) in the fully grown amylin aggregates.
Acknowledgments
This work was supported in part by a grant (CHE-0832584) from the National Science Foundation (NSF) to J.L.S. and M.T.Z. J.L.S also thanks the NSF for support of this work through grant CHE-0750307. M.T.Z. thanks the National Institutes of Health (DK79895) for support. J.J.d.P. thanks the NSF for support of this work through the University of Wisconsin Materials Research Science and Engineering Center on Nanostructured Interfaces and grant CBET-0755730. J.J.d.P. and J.L.S. are grateful for support from the National Institutes of Health (1R01DK088184).
Supporting Material
References
- 1.Dobson C.M. Protein misfolding, evolution and disease. Trends Biochem. Sci. 1999;24:329–332. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 2.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 3.Walsh D.M., Selkoe D.J. Aβ oligomers—a decade of discovery. J. Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 4.Klein W.L., Stine W.B., Jr., Teplow D.B. Small assemblies of unmodified amyloid β-protein are the proximate neurotoxin in Alzheimer's disease. Neurobiol. Aging. 2004;25:569–580. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 5.Bucciantini M., Giannoni E., Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 6.Silveira J.R., Raymond G.J., Caughey B. The most infectious prion protein particles. Nature. 2005;437:257–261. doi: 10.1038/nature03989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green J., Goldsbury C., Aebi U. Full-length rat amylin forms fibrils following substitution of single residues from human amylin. J. Mol. Biol. 2003;326:1147–1156. doi: 10.1016/s0022-2836(02)01377-3. [DOI] [PubMed] [Google Scholar]
- 8.Reddy A.S., Wang L., De Pablo J.J. Solution structures of rat amylin peptide: simulation, theory, and experiment. Biophys. J. 2010;98:443–451. doi: 10.1016/j.bpj.2009.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kayed R., Head E., Glabe C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 10.Clark A., Cooper G.J.S., Turner R.C. Islet amyloid formed from diabetes-associated peptide may be pathogenic in type-2 diabetes. Lancet. 1987;2:231–234. doi: 10.1016/s0140-6736(87)90825-7. [DOI] [PubMed] [Google Scholar]
- 11.Lorenzo A., Razzaboni B., Yankner B.A. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature. 1994;368:756–760. doi: 10.1038/368756a0. [DOI] [PubMed] [Google Scholar]
- 12.Sumner-Makin O., Serpell L.C. Structural characterization of islet amyloid polypeptide fibrils. J. Mol. Biol. 2004;335:1279–1288. doi: 10.1016/j.jmb.2003.11.048. [DOI] [PubMed] [Google Scholar]
- 13.Jayasinghe S.A., Langen R. Identifying structural features of fibrillar islet amyloid polypeptide using site-directed spin labeling. J. Biol. Chem. 2004;279:48420–48425. doi: 10.1074/jbc.M406853200. [DOI] [PubMed] [Google Scholar]
- 14.Strasfeld D.B., Ling Y.L., Zanni M.T. Strategies for extracting structural information from 2D IR spectroscopy of amyloid: application to islet amyloid polypeptide. J. Phys. Chem. B. 2009;113:15679–15691. doi: 10.1021/jp9072203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shim S.H., Gupta R., Zanni M.T. Two-dimensional IR spectroscopy and isotope labeling defines the pathway of amyloid formation with residue-specific resolution. Proc. Natl. Acad. Sci. USA. 2009;106:6614–6619. doi: 10.1073/pnas.0805957106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luca S., Yau W.M., Tycko R. Peptide conformation and supramolecular organization in amylin fibrils: constraints from solid-state NMR. Biochemistry. 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cecchini M., Curcio R., Caflisch A. A molecular dynamics approach to the structural characterization of amyloid aggregation. J. Mol. Biol. 2006;357:1306–1321. doi: 10.1016/j.jmb.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 18.Zanuy D., Nussinov R. The sequence dependence of fiber organization. A comparative molecular dynamics study of the islet amyloid polypeptide segments 22–27 and 22–29. J. Mol. Biol. 2003;329:565–584. doi: 10.1016/s0022-2836(03)00491-1. [DOI] [PubMed] [Google Scholar]
- 19.Apostolidou M., Jayasinghe S.A., Langen R. Structure of α-helical membrane-bound human islet amyloid polypeptide and its implications for membrane-mediated misfolding. J. Biol. Chem. 2008;283:17205–17210. doi: 10.1074/jbc.M801383200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knight J.D., Hebda J.A., Miranker A.D. Conserved and cooperative assembly of membrane-bound α-helical states of islet amyloid polypeptide. Biochemistry. 2006;45:9496–9508. doi: 10.1021/bi060579z. [DOI] [PubMed] [Google Scholar]
- 21.Ling Y.L., Strasfeld D.B., Zanni M.T. Two-dimensional infrared spectroscopy provides evidence of an intermediate in the membrane-catalyzed assembly of diabetic amyloid. J. Phys. Chem. B. 2009;113:2498. doi: 10.1021/jp810261x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abedini A., Raleigh D.P. A role for helical intermediates in amyloid formation by natively unfolded polypeptides? Phys. Biol. 2009;6:015005. doi: 10.1088/1478-3975/6/1/015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manor J., Mukherjee P., Arkin I.T. Gating mechanism of the influenza A M2 channel revealed by 1D and 2D IR spectroscopies. Structure. 2009;17:247–254. doi: 10.1016/j.str.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woys A.M., Lin Y.-S., Zanni M.T. 2D IR line shapes probe ovispirin peptide conformation and depth in lipid bilayers. J. Am. Chem. Soc. 2010;132:2832–2838. doi: 10.1021/ja9101776. [DOI] [PubMed] [Google Scholar]
- 25.Jaikaran E.T.A.S., Higham C.E., Fraser P.E. Identification of a novel human islet amyloid polypeptide β-sheet domain and factors influencing fibrillogenesis. J. Mol. Biol. 2001;308:515–525. doi: 10.1006/jmbi.2001.4593. [DOI] [PubMed] [Google Scholar]
- 26.Kayed R., Bernhagen J., Kapurniotu A. Conformational transitions of islet amyloid polypeptide (IAPP) in amyloid formation in vitro. J. Mol. Biol. 1999;287:781–796. doi: 10.1006/jmbi.1999.2646. [DOI] [PubMed] [Google Scholar]
- 27.Dunker A.K., Lawson J.D., Obradovic Z. Intrinsically disordered protein. J. Mol. Graph. Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 28.Jaikaran E.T.A.S., Clark A. Islet amyloid and type 2 diabetes: from molecular misfolding to islet pathophysiology. Biochim. Biophys. Acta. 2001;1537:179–203. doi: 10.1016/s0925-4439(01)00078-3. [DOI] [PubMed] [Google Scholar]
- 29.Padrick S.B., Miranker A.D. Islet amyloid polypeptide: identification of long-range contacts and local order on the fibrillogenesis pathway. J. Mol. Biol. 2001;308:783–794. doi: 10.1006/jmbi.2001.4608. [DOI] [PubMed] [Google Scholar]
- 30.Jha S., Sellin D., Winter R. Amyloidogenic propensities and conformational properties of ProIAPP and IAPP in the presence of lipid bilayer membranes. J. Mol. Biol. 2009;389:907–920. doi: 10.1016/j.jmb.2009.04.077. [DOI] [PubMed] [Google Scholar]
- 31.Cort J., Liu Z., Andersen N.H. Beta-structure in human amylin and two designer β-peptides: CD and NMR spectroscopic comparisons suggest soluble β-oligomers and the absence of significant populations of β-strand dimers. Biochem. Biophys. Res. Commun. 1994;204:1088–1095. doi: 10.1006/bbrc.1994.2574. [DOI] [PubMed] [Google Scholar]
- 32.Breeze A.L., Harvey T.S., Campbell I.D. Solution structure of human calcitonin gene-related peptide by 1H NMR and distance geometry with restrained molecular dynamics. Biochemistry. 1991;30:575–582. doi: 10.1021/bi00216a036. [DOI] [PubMed] [Google Scholar]
- 33.Yonemoto I.T., Kroon G.J.A., Kelly J.W. Amylin proprotein processing generates progressively more amyloidogenic peptides that initially sample the helical state. Biochemistry. 2008;47:9900–9910. doi: 10.1021/bi800828u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cort J.R., Liu Z., Andersen N.H. Solution state structures of human pancreatic amylin and pramlintide. Protein Eng. Des. Sel. 2009;22:497–513. doi: 10.1093/protein/gzp029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reference deleted in proof.
- 36.Patil S.M., Xu S., Alexandrescu A.T. Dynamic α-helix structure of micelle-bound human amylin. J. Biol. Chem. 2009;284:11982–11991. doi: 10.1074/jbc.M809085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jayasinghe S.A., Langen R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry. 2005;44:12113–12119. doi: 10.1021/bi050840w. [DOI] [PubMed] [Google Scholar]
- 38.Jayasinghe S.A., Langen R. Membrane interaction of islet amyloid polypeptide. Biochim. Biophys. Acta. Biomembranes. 2007;1768:2002–2009. doi: 10.1016/j.bbamem.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 39.Engel M.F.M., Yigittop H., Antoinette Killian J. Islet amyloid polypeptide inserts into phospholipid monolayers as monomer. J. Mol. Biol. 2006;356:783–789. doi: 10.1016/j.jmb.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 40.Williamson J.A., Miranker A.D. Direct detection of transient α-helical states in islet amyloid polypeptide. Protein Sci. 2007;16:110–117. doi: 10.1110/ps.062486907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nanga R.P., Brender J.R., Ramamoorthy A. Three-dimensional structure and orientation of rat islet amyloid polypeptide protein in a membrane environment by solution NMR spectroscopy. J. Am. Chem. Soc. 2009;131:8252–8261. doi: 10.1021/ja9010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dupuis N.F., Wu C., Bowers M.T. Human islet amyloid polypeptide monomers form ordered β-hairpins: a possible direct amyloidogenic precursor. J. Am. Chem. Soc. 2009;131:18283–18292. doi: 10.1021/ja903814q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berendsen H.J.C., Postma J.P.M., Hermans J. Interaction models for water in relation to protein hydration. In: Pullman B., editor. Intermolecular Forces. Reidel, Dordrecht; The Netherlands: 1981. [Google Scholar]
- 44.Lindahl E., Hess B., van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 45.van der Spoel D., Lindahl E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 46.Darden T., York D., Pedersen L.G. Particle mesh Ewald: an N· log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089. [Google Scholar]
- 47.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 48.Berendsen H.J.C., Postma J.P.M., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684. [Google Scholar]
- 49.Sugita Y., Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999;314:141–151. [Google Scholar]
- 50.Sanbonmatsu K.Y., García A.E. Structure of Met-encephalin in explicit aqueous solution using replica exchange molecular dynamics. Proteins. 2002;46:225–234. doi: 10.1002/prot.1167. [DOI] [PubMed] [Google Scholar]
- 51.Rathore N., Chopra M., de Pablo J.J. Optimal allocation of replicas in parallel tempering simulations. J. Chem. Phys. 2005;122:024111. doi: 10.1063/1.1831273. [DOI] [PubMed] [Google Scholar]
- 52.Balsera M., Wriggers W., Schulten K. Principal component analysis and long time protein dynamics. J. Phys.Chem. 1996;100:2567–2572. [Google Scholar]
- 53.Emberly E.G., Mukhopadhyay R., Wingreen N.S. Flexibility of β-sheets: principal component analysis of database protein structures. Proteins. 2004;55:91–98. doi: 10.1002/prot.10618. [DOI] [PubMed] [Google Scholar]
- 54.Maisuradze G.G., Liwo A., Scheraga H.A. Principal component analysis for protein folding dynamics. J. Mol. Biol. 2009;385:312–329. doi: 10.1016/j.jmb.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Müller M., Daoulas K.C. Calculating the free energy of self-assembled structures by thermodynamic integration. J. Chem. Phys. 2008;128:024903. doi: 10.1063/1.2818565. [DOI] [PubMed] [Google Scholar]
- 56.Lin Y.-S., Shorb J.M., Skinner J.L. Empirical amide I vibrational frequency map: application to 2D-IR line shapes for isotope-edited membrane peptide bundles. J. Phys. Chem. B. 2009;113:592–602. doi: 10.1021/jp807528q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.la Cour Jansen T., Dijkstra A.G., Knoester J. Modeling the amide I bands of small peptides. J. Chem. Phys. 2006;125:44312. doi: 10.1063/1.2218516. [DOI] [PubMed] [Google Scholar]
- 58.Krimm S., Abe Y. Intermolecular interaction effects in the amide I vibrations of polypeptides. Proc. Natl. Acad. Sci. USA. 1972;69:2788–2792. doi: 10.1073/pnas.69.10.2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moore W.H., Krimm S. Transition dipole coupling in amide I modes of β-polypeptides. Proc. Natl. Acad. Sci. USA. 1975;72:4933–4935. doi: 10.1073/pnas.72.12.4933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Torii H., Tasumi M. Model calculations on the amide-I infrared bands of globular proteins. J. Chem. Phys. 1992;96:3379. [Google Scholar]
- 61.Gorbunov R.D., Stock G. Ab initio based building block model of amide I vibrations in peptides. Chem. Phys. Lett. 2007;437:272–276. doi: 10.1063/1.1898215. [DOI] [PubMed] [Google Scholar]
- 62.Torii H., Tasumi M. Ab initio molecular orbital study of the amide I vibrational interactions between the peptide groups in di- and tripeptides and considerations on the conformation of the extended helix. J. Raman Spectrosc. 1998;29:81–86. [Google Scholar]
- 63.Howitt S.G., Kilk K., Poyner D.R. The role of the 8–18 helix of CGRP8-37 in mediating high affinity binding to CGRP receptors; Coulombic and steric interactions. Br. J. Pharmacol. 2003;138:325–332. doi: 10.1038/sj.bjp.0705040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van der Spoel D., van Maaren P.J., Tîmneanu N. Thermodynamics of hydrogen bonding in hydrophilic and hydrophobic media. J. Phys. Chem. B. 2006;110:4393–4398. doi: 10.1021/jp0572535. [DOI] [PubMed] [Google Scholar]
- 65.Modig K., Pfrommer B.G., Halle B. Temperature-dependent hydrogen-bond geometry in liquid water. Phys. Rev. Lett. 2003;90:075502. doi: 10.1103/PhysRevLett.90.075502. [DOI] [PubMed] [Google Scholar]
- 66.Yu L., Edalji R., Olejniczak E.T. Structural characterization of a soluble amyloid β-peptide oligomer. Biochemistry. 2009;48:1870–1877. doi: 10.1021/bi802046n. [DOI] [PubMed] [Google Scholar]
- 67.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 68.Xu W., Ping J., Mu Y. Assembly dynamics of two-β sheets revealed by molecular dynamics simulations. J. Chem. Phys. 2009;130:164709. doi: 10.1063/1.3123532. [DOI] [PubMed] [Google Scholar]
- 69.Rivera E., Straub J., Thirumalai D. Sequence and crowding effects in the aggregation of a 10-residue fragment derived from islet amyloid polypeptide. Biophys. J. 2009;96:4552–4560. doi: 10.1016/j.bpj.2009.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.