

Abstract
Cochlear outer hair cells undergo reversible changes in shape when externally stimulated. This response, known as OHC motility, is a central component of the cochlear amplifier, the mechanism responsible for the high sensitivity of mammalian hearing. We report that actin depolymerization, as regulated by activation/inhibition of LIMK/cofilin-mediated pathways, has a pivotal role in OHC motility. LIMK-mediated cofilin phosphorylation, which inhibits the actin depolymerizing activity of this protein, increases both electromotile amplitude and total length of guinea pig OHCs. In contrast, a decrease in cofilin phosphorylation reduces both OHC electromotile amplitude and OHC length. Experiments with acetylcholine and lysophosphatidic acid indicate that the effects of these agents on OHC motility are associated with regulation of cofilin phosphorylation via different signaling cascades. On the other hand, nonlinear capacitance measurements confirmed that all observed changes in OHC motile response were independent of the performance of the motor protein prestin. Altogether, these results strongly support the hypothesis that the cytoskeleton has a major role in the regulation of OHC motility, and identify actin depolymerization as a key process for modulating cochlear amplification.
Introduction
Outer hair cells (OHCs) elongate and shorten in response to electrical stimulation by activating a plasma membrane-based force generator mechanism associated with conformational changes in the integral membrane protein prestin (1,2). A variety of mechanical and chemical stimuli, on the other hand, induce changes in OHCs' length by activating a prestin-independent mechanism associated with cytoskeletal reorganization (1,3). The prestin-dependent (electromotility) and the prestin-independent (slow motility) mechanisms, working alone or in combination and perhaps in association with an active hair bundle motion, are part of the cochlear amplifier, the active mechanism enhancing sensitivity and frequency discrimination of the mammalian ear (1).
OHCs possess a cortical cytoskeleton lying underneath the lateral plasma membrane. It is mainly composed of circumferentially oriented actin filaments cross-linked by spectrin tetramers and linked to the plasma membrane by thousands of ∼25-nm long, 10-nm diameter pillars (3). It has been suggested that the cortical cytoskeleton provides the vectorial component to the forces generated by prestin molecules in the lateral plasma membrane of OHCs (3,4), and that it could be involved in the regulation of their motile responses (5,6).
Rho GTPases are crucial regulators of the actin cytoskeleton known to mediate in different types of cell motility. Previous results from our laboratory suggested that cytoskeletal changes mediated by Rho GTPases are part of a cellular mechanism of homeostatic control of OHC motility (5,6). Acetylcholine (ACh), the major neurotransmitter released by efferent terminals at the base of OHCs (7), was reported to affect OHC motility (8,9) by activating a Rho Kinase (ROCK)-independent pathway (5,6). Lysophosphatidic acid (LPA)—a lipid mediator with diverse biological activities—is known to influence cell motility in several cell systems by activating RhoA-, Rac1-, and Cdc42-mediated pathways (10). Thus, ACh and LPA are two important tools to investigate the role of the cytoskeleton and Rho-mediated signals in the regulation of OHC motility.
As potent regulators of actin dynamics and recognized targets of Rho GTPases, LIM-kinases (LIMK) are potential cytoskeletal effectors of signaling cascades involved in the regulation of OHC motility (11). The two known members of the LIMK family, LIMK1 and LIMK2, display cell type-specific expression levels and different subcellular localization (12,13). LIMK are phosphorylated by RhoA via ROCK-mediated signals, and by Cdc42 through the myotonic dystrophy kinase-related Cdc42-binding kinase (MRCKα) (11). In addition, Rac1 and Cdc42 phosphorylate LIMK1 through p21-activated kinases (11). Both LIMK exert their effect via phosphorylating—and thus inactivating—cofilin, a small protein with actin depolymerizing activity (14). Cells lacking cofilin have impaired locomotion; those overexpressing cofilin are more motile (15).
We have investigated the expression and functional role of LIMK and cofilin in guinea pig OHCs. LIMK2 was found expressed both at the cell body and the hair bundle of isolated OHCs, whereas LIMK1 was abundant at the cell body but absent at the OHCs' stereocilia. We also found that both ACh and LPA stimulate cofilin phosphorylation in OHCs. In addition, our findings indicate LIMK-mediated cofilin phosphorylation, and hence actin depolymerization, would regulate different aspects of OHC motility and cochlear amplification. Thus, any disruption in the signaling pathways involving these molecules could result in extreme physiological responses such as hyperacusis or deafness.
Materials and Methods
Isolation of outer hair cells
Cochleae were obtained from young guinea pigs (Cavia porcellus, 200–300 g) euthanized with CO2 with procedures approved by the Institutional Animal Care and Use Committee. Cochlear spirals were placed in Leibowitz L-15 (Gibco-Invitrogen, Grand Island, NY) containing 1 mg/mL collagenase (type IV; Sigma, St. Louis, MO), and incubated at 31°C for 3 min. The organ of Corti was removed from the bone with fine needles and OHCs were mechanically dissociated by reflux through the needle of a 50 μL syringe (Hamilton, Reno, NV). Isolated OHCs were then moved to a recording chamber (PCCS1; Bioscience Tools, San Diego, CA) filled with L-15, and observed with Nomarski differential interference contrast optics on an Axiovert 135TV inverted microscope with a 63×/1.2 C-Apochromat objective (Carl Zeiss, Jena, Germany). Only OHCs meeting established health criteria (16) were used in this study.
Immunolabeling
Excised cochlear spirals were incubated for variable periods in either L-15 alone (control) or with the following agents (alone or combined): 10 μM LPA (Sigma), 100 μM ACh (Sigma), 2 μg/mL cell permeable C3-transferase (Cytoskeleton, Denver, CO), 10 μM Y27632 (Upstate Biotechnology, Lake Placid, NY). OHCs dissociated from cochlear spirals were moved to a petri dish with an uncoated glass bottom (MatTek, Ashland, MA), fixed in 4% paraformaldehyde (EMS, Fort Washington, PA) in phosphate-buffered saline for 2 h and processed for confocal microscopy. Anti-LIMK1 (C-18), anti-LIMK2 (H-78 and C-19), anti-cofilin (N-19), anti-phosphorylated cofilin (p-cofilin @ Ser3), anti-LPA2 (EDG-4, C-16), anti-LPA3 (EDG-7, K-19) and anti-LPA4 (P2Y9, N-20) from Santa Cruz Biotechnologies (Santa Cruz, CA), anti-p-LIMK1/LIMK2 (Thr508/Thr505) from Cell Signaling Technology (Beverly, MA), and anti-LPA1 (EDG-2) from Upstate Biotechnology were used as primary antibodies at 1:100/1:200 dilutions. Cy3 and Cy2 from Jackson ImmunoResearch (West Grove, PA), and Alexa 488 (Molecular Probes-Invitrogen, Eugene, OR) were used as secondary antibodies at 1:500/1:1000 dilutions. Actin was stained with rhodamine phalloidin (Molecular Probes). Samples were observed with LSM-410 (Carl Zeiss) and TSC-SP5 Broadband Spectra (Leica, Wetzlar, Germany) laser confocal microscopes, with C-Apo 63×/1.2 and HCX-PL 63×/1.2 objectives, respectively. For relative quantification of cofilin phosphorylation, green (p-cofilin) and red (cofilin) channels were evaluated separately for every OHC with the Analysis feature in the Photoshop CS4 Extended software (Adobe, San Jose, CA). By using the Lasso Tool to define the borders of the OHC image and the integrated density (ID) Tool from Photoshop to calculate the sum of the values of the pixels in each separated channel in the selected OHC image, we were able to define the ratio ID green/ID red as an estimation of the ratio p-cofilin-associated/cofilin-associated fluorescence in each single cell in every experimental condition. Values of at least 20 OHCs per condition were statistically analyzed using analysis-of-variance techniques.
Electrophysiology
Whole-cell voltage-clamp was achieved with either conventional or nystatin-perforated patch techniques (17,18) at room temperature using an EPC-9 patch-clamp amplifier (HEKA, Lambrecht, Germany). Both techniques provided similar results, confirming OHC motility experiments were not affected by washout of cytoplasmic components. Patch electrodes were made from borosilicate capillary glass (G-1.5; Narishige, Tokyo, Japan) using a P-97 micropipette puller (Sutter Instruments, Novato, CA). Continuous perfusion of an external solution consisting of L-15 adjusted to 305–310 mOsm with distilled water was provided at a rate of 0.3 mL/min using a syringe pump. Internal (intrapipette) solution was composed of 150 mM KCl, 1 mM MgCl2, 0.1 mM EGTA, 2 mM ATP-Mg, 0.1 mM GTP-Na, and 10 mM HEPES; pH was adjusted to 7.2 with Tris. Nystatin (Sigma) was initially dissolved in acidified methanol at 10 mg/mL, and this stock solution was added to the internal solution at a final concentration of 200 μg/mL just before use. Osmolarity of the intrapipette solution was adjusted to the same value of the control extracellular solution by adding glucose. The resistance between the patch electrode filled with this solution and the bath solution was 4–6 MΩ. Measurements of voltage-dependent nonlinear capacitance (NLC), the electrical signature of prestin (19), were performed using the software lock-in amplifier function included in the Pulse software (HEKA). Capacitance function was fitted to the first derivative of a two-state Boltzmann function relating nonlinear charge to membrane voltage (19). For capacitance measurements, OHCs were perfused with a blocking solution containing 100 mM NaCl, 20 mM CsCl, 20 mM tetraethylammonium-Cl, 2 mM CoCl2, 1.52 mM MgCl2, 5 mM glucose, and 10 mM HEPES, adjusted to pH 7.2 with Tris. The intrapipette solution consisted of 140 mM CsCl, 2 mM MgCl2, 10 mM EGTA, and 10 mM HEPES, adjusted to pH 7.2 with Tris. Osmolarity of these solutions was adjusted to 305–310 mOsm with glucose. Analysis-of-variance techniques were used to evaluate the effects of pharmacological agents.
Pharmacological agents
ACh and LPA were used as activator of Rho GTPases, cell permeable C3-tranferase as RhoA inhibitor, Y27632 as ROCK inhibitor, and peptides S3 (MASGVAVSDGVIKVFN) and S3-RV (NFVKIVGDSVAVGSAM) (Sigma Genosys, Woodlands, TX), as a competitive inhibitor of cofilin phosphorylation and its control, respectively (20). In addition, latrunculin-A (Sigma) was used as an agent to prevent actin polymerization. For motility experiments, Y27632 (5 μM), S3 (5 μg/mL), and S3-RV (5 μg/mL) were included in the pipette solution, thus these drugs started causing effect only after the whole-cell patch-clamp was obtained. LPA (10 μM) and ACh (100 μM), in contrast, were added to the cells' external media through the perfusion system typically 4 min before membrane rupture (except where specifically indicated). OHCs were electrically stimulated every 2 min, with the first pulse given 60 s after membrane rupture. We set the time of the first electrical stimulation as time 0 for electromotility measurements.
Video analysis and data handling
Images of isolated OHCs were captured and analyzed by using two different experimental approaches. In a first approach, images were captured in QuickTime (Apple, Cupertino, CA) video format at standard video resolution (720 × 480 pixels) and frame rate (30 fps), and analyzed off-line using DIAS software (Soll Technologies, Iowa City, IA) as previously described (21,22). In a second approach, images were captured in AVI format at higher resolution (1024 × 1024 pixels) and frequencies (60–1000 fps) using the high speed Photron Fastcam X 1024 PCI camera (Photron, San Diego, CA) and the High-Power LED Illumination System-36AD3500 (Lightspeed Technologies, Campbell, CA). Images were analyzed off-line using ProAnalyst software (Xcitex, Cambridge, MA). Data was further processed using Excel (Microsoft, Redmond, WA) and IGOR Pro (WaveMetrics, Lake Oswego, OR) software.
Analysis of electromotility and slow OHC motility
Electromotility and slow OHC motility were investigated with separate protocols. For electromotility experiments, isolated OHCs underwent electrical stimulations under the voltage-clamp protocol (Fig. S1 Aa in the Supporting Material). The overlap of current traces (Fig. S1 B, inset) indicated absence of electrophysiological degradation during the course of experiments. The absence of a significant contribution of prestin-independent mechanisms was confirmed by extraction analysis (21) (Fig. S1 D). For analysis of slow OHC motility, cell length was monitored at a constant VH = −70 mV value to guarantee the absence of voltage-dependent contributions. Values were expressed as a ratio to the cell length just before the disruption of the membrane for whole-cell voltage clamp (Fig. S1 C). Although in individual cases (e.g., Fig. S1 C, inset) cell length remained near constant for 20 or more min, variability increased with time suggesting cell deterioration (Fig. S1 Cb). Our studies indicated that, under our experimental conditions, cell length data collected more than 10 min after membrane rupture was less reliable. Therefore, only results corresponding to the first 10 min of every experiment are being reported in this study.
Results
Guinea pig OHCs express LIMK1, LIMK2, cofilin, LPA1, and LPA4
Strong immunolabeling of LIMK1, LIMK2, and cofilin was observed in guinea pig OHCs by confocal microscopy (Fig. 1 A). LIMK2 labeling was present throughout the cell body, including the hair bundle. LIMK1 labeling, in contrast, was abundant in the cell body but not evident at the hair bundle. Cofilin and phosphorylated cofilin (p-cofilin) distributions were similar throughout the OHCs, colocalizing along the body of untreated cells and labeling distinctly the hair bundle, the cuticular plate, the lateral cortex, and the actin-rich infracuticular structure. Antibody specificity was evaluated by preadsorption with specific peptides (Fig. S2).
Figure 1.
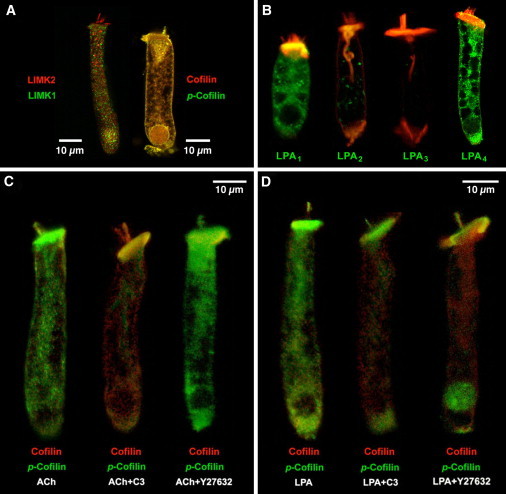
Immunolocalization of LIMK1, LIMK2, cofilin, and LPA receptors in isolated guinea pig OHCs. (A) LIMK1 and LIMK2 labeling was strong in the cell soma. In the hair bundle, in contrast, only LIMK2 was detected. Cofilin and p-cofilin showed a similar, widespread distribution. (B) LPA1 and LPA4, but not LPA2 and LPA3, are abundantly expressed in guinea pig OHCs. (Red, rhodamine phalloidin.) (C) Exposure to ACh increased cofilin phosphorylation (left). ACh effect was inhibited by C3 (center) but not by Y27632 (right). (D) LPA induced a generalized increase in cofilin phosphorylation (left). Exposure to LPA+C3 (center) and LPA+Y27632 (right) resulted in redistribution of p-cofilin labeling, which concentrate at the hair bundle, cuticular plate, nucleus, and infranuclear region.
All LPA activities in mammals are mediated by, at least, five G-protein-coupled receptors: LPA1, LPA2, LPA3, LPA4, and LPA5 (23). In OHCs we detected strong reactivity to antibodies against LPA1 and LPA4, but not LPA2 and LPA3 (Fig. 1 B). Expression of LPA5 was not investigated because of lack of reliable antibodies. Labeling of LPA1 was observed at the lateral surface of OHCs and the cytoplasm, but not at the apical plasma membrane, hair bundle, or nucleus. LPA4 labeling, in contrast, was also observed at the apical plasma membrane and nucleus (Fig. 1 B).
ACh and LPA stimulate LIMK-mediated cofilin phosphorylation
We used anti-cofilin and anti-p-cofilin antibodies in OHCs exposed to ACh and LPA. Exposure of isolated OHCs to ACh for 30 min significantly increased the ratio p-cofilin/cofilin immunoreactivity respect to the same ratio in nonexposed (control) cells (ACh = 12.7 ± 2.9 versus control = 1.8 ± 0.2, p ≤ 0.0001. Fig. 1 C, left). ACh-induced increase in p-cofilin/cofilin ratio was prevented by pre- and/or simultaneous incubation with the RhoA inhibitor C3-transferase (0.22 ± 0.03, p ≤ 0.16 versus control. Fig. 1 C, center) but not by the ROCK inhibitor Y27632 (6.7 ± 0.5, p ≤ 0.0001 versus control. Fig. 1 C, right).
LPA also increased the p-cofilin/cofilin ratio (Fig. 1 D, left), but not as much as ACh (5.3 ± 0.4, p ≤ 0.0015 versus control, p ≤ 0.0001 versus ACh). LPA effect, however, was not affected by C3 but reduced by Y27632 (LPA+C3 = 5.3 ± 1.4, p ≤ 0.97 versus LPA, p ≤ 0 .01 versus control; LPA+Y27632= 2.1 ± 0.3, p ≤ 0.07 versus LPA. P ≤ 0.8 versus control). Importantly, C3 and Y27632 induced redistribution of p-cofilin labeling, with reactivity concentrated in the apical (hair bundle and cuticular plate) and basal (nucleus and Retzius body, immediately below the nucleus) regions of the cells (Fig. 1 D, center and right).
ROCK/LIMK/cofilin-mediated signals regulate OHC motility
To investigate the role of ROCK/LIMK/cofilin-mediated signals in OHC motility we used the ROCK inhibitor Y27632 and S3, a peptide corresponding to the first 16 amino acids of cofilin containing the LIMK phosphorylation site at serine 3. This peptide binds to both LIMK1 and LIMK2 competitively, inhibiting their ability to phosphorylate cofilin (20).
A pure electromotile response, as confirmed by motility analysis (21), was elicited by stimulating isolated OHCs with single electrical pulses repeated every 2 min (Fig. 2 A). In contrast to control cells, which exhibited a near-constant response, OHCs exposed to Y27632 showed a significant reduction in electromotile amplitude (p ≤ 0.003, Y27632 group versus control group), with progressive impairment starting <4 min after the start of the treatment (Fig. 2 Aa). S3-loaded OHCs showed a similar decrease in electromotile amplitude over time (p ≤ 0.05, S3 versus control; see Fig. 2 Ab). Electromotile amplitude did not decrease when S3RV, a peptide with the same 16 amino acids but with reverse sequence of S3, was used, confirming the effect was specific. NLC measurements showed no effects of Y27632 and S3 on prestin performance, indicating that the changes in electromotile amplitude induced by the inhibitors were not associated with the motor proteins (Fig. 2 Ac and Table 1).
Figure 2.
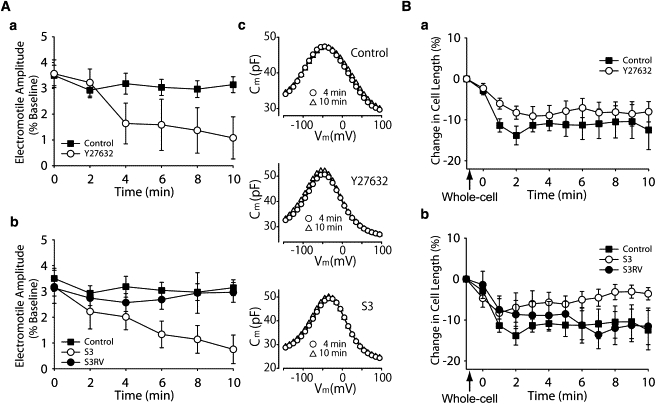
Role of cofilin phosphorylation on OHC electromotility (A) and slow OHC motility (B). (Aa) Electromotile amplitude in control (n = 5) and Y27632-exposed (n = 5) OHCs. (Ab) Electromotile amplitude in S3 (n = 7) and S3RV-treated (n = 4) OHCs. Control values are the same that in panel Aa. (Ac) NLC measurements, corresponding to single, representative cells, showing no effects of treatment on Prestin performance. (Ba) Slow changes in total length in control (n = 10) and Y27632-exposed (n = 14) OHCs. (Bb) Slow length changes in OHCs exposed to S3 (n = 6) and S3RV (n = 5). Control values are the same that in panel Ba, and were included here for comparison. (Points and vertical bars represent mean ± SE.)
Table 1.
Effects of Y27632 and S3 on OHC's nonlinear capacitance
| 0 min |
4 min |
10 min |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Y27632 (n = 6) | Control (n = 6) | S3 (n = 3) | Y27632 (n = 6) | Control (n = 6) | S3 (n = 3) | Y27632 (n = 6) | Control (n = 6) | S3 (n = 3) | |||||||
| Vmax(mV) | −52.1 ± 6.8 | NS (p = 0.939) | −51.7 ± 7.8 | NS (p = 0.359) | −46.3 ± 8.8 | −58.0 ± 6.7 | NS (p = 0.954) | −58.2 ± 8.3 | NS (p = 0.184) | −50.3 ± 8.9 | −59.6 ± 6.7 | NS (p = 0.981) | −59.5 ± 9.1 | NS (p = 0.200) | −51.9 ± 8.4 |
| Cmax(pF) | 47.0 ± 2.4 | NS (p = 0.309) | 42.0 ± 1.2 | NS (p = 0.519) | 45.9 ± 1.2 | 47.6 ± 2.4 | NS (p = 0.362) | 43.1 ± 1.5 | NS (p = 0.548) | 46.7 ± 1.7 | 47.5 ± 2.5 | NS (p = 0.413) | 43.5 ± 1.6 | NS (p = 0.554) | 47.1 ± 2.0 |
| z | 0.80 ± 0.07 | NS (p = 0.987) | 0.73 ± 0.03 | NS (p = 0.978) | 0.89 ± 0.03 | 0.79 ± 0.07 | NS (p = 0.988) | 0.72± 0.03 | NS (p = 0.973) | 0.92 ± 0.03 | 0.76 ± 0.07 | NS (p = 0.991) | 0.70 ± 0.03 | NS (p = 0.970) | 0.93 ± 0.03 |
| Qmax(pC) | 2.93 ± 0.19 | NS (p = 0.958) | 2.67 ± 0.08 | NS (p = 0.970) | 2.45 ± 0.13 | 2.98 ± 0.25 | NS (p = 0.956) | 2.71 ± 0.12 | NS (p = 0.968) | 2.47 ± 0.20 | 3.07 ± 0.23 | NS (p = 0.949) | 2.76 ± 0.13 | NS (p = 0.960) | 2.46 ± 0.24 |
| Clin(pF) | 24.5 ± 0.7 | NS (p = 0.766) | 23.1 ± 0.9 | NS (p = 0.858) | 24.1 ± 1.5 | 24.3 ± 0.6 | NS (p = 0.838) | 23.4 ± 0.9 | NS (p = 0.918) | 24.0 ± 1.5 | 24.0 ± 0.8 | NS (p = 0.892) | 23.4 ± 0.8 | NS (p = 0.915) | 24.0 ± 1.4 |
Vmax, voltage at peak capacitance; Cmax, peak capacitance; z, valence; Qmax, maximum nonlinear charge moved; Clin, linear membrane capacitance.
Cofilin inhibition also affected OHC slow motility (Fig. 2 Ba). Control cells shortened rapidly and then stabilized ∼3 min after the establishment of the whole-cell configuration, never recovering their original length. Cells exposed to Y27632 showed similar changes, with no significant differences between both groups (Fig. 2 Ba). Exposure to S3, in contrast, abolished cell shortening associated with establishment of whole-cell configuration, with OHCs reaching approximately their original length at the end of the experiment (p ≤ 0.02 versus control. Fig. 2 Bb). The small, transient cell contraction (<5%) observed immediately after membrane disruption was probably related to the time required for S3 to diffuse into the cells. No effects were observed in cell exposed to S3RV.
LIMK and cofilin mediate ACh and LPA effects on OHC electromotility
Next, we examined the effects of ACh and LPA on OHCs that were either untreated or exposed to Y27632 and S3. Control cells showed an average electromotile amplitude of 3.12% (relative to total cell length. Range 2.92–3.49%), ACh increased this amplitude by 14–27%, with values slightly fluctuating around an average of 3.84% (range 3.71–3.99%) over the 10 min experiment (Fig. 3 Aa). As described in a previous report (6), ACh effect on OHCs exposed to Y27632 consisted of a slight decrease in electromotile amplitude in the first 4 min, followed by a partial recovery (Fig. 3 Aa). The response elicited by ACh in cells exposed to S3, in contrast, was similar to that elicited by S3 alone, a continuous decrease in electromotile amplitude with time (from 3.8% to 0.7%. Fig. 3 Aa). We also confirmed previous reports (6,9) showing that ACh does not affect prestin function, as indicated by lack of changes in NLC (inset, Fig. 3 Aa and Table 2).
Figure 3.
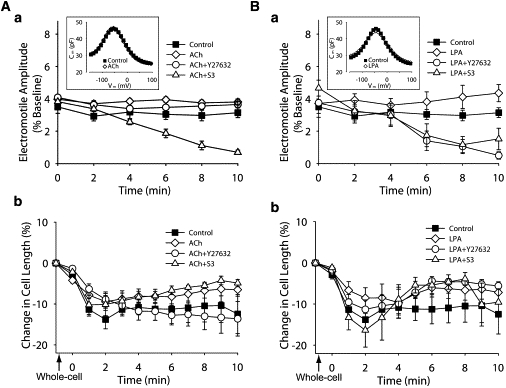
Effect of ACh and LPA on OHCs exposed to Y27632 and S3. (Aa) ACh effect on electromotile amplitude of untreated cells (ACh, n = 8) and cells exposed to Y27632 (n = 4) and S3 (n = 4). Data for control condition is the same used in Fig. 2Aa, and was included here for comparison. (Inset) NLC measurements. (Ab) ACh effect on slow motility of untreated cells (ACh, n = 8) and cells exposed to Y27632 (n = 4) and S3 (n = 4). Data for control condition is the same used in Fig. 2Ba, and was included here for comparison. (Ba and Bb) Same as for panels Aa and Ab, but using LPA (n = 5), Y27632+LPA (n = 6), and S3+LPA (n = 7). (Points and vertical bars represent mean ± SE.)
Table 2.
Effects of ACh and LPA on OHC's nonlinear capacitance
| Control (n = 4) | ACh (n = 4) | Control (n = 5) | LPA(n = 5) | |||
|---|---|---|---|---|---|---|
| Vmax(mV) | −38.4 ± 6.8 | NS (p = 0.635) | −40.2 ± 6.6 | −44.3 ± 4.2 | NS (p = 0.825) | −45.1 ± 3.0 |
| Cmax(pF) | 45.3 ± 2.4 | NS (p = 0.939) | 45.0 ± 1.8 | 43.3 ± 2.2 | NS (p = 0.958) | 43.2 ± 2.6 |
| z | 0.83 ± 0.04 | NS (p =0.999) | 0.82 ± 0.05 | 0.85 ± 0.04 | NS (p = 0.996) | 0.83 ± 0.04 |
| Qmax(pC) | 2.45 ± 0.16 | NS (p = 0.996) | 2.47 ± 0.09 | 2.31 ± 0.19 | NS (p = 0.994) | 2.29 ± 0.20 |
| Clin(pF) | 24.1 ± 0.9 | NS (p = 0.959) | 23.9 ± 0.8 | 22.8 ± 0.7 | NS (p = 0.930) | 23.1 ± 0.7 |
Vmax, voltage at peak capacitance; Cmax, peak capacitance; z, valence; Qmax, maximum nonlinear charge moved; Clin, linear membrane capacitance.
Except for a dip at 4 min, exposure to LPA increased OHC's electromotile amplitude by 13–38% in a progressive manner (average 4.1%, range 3.8–4.6%. Fig. 3 Ab). The response of OHCs to LPA+Y27632 or LPA+S3, in turn, was similar to that elicited by Y27632 or S3 alone, indicating that either ROCK inhibition or cofilin inhibition were sufficient to completely block the stimulatory effect of LPA on OHC electromotile amplitude. Interestingly, statistical analysis indicated that OHCs responses to ACh+S3 and LPA+S3 were similar, whereas responses to ACh+Y27632 and LPA+Y27632 showed a significant difference (p ≤ 0.0001). NLC measurements confirmed that neither LPA alone nor combined with Y27632 or S3 influenced prestin motor response, supporting the idea that the observed effects were mediated by cytoskeletal changes (inset, Fig. 3 Ba and Table 2).
LIMK and cofilin also mediate ACh and LPA effects on slow OHC motility
OHCs exposed to ACh shortened during the first 2 min, just like control cells, but the peak contraction lasted only ∼1 min and was followed by recovery (Fig. 3 Ab). This response resembled the time course of the effects of ACh perfusion on the displacement of the basilar membrane measured at the basal turn of the guinea pig cochlea (24). Interestingly, cells treated with ACh+Y27632 showed the same response as control-cells, whereas cells exposed to ACh+S3 responded just like those treated with ACh alone. Statistical analysis showed that the responses elicited by ACh and ACh+S3 were, on average, significantly different (p ≤ 0.05) to those observed in control and ACh+Y27632-exposed cells (Fig. 3 Ab). OHCs exposed to LPA, LPA+Y27632, and LPA+S3 showed a significant rebound effect (p ≤ 0.02 versus control) similar to that observed in OHCs exposed to ACh and ACh+S3 (Fig. 3 Ba). This rebound effect was clearly distinct from the near asymptotic decrease in length observed in cells exposed to Y27632 alone and the slight, but continuous increase in length displayed by OHCs exposed to S3 alone.
Effect of latrunculin-A on isolated OHCs
We exposed isolated OHCs to latrunculin-A, an agent that prevents actin monomers from polymerizing into filaments (25), to investigate whether inhibition of actin polymerization could ameliorate the effects of Y27632 and S3 on OHC motility. We speculated that exposure to latrunculin-A (decreasing actin polymerization) could somewhat compensate the effects of Y27632 and S3 (decreasing actin depolymerization). Interestingly, addition of nanomolar concentrations of latrunculin-A to the external medium induced a fast and irreversible failure of the OHC's lateral wall, with disruption of the plasma membrane followed by cell death (Fig. 4 A). Perfusion experiments consistently showed a fast disruption of cytoplasmic structures, followed by cell elongation and ballooning of the basal region of the OHCs. The plasma membrane was disrupted at the basal region before the effects of the drug on the lateral wall were apparent (Fig. 4, B and C). Latrunculin effects were not prevented by previous perfusion of OHCs with S3 through the patch pipette. Thus, whereas actin depolymerization may be important for regulating OHC motility, actin polymerization seems to be crucial for maintaining OHC shape and functionality.
Figure 4.
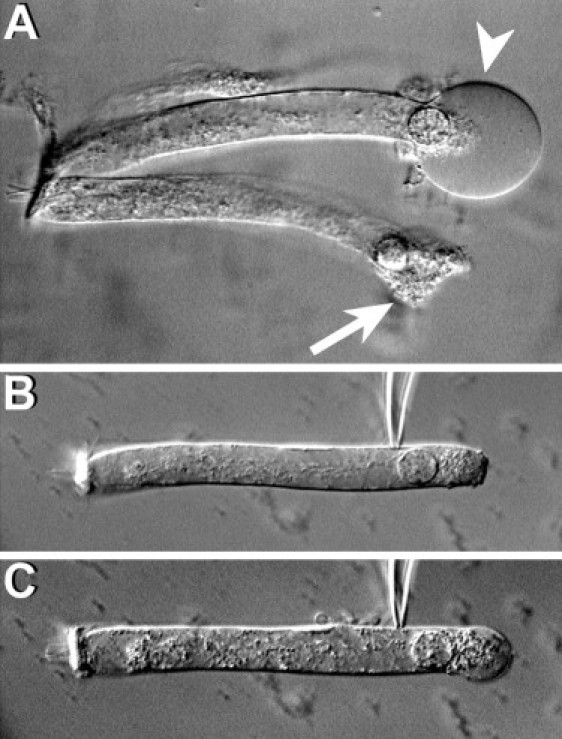
Effect of latrunculin. (A) After the addition of 0.2 μM of latrunculin-A to the external medium, the basal region of the OHCs expands quickly, adopting a spherical shape (arrowhead), and then bursts, killing the cell (arrow). (B) A typical cell immediately after attachment of a patch pipette filled with latrunculin, and (C) moments later, showing latrunculin-induced disruption of cytoplasmic structures, cell elongation, and progressive ballooning of the basal part. All cells died before the membrane was ruptured to establish whole-cell configuration.
Discussion
Previous studies have suggested that OHC motility is regulated by cytoskeletal changes mediated by Rho GTPases, but no details about the molecular targets of the Rho-mediated signals were provided (5,6). Here, we present evidence indicating that the regulatory mechanism relies, at least in part, on the control of the depolymerization rate of actin filaments by LIMK/cofilin-mediated signals.
ACh and LPA induce cofilin phosphorylation via different pathways
Our results indicate both ACh and LPA induce LIMK-mediated-cofilin phosphorylation, therefore decreasing the rate of actin depolymerization, but they achieve this effect by activating different signaling pathways. Whereas ACh uses a RhoA-dependent (C3-sensitive), ROCK-independent (Y27632-insensitive) signaling cascade, LPA would phosphorylate cofilin via either RhoA/ROCK or a different pathway involving neither RhoA nor ROCK (Fig. 1, C and D).
Activation of ACh receptors in OHCs leads to an increase in intracellular Ca2+ and the opening of small conductance Ca2+-activated K+ channels (26). It has already been suggested that a Ca2+-dependent pathway activated by ACh would target proteins in the OHC cytoskeleton (9). Interestingly, studies in endothelial (27) and smooth muscle cells (28,29) suggest that increasing levels of intracellular Ca2+ induced by agonist stimulation could led to direct activation of Rho GTPases by facilitating selective translocation of inactivated RhoA to the plasma membrane, where it becomes available for activation (29). Thus, we speculate that a similar mechanism could be at work in OHCs, with ACh-induced RhoA signals activating LIMK either via ROCK- or Rac1/Cdc42-mediated pathways (Fig. 5).
Figure 5.
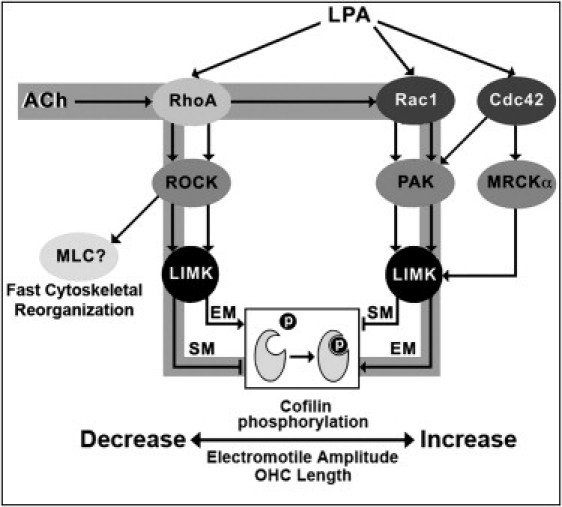
Schematic model for the signaling pathways targeting cofilin phosphorylation activated by ACh and LPA as suggested by results in this article. The pathways putatively stimulated by ACh are highlighted in gray. (EM, electromotility; SM, slow motility.)
The effects of C3 and Y27632 on LPA-induced cofilin phosphorylation (Fig. 1 D) could be explained by our finding that guinea pig OHCs express two types of LPA receptors, LPA1 and LPA4 (Fig. 1 B). It has been reported that, in a rat neuronal cell line, LPA could stimulate a Rac-dependent pathway via LPA1 and a RhoA/ROCK-dependent pathway via LPA1/LPA4 (30). Thus, we speculate something similar could be occurring in OHCs, with LPA-induced cofilin phosphorylation being mediated by different signaling pathways associated, respectively, with LPA1 and LPA4.
Subcellular distribution suggests different functional roles for LIMK1 and LIMK2 in guinea pig OHCs
Although LIMK1 and LIMK2 are very homologous, there is increasing evidence that each may be subject to different regulatory pathways and may contribute to both distinct and overlapping cellular functions (11). The different distribution of LIMK1 and LIMK2 in guinea pig OHCs, and the existence of at least two specific signaling pathways able to induce LIMK-mediated cofilin phosphorylation, strongly supports this idea. The presence of cofilin and LIMK2—but not LIMK1—in the hair bundle of isolated OHCs, and the failure of C3 and Y27632 in preventing LPA-induced cofilin phosphorylation there (Fig. 1 D), suggests that cofilin-induced actin depolymerization in the stereocilia would be controlled by a RhoA/ROCK-independent, LIMK2-mediated signals. LIMK1 and RhoA/ROCK-dependent signals, on the other hand, could be more directly associated with OHC motility, because both electromotility and slow motility are mechanical responses involving the OHC's main body.
Cofilin phosphorylation is a major regulator of OHC electromotility
The decrease in electromotile amplitude induced by Y27632 in nontreated and LPA-exposed OHCs (Figs. 2 Aa and 3 Ba) confirmed previous results from our laboratory, suggesting involvement of a RhoA/ROCK-mediated pathway in the regulation of prestin-dependent OHC motility (6). We have further extended these results by demonstrating that complete inhibition of cofilin phosphorylation with S3 induced an identical response in cells stimulated with LPA and ACh, whereas Y27632 effects were overridden by ACh but not by LPA. These results lead to two significant conclusions: first, signals mediated by the RhoA/ROCK/LIMK/cofilin pathway are sufficient to explain all observed effects of LPA on OHC electromotility, and second, cofilin phosphorylation induced by activation of a ROCK-independent pathway is sufficient to explain all observed effects of ACh on OHC electromotility.
We can envision a scenario where OHC electromotile amplitude is maintained at an optimal level by a very sensitive mechanism largely based on cofilin-regulated actin depolymerization. A large pool of cofilin would be normally phosphorylated (see Fig. 1 A), keeping the rate of actin depolymerization low and electromotile amplitude high. LPA stimulation, however, would still be able to increase electromotility amplitude via overactivation of an already functional RhoA/ROCK/LIMK/cofilin signaling pathway. The signals generated by LPA would phosphorylate more cofilin, increasing stability of actin filaments and the actin-based cortical cytoskeleton by inhibiting actin depolymerization. Inhibition of ROCK, in turn, would increase the rate of actin depolymerization by inhibiting cofilin phosphorylation, with a consequent disruption in cortical cytoskeleton structure and significant decrease in electromotile amplitude because of inefficient harnessing of the forces generated by prestin molecules in the lateral plasma membrane. ACh, on the other hand, would be able to inhibit actin depolymerization by phosphorylating cofilin via ROCK-independent pathway/s, returning electromotile amplitude to normal values. Thus, ACh would be working as a fast-reset mechanism as suggested in a previous model (6). In contrast to the previous model, however, rather than inhibiting a putative Rac-mediated signaling pathway aimed at decreasing OHC electromotile amplitude, ACh would be stimulating that pathway to phosphorylate cofilin, bypassing the ROCK-dependent regulatory mechanism (Fig. 5). In addition, our results also indicate a minimal—if any—contribution of alternative ROCK-mediated signals targeting the cytoskeleton (not involving LIMK-mediated cofilin phosphorylation, e.g., a RhoA/ROCK/adducin cascade (6)) to the regulation of OHC electromotility.
Cofilin phosphorylation is also involved in the regulation of OHC slow motility
Exposure to S3, but not to Y27632, abolished cell shortening linked to the establishment of the whole-cell patch configuration (Fig. 2, Aa and Ab). Thus, we can hypothesize that this contraction is associated with cofilin phosphorylation mediated by a ROCK-independent mechanism. Moreover, the similar inhibition of cell shortening induced by LPA and LPA+Y27632 (Fig. 3 Bb) suggest that LPA would also be able to inhibit cofilin phosphorylation via a ROCK-independent mechanism. Thus, our results not only provide evidence that LPA could be able to stimulate two different signaling pathways in guinea pig OHCs, but suggest that LPA would be regulating OHC electromotility by stimulating cofilin phosphorylation via a ROCK-dependent pathway, and regulating OHC slow motility by inhibiting cofilin phosphorylation via a ROCK-independent pathway (Fig. 5).
ACh, on the other hand, would affect OHC motility just like LPA but via different pathways. The similar effects of ACh alone and ACh+S3 on slow motility suggest that ACh inhibits cofilin phosphorylation, whereas the response to ACh+Y27632 suggests that this effect would be mediated by ROCK. Thus, ACh would be regulating OHC electromotility by stimulating cofilin phosphorylation via a ROCK-independent pathway, and regulating OHC slow motility by inhibiting cofilin phosphorylation via a ROCK-dependent pathway (Fig. 5).
How does cofilin phosphorylation translate into regulation of OHC motor function?
The results here, showing that S3, LPA, and ACh do not change the capacitance-voltage function of prestin, make unlikely a direct effect of LIMK/cofilin-mediated signals on the motor protein. The amplitude of OHC electromotility, however, relies not only on prestin performance but also on the effective harnessing of the individual forces generated by prestin molecules in the plane of the membrane (3,4). Thus, the well-known effects of LIMK/cofilin-mediated signals on actin dynamics strongly support the idea that the actin-based cytoskeleton could play a central role in the regulation of prestin-dependent OHC electromotility. We speculate that LIMK/cofilin-mediated signals would regulate the length and distribution of actin filaments in the OHC cortical cytoskeleton, modulating the magnitude of the vectorial component of the prestin-generated forces in the plasma membrane along the longitudinal axis of the OHCs.
While prestin-dependent motility is unique to OHCs, every cell is able to change its shape in response to specific signals in a process involving active reorganization of the actin cytoskeleton. Slow OHC motility requires a correct spatial and temporal assembly and disassembly of actin. Thus, control of LIMK-mediated cofilin phosphorylation is a natural regulatory mechanism of OHC shape. Because the micromechanics of the cochlear partition depends critically on the length of the OHCs, slow motility regulation could have a profound influence on cochlear amplification. In particular, the proposed ability of the efferent feedback to protect the cochlea from noise trauma (31) could be associated with the effect of ACh on OHC slow motility rather than electromotility. Electrical stimulation of medial olivocochlear fibers result in release of ACh from the end terminals at the base of cochlear OHCs, and the major effect is a reduction in cochlear amplification by some 20 dB (7). However, the reported increase in OHC electromotile amplitude induced by ACh ((8,9) and this report) is contrary to the expected inhibitory response. These results could be conciliated, assuming ACh-induced slow changes in OHC length as the most important mechanical effect of efferent stimulation. OHC elongation, for instance, might increase the depth of the subtectorial space, changing its hydrodynamic properties and diminishing the gain of the cochlear amplifier by affecting the stimulation of the IHC's stereocilia by fluid flow (32). This speculation is consistent with previous results from other laboratories showing that ACh stimulates a Ca2+-dependent pathway targeting the OHC cytoskeleton (9), and that in vivo perfusion of ACh in the guinea pig cochlea induces a measurable displacement of the basilar membrane with the same time course of the slow changes in OHC length reported in this study (24).
Conclusions
The rate of actin depolymerization, as regulated by LIMK-mediated cofilin phosphorylation, would be pivotal for the motile response of cochlear OHCs. The amplitude of the electromotile response is associated to the available amount of nonphosphorylated (active) cofilin. LIMK-mediated control of cofilin phosphorylation would regulate the rate of depolymerization of actin filaments, the stability of the cortical cytoskeleton, and, consequently, OHC electromotile amplitude and total length. LPA works as an exogenous agonist for the electromotile process by overactivating a RhoA/ROCK/LIMK-mediated pathway able to induce cofilin phosphorylation, and influencing OHC slow motility by stimulating a ROCK-independent pathway inhibiting cofilin phosphorylation. ACh is able to stimulate cofilin phosphorylation and OHC electromotility via a RhoA-dependent, ROCK-independent signaling cascade, and modulate OHC slow motility by inhibiting cofilin phosphorylation via RhoA/ROCK. This regulation of cell length and electromotile amplitude could have profound influence on the gain of the cochlear amplifier.
Acknowledgments
The authors thank Mrs. Shanping Chen for technical assistance, and declare no existing or potential conflict of interest.
This work was supported by National Institutes of Health grant No. DC010146 and House Ear Institute. Its content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the House Ear Institute.
Supporting Material
References
- 1.Ashmore J. Cochlear outer hair cell motility. Physiol. Rev. 2008;88:173–210. doi: 10.1152/physrev.00044.2006. [DOI] [PubMed] [Google Scholar]
- 2.Zheng J., Shen W., Dallos P. Prestin is the motor protein of cochlear outer hair cells. Nature. 2000;405:149–155. doi: 10.1038/35012009. [DOI] [PubMed] [Google Scholar]
- 3.Holley M.C. Outer hair cell motility. In: Dallos P., Popper A.N., Fay R.R., editors. The Cochlea. Springer; New York: 1996. pp. 386–434. [Google Scholar]
- 4.Holley M.C., Kalinec F., Kachar B. Structure of the cortical cytoskeleton in mammalian outer hair cells. J. Cell Sci. 1992;102:569–580. doi: 10.1242/jcs.102.3.569. [DOI] [PubMed] [Google Scholar]
- 5.Kalinec F., Zhang M., Kalinec G. Rho GTPases mediate the regulation of cochlear outer hair cell motility by acetylcholine. J. Biol. Chem. 2000;275:28000–28005. doi: 10.1074/jbc.M004917200. [DOI] [PubMed] [Google Scholar]
- 6.Zhang M., Kalinec G.M., Kalinec F. ROCK-dependent and ROCK-independent control of cochlear outer hair cell electromotility. J. Biol. Chem. 2003;278:35644–35650. doi: 10.1074/jbc.M301668200. [DOI] [PubMed] [Google Scholar]
- 7.Guinan J.J. Physiology of cochlear efferents. In: Dallos P., Popper A.N., Fay R.R., editors. The Cochlea. Springer-Verlag; New York: 1996. pp. 435–502. [Google Scholar]
- 8.Dallos P., He D.Z.Z., Evans B.N. Acetylcholine, outer hair cell electromotility, and the cochlear amplifier. J. Neurosci. 1997;17:2212–2226. doi: 10.1523/JNEUROSCI.17-06-02212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frolenkov G.I., Mammano F., Kachar B. Two distinct Ca2+-dependent signaling pathways regulate the motor output of cochlear outer hair cells. J. Neurosci. 2000;20:5940–5948. doi: 10.1523/JNEUROSCI.20-16-05940.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moolenaar W.H., van Meeteren L.A., Giepmans B.N. The ins and outs of lysophosphatidic acid signaling. Bioessays. 2004;26:870–881. doi: 10.1002/bies.20081. [DOI] [PubMed] [Google Scholar]
- 11.Scott R.W., Olson M.F. LIM kinases: function, regulation and association with human disease. J. Mol. Med. 2007;85:555–568. doi: 10.1007/s00109-007-0165-6. [DOI] [PubMed] [Google Scholar]
- 12.Acevedo K., Moussi N., Bernard O. LIM kinase 2 is widely expressed in all tissues. J. Histochem. Cytochem. 2006;54:487–501. doi: 10.1369/jhc.5C6813.2006. [DOI] [PubMed] [Google Scholar]
- 13.Foletta V.C., Moussi N., Bernard O. LIM kinase 1, a key regulator of actin dynamics, is widely expressed in embryonic and adult tissues. Exp. Cell Res. 2004;294:392–405. doi: 10.1016/j.yexcr.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 14.Bamburg J.R. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu. Rev. Cell Dev. Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- 15.dos Remedios C.G., Chhabra D., Nosworthy N.J. Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol. Rev. 2003;83:433–473. doi: 10.1152/physrev.00026.2002. [DOI] [PubMed] [Google Scholar]
- 16.Zajic G., Schacht J. Comparison of isolated outer hair cells from five mammalian species. Hear. Res. 1987;26:249–256. doi: 10.1016/0378-5955(87)90061-x. [DOI] [PubMed] [Google Scholar]
- 17.Horn R., Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. J. Gen. Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akaike N., Harata N. Nystatin perforated patch recording and its applications to analyses of intracellular mechanisms. Jpn. J. Physiol. 1994;44:433–473. doi: 10.2170/jjphysiol.44.433. [DOI] [PubMed] [Google Scholar]
- 19.Santos-Sacchi J. Reversible inhibition of voltage-dependent outer hair cell motility and capacitance. J. Neurosci. 1991;11:3096–3110. doi: 10.1523/JNEUROSCI.11-10-03096.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishita M., Aizawa H., Mizuno K. Stromal cell-derived factor 1α activates LIM kinase 1 and induces cofilin phosphorylation for T-cell chemotaxis. Mol. Cell. Biol. 2002;22:774–783. doi: 10.1128/MCB.22.3.774-783.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsumoto N., Kalinec F. Extraction of prestin-dependent and prestin-independent components from complex motile responses in guinea pig outer hair cells. Biophys. J. 2005;89:4343–4351. doi: 10.1529/biophysj.105.064626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsumoto N., Kalinec F. Prestin-dependent and prestin-independent motility of guinea pig outer hair cells. Hear. Res. 2005;208:1–13. doi: 10.1016/j.heares.2005.03.030. [DOI] [PubMed] [Google Scholar]
- 23.Choi J.W., Herr D.R., Chun J. LPA receptors: subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 2010;50:157–186. doi: 10.1146/annurev.pharmtox.010909.105753. [DOI] [PubMed] [Google Scholar]
- 24.Murugasu E., Russell I.J. The role of calcium on the effects of intracochlear acetylcholine perfusion on basilar membrane displacement in the basal turn of the guinea pig cochlea. Aud. Neurosci. 1996;2:363–376. doi: 10.1523/JNEUROSCI.16-01-00325.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morton W.M., Ayscough K.R., McLaughlin P.J. Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nat. Cell Biol. 2000;2:376–378. doi: 10.1038/35014075. [DOI] [PubMed] [Google Scholar]
- 26.Oliver D., Klöcker N., Fakler B. Gating of Ca2+-activated K+ channels controls fast inhibitory synaptic transmission at auditory outer hair cells. Neuron. 2000;26:595–601. doi: 10.1016/s0896-6273(00)81197-6. [DOI] [PubMed] [Google Scholar]
- 27.Masiero L., Lapidos K.A., Kohn E.C. Regulation of the RhoA pathway in human endothelial cell spreading on type IV collagen: role of calcium influx. J. Cell Sci. 1999;112:3205–3213. doi: 10.1242/jcs.112.19.3205. [DOI] [PubMed] [Google Scholar]
- 28.Sakurada S., Takuwa N., Takuwa Y. Ca2+-dependent activation of Rho and Rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circ. Res. 2003;93:548–556. doi: 10.1161/01.RES.0000090998.08629.60. [DOI] [PubMed] [Google Scholar]
- 29.Gong M.C., Fujihara H., Somlyo A.P. Translocation of rhoA associated with Ca2+ sensitization of smooth muscle. J. Biol. Chem. 1997;272:10704–10709. doi: 10.1074/jbc.272.16.10704. [DOI] [PubMed] [Google Scholar]
- 30.Yanagida K., Ishii S., Shimizu T. LPA4/p2y9/GPR23 mediates rho-dependent morphological changes in a rat neuronal cell line. J. Biol. Chem. 2007;282:5814–5824. doi: 10.1074/jbc.M610767200. [DOI] [PubMed] [Google Scholar]
- 31.Taranda J., Maison S.F., Elgoyhen A.B. A point mutation in the hair cell nicotinic cholinergic receptor prolongs cochlear inhibition and enhances noise protection. PLoS Biol. 2009;7:e18. doi: 10.1371/journal.pbio.1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lukashkin A.N., Richardson G.P., Russell I.J. Multiple roles for the tectorial membrane in the active cochlea. Hear. Res. 2010;266:26–35. doi: 10.1016/j.heares.2009.10.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.