

Abstract
Parkinson's disease is characterized by the presence of intracellular aggregates composed primarily of the neuronal protein α-synuclein (αS). Interactions between αS and various cellular membranes are thought to be important to its native function as well as relevant to its role in disease. We use fluorescence correlation spectroscopy to investigate binding of αS to lipid vesicles as a function of the lipid composition and membrane curvature. We determine how these parameters affect the molar partition coefficient of αS, providing a quantitative measure of the binding energy, and calculate the number of lipids required to bind a single protein. Specific anionic lipids have a large effect on the free energy of binding. Lipid chain saturation influences the binding interaction to a lesser extent, with larger partition coefficients measured for gel-phase vesicles than for fluid-phase vesicles, even in the absence of anionic lipid components. Although we observe variability in the binding of the mutant proteins, differences in the free energies of partitioning are less dramatic than with varied lipid compositions. Vesicle curvature has a strong effect on the binding affinity, with a >15-fold increase in affinity for small unilamellar vesicles over large unilamellar vesicles, suggesting that αS may be a curvature-sensing protein. Our findings provide insight into how physical properties of the membrane may modulate interactions of αS with cellular membranes.
Introduction
α-Synuclein (αS) is a small, normally highly soluble neuronal protein whose aggregation and deposition as fibrillar amyloidogenic deposits is one of the hallmarks of Parkinson's disease (PD). Although its precise role in disease progression is still unclear, three point mutations to αS (A30P, E46K, and A53T) and gene multiplication have been linked to early-onset PD (1–5), establishing that it is critical to the pathology of the disease.
The sequence of αS is divided into three domains: a positively charged N-terminal region that contains repeats of the highly conserved KTKEGV sequence, a central region known as the non-amyloid-β component, and an acidic C-terminal region (Fig. 1). Although intrinsically disordered in solution, the N-terminal 100 residues form an amphipathic α-helix upon membrane binding, whereas the C-terminal region remains largely unstructured (6). Natively, αS is localized to presynaptic terminals (7) and there is evidence that it is involved in synaptic vesicle trafficking and dopamine neurotransmission (8,9). In transgenic mice, αS knockout is nonlethal, although studies of these animals and in neuronal culture have reported altered dopamine release and uptake (8,10), a decreased synaptic vesicle pool (11), and changes in synaptic vesicle recycling (12). PD pathology may be associated with a change or disruption of native αS-lipid interactions, and in vitro studies indicate that early-onset PD mutations modify αS lipid-binding and aggregation propensities (13–16). It has also been suggested that the mechanism of toxicity in PD involves direct, disruptive interactions between αS and cellular membranes (17). Because of their potential relevance to both the native and pathological roles of αS, characterization of the interactions between αS and lipid membranes is of great interest.
Figure 1.
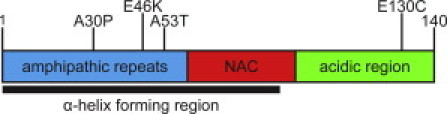
Schematic of αS illustrating three major regions. Shown are the three PD-associated mutations (A30P, E46K, and A53T) and the site of the cysteine mutation used for labeling (E130C).
Additional motivation for studying the interaction of αS with various model membranes comes from the observation that the physical properties of the membrane, such as alteration of the lipid composition, may influence αS function or dysfunction. For example, early studies found that αS has a much greater affinity for highly curved vesicles (18–24), and it was suggested that this preference may be related to the proposed function of αS in binding to similarly highly curved synaptic vesicles (20). Membrane curvature has the ability to modulate the conformation of the helical region of αS (25–29), providing a possible explanation for selective binding to highly curved membranes. Furthermore, the ability of αS to disrupt lipid bilayers or induce aggregation in vitro, which has been suggested to be a mechanism for αS-associated toxicity in PD (21,22,30–37), appears to be dependent on lipid composition.
Although there exists a significant body of work aimed at understanding the nature of interactions between αS and lipid membranes, much of this work is qualitative and often contradictory. Quantitative measurements of interaction energies and factors contributing to membrane affinities are largely lacking. For example, although there is a general consensus that αS interacts preferentially with negatively charged membranes, there is a lack of agreement as to whether specific anionic lipids are required or preferred (13,20–23,35,38–40). There are further conflicts as to whether αS binds to membranes composed of zwitterionic lipids, with varied reports of strong, weak, or no binding (20,21,23,35,38–40). In contrast to early studies that suggested that binding requires highly curved vesicles (19,20,23,24), more recent studies have shown binding to model membranes of a variety of curvatures (18,21,22,34,36,38–41), including planar supported bilayers (42,43).
In this work, fluorescence correlation spectroscopy (FCS) is used to measure the interactions between αS and lipid vesicles. We extract partition coefficients for αS using vesicles of varying lipid compositions and sizes. We find that specific anionic lipids have a dramatic effect on the binding affinity, and αS binds with higher affinity to gel-phase vesicles than to fluid-phase vesicles. In contrast, there are relatively minor differences in the free energies of partitioning between the wild-type (WT) and disease-associated mutants. Binding exhibits a nonlinear dependence on curvature, resulting in a moderate range of partitioning free energies. The number of lipids in the binding site of a single αS is independent of curvature and composition, and corresponds well to the estimated size of the membrane-bound α-helical region of αS. For context, we compare our measured partition coefficients to published values for αS.
Materials and Methods
The value αS was expressed, purified, and fluorescently labeled as described previously (25) (see Supporting Material).
Vesicle preparation
The lipids used were 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (POPS), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate (POPA), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), and 1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol (DPPG) (Avanti Polar Lipids, Alabaster, AL). Lipids were dissolved to 10–20 mg/mL in chloroform and stored at −20°C. Aliquots were removed, dried with nitrogen, and dehydrated in a vacuum-evacuated desiccator overnight. The resulting lipid film was rehydrated in MOPS buffer (20 mM MOPS, 147 mM NaCl, 2.7 mM KCl, pH 7.4) for 1 h. For preparation of large unilamellar vesicles (LUVs), aqueous lipid solutions were extruded 21 times through two stacked membranes (Whatman Nucleopore Track-Etch, Maidstone, UK) with 30 nm, 50 nm, 100 nm, or 200 nm diameter pores in a Liposofast extruder (Avestin, Ottawa, Canada). Lipid solutions containing DPPC or DPPG were heated to 55°C (above the melting temperatures) before extrusion. For preparation of small unilamellar vesicles (SUVs), rehydrated lipids were sonicated for 30 min on ice using a flat tip on a Misonix sonicator 3000 at 50% duty (Farmingdale, NY). SUVs were centrifuged at 8000 g for 15 min to remove particulate matter. All vesicles were assayed for inorganic phosphate to determine the total lipid concentration (44,45) and characterized by dynamic light scattering (DLS; see Supporting Material). All lipid compositions are given as molar ratios of components, and all protein/lipid ratios are given as molar ratios of protein to accessible lipid (see Supporting Material for calculation of accessible lipid).
Fluorescence correlation spectroscopy
FCS measurements were made on an instrument bulit in house based on an inverted Olympus IX-71 microscope as described previously (46). The vesicles were smaller than the dimensions of the focused laser beam, and both vesicles and protein freely diffused within the focal volume. Measurements were made in eight-well chambered coverglasses (Nunc, Rochester, NY) that were first treated to prevent αS from adsorbing to the chamber surfaces (Supporting Material).
Binding studies were made using a constant concentration of αS and varying the lipid concentrations. The samples were allowed to equilibrate for 5 min after mixing before measurement, and all measurements were at 20.5 ± 0.1°C in MOPS buffer. A complementary binding curve was obtained by adding increasing amounts of αS to vesicles in pH 5.0 MOPS buffer until the lipid/bound protein ratio reached a saturating value. All data shown are from at least two separate binding curves under identical conditions. For all studies, each autocorrelation curve was 10 s, repeated 25–30 times. Increasing the measurement time to 30 s for larger vesicles (47) did not results in any change in the autocorrelation curves. The curves were averaged together to obtain statistical variations used for weighting in the fitting outlined below. Fitting was done with the use of MATLAB (The MathWorks, Natick, MA).
The average curve was fit to Eq. 1 for multiple species of differing brightness diffusing in a three-dimensional Gaussian volume (48,49):
| (1) |
The only free parameters in Eq. 1 are N, the number of proteins, FF, the fraction of αS free in solution, and Q, the average brightness of the vesicles relative to a single αS (Supporting Material). The fraction of αS bound to vesicles, FB, is 1 − FF. The structure factor s, the ratio of the radial to axial dimensions of the focal volume, was determined as a free parameter for solutions of Alexa488 hydrazide dye and then fixed to the experimentally determined value of 0.2 for all subsequent fitting. The diffusion times ταS and τvesicle (for αS and vesicles, respectively) were measured independently and fixed for binding measurements. The vesicles were not completely homogeneously sized, but were distributed around the average value measured by FCS and DLS. Simulations by our group and others (50) illustrate that the diffusion time returned is a population-weighted average, and that a single τvesicle is an accurate representation of the average properties of the true distribution (Supporting Material for fit equation and details).
Partition coefficient and ΔG calculations
FB extracted from fitting the autocorrelation curve was plotted as a function of accessible lipid to obtain binding curves. The curves were fit by means of a hyperbolic equation using Origin (OriginLab, Northampton, MA):
| (2) |
where FB is the fraction αS bound from fitting to Eq. 1, x is the concentration of accessible lipids (see Supporting Material for calculation), and Kd,app (the apparent dissociation constant) is the midpoint of the binding curve.
A molar partition coefficient (Kp) for each sample was calculated from Kd,app using Kp = [αSlipid]/[αSfree] (51), where [αSlipid] is the moles of αS bound per volume of lipid, and [αSfree] is the moles of αS free per volume of aqueous solution. [αSlipid] can be calculated from [αSbound], a parameter calculated from the FB extracted from fitting FCS data, as [αSlipid] = [αSbound] × (total volume / lipid volume). Since Kd,app is the concentration of lipid when [αSbound] = [αSfree], Kp is calculated according to Kp = [Kd,app × NA × vlipid]−1, where vlipid is the volume of one lipid molecule and NA is Avogadro's number. Individual Kp values were averaged and the error of the fit to a hyperbolic curve was propagated for uncertainty in all calculations. ΔG of partitioning was calculated as −RTlnKp.
Results
Characterization of vesicles
During extrusion, vesicles can undergo fragmentation, reforming vesicles with a diameter characteristic of the bending modulus of the lipid and limiting the smallest attainable diameter (52). In our experiments, an accurate measurement of the vesicle size is important both for data fitting and for achieving a meaningful understanding of the role of curvature in binding. By DLS, vesicles composed of 1:1 POPS/POPC extruded through membranes with 30 nm, 50 nm, 100 nm, and 200 nm pores were found to have diameters of 77 ± 11 nm, 93 ± 17 nm, 116 ± 22 nm, and 182 ± 11 nm, respectively, and thus can be described as LUVs. SUVs exhibited the higher polydispersity characteristic of sonicated vesicles and had an average diameter of 46 nm, making them a good model for ∼40 nm synaptic vesicles (53). The relationship between vesicle diffusion times measured by FCS and the diameters determined by DLS was linear (Fig. S2). We saw no evidence of protein or vesicle aggregation, even at protein/lipid ratios much higher than those used in our studies, by either FCS or DLS (Supporting Material and Fig. S3).
The vesicles used in this study are simplified membranes that allow us to measure the effects of certain parameters on αS-membrane binding in the absence of other membrane-associated proteins. Although they do not closely resemble physiological membranes, many conditions were controlled to mimic cellular membranes whenever possible. Palmitoyl/oleoyl tails were chosen to mimic the tail asymmetry and monounsaturation of physiological lipids, and the PC headgroup is the most common zwitterionic headgroup found in vivo. Although the anionic content of the vesicles is higher (50%) than that of synaptic vesicles (∼30%), and specific lipids such as PA are present only in very small amounts in cellular membranes, our choice of compositions allowed measurement of complete binding curves and a closer comparison with previous qualitative and quantitative studies that overwhelmingly used vesicles with a high anionic content.
αS binds as a monomer
Binding curves were measured by FCS. As vesicles were added to a constant concentration of αS, the autocorrelation curve shifted to longer decay times reflecting binding of αS to the vesicles (Fig. 2 A). At each lipid concentration, the fraction of αS bound was found with the use of Eq. 1. Binding curves were fit to Eq. 2 to obtain Kd,app, which was then used to calculate thermodynamic parameters Kp and ΔG of partitioning for comparison with other measurements.
Figure 2.
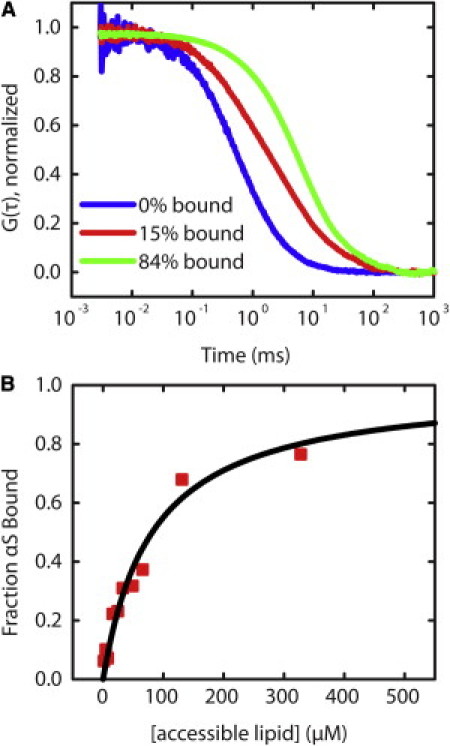
Binding of αS to vesicles. (A) Normalized autocorrelation curves show a shift to longer times as αS binds to vesicles (from left to right with increasing lipid concentration). (B) αS binding curve derived from FCS measurements using Eq. 1 in the text (squares) is fit well by a hyperbolic equation (Eq. 2 in the text; line).
The hyperbolic shape of the binding curve (Fig. 2 B) is consistent with a noncooperative binding model wherein monomeric αS binds to identical binding sites on the lipid bilayer. It also suggests that αS does not oligomerize upon binding, which may manifest as a sigmoidal or otherwise nonhyperbolic binding curve. Although sigmoidal binding curves and clustering of αS bound to a membrane were observed in previous studies (20,31,38,42,43), differences between those studies and our own may be explained by differences in experimental conditions, including the relative amounts of protein/lipid and the observation method used. The low protein concentrations and short measurement times employed for FCS are expected to reduce the propensity of αS to aggregate. Of note, we observed a correlation between low labeling efficiency of αS and the appearance of sigmoidal binding curves (data not shown), which were reported in an earlier FCS study of αS binding (38). The hyperbolic shape of the curve can be recovered by including a reducing agent during measurements (data not shown). This finding suggests that unlabeled αS has an altered interaction with the bilayer when it has a free cysteine, possibly as a result of forming covalent dimers, which have been reported to bind lipid bilayers with higher affinity than monomer αS and increase cellular toxicity (14,33,54).
Anionic headgroups and chain saturation
Using 93 nm vesicles composed of anionic lipids POPS, POPG, or POPA in 1:1 mixtures with zwitterionic POPC, we measured the binding affinity of WT αS. Although there are mixed reports for many individual lipids, the trend suggested by our data, PA>PS≈PG (Fig. 3 A), is in agreement with and unifies many of the qualitative observations of various lipids that have been reported (13,21–23,29,36,38). Whereas αS binds to POPG and POPS with similar affinities, it binds to POPA with >60 times greater affinity (Fig. 3 A and Table 1). In comparison with an earlier FCS study that found a Kd,app of ∼50 μM for WT binding 1:1 POPS/POPC LUVs with ∼120 nm diameter (38), our Kd,app of 169 μM for 116 nm LUVs is somewhat higher, reflecting both the higher salt concentration used in our work and differences in the equations used to fit the FCS binding curves. The equation used in this work takes into account the brightness of vesicles with αS bound, which tends to result in a higher calculated Kd,app (Fig. S1).
Figure 3.
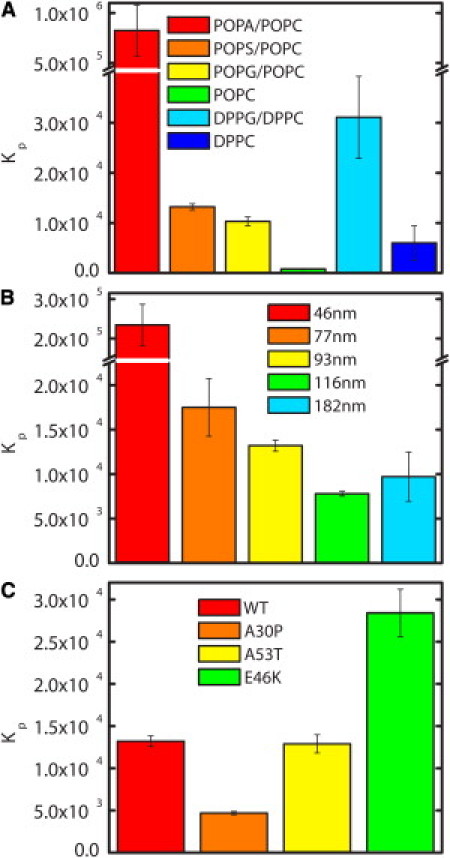
Molar partition coefficients (Kp) of αS. Comparison of Kp values measured for (A) WT αS with 93 nm LUVs of six different lipid compositions, (B) WT with 1:1 POPS/POPC vesicles covering a range of diameters, and (C) WT and disease-associated αS mutants with 93 nm 1:1 POPS/POPC vesicles.
Table 1.
Apparent dissociation constants (Kd,app), molar partition coefficients (Kp), and free energies of partitioning (ΔG) for WT and mutant αS with various LUVs
| Kd,app (μM) | Kp | ΔG (kcal/mol) | |
|---|---|---|---|
| (A) Lipids | |||
| POPA/POPC | 1.60 ± 0.49 | 825,000 ± 254,000 | −7.94 ± 0.18 |
| POPS/POPC | 100 ± 5 | 13,200 ± 630 | −5.53 ± 0.03 |
| POPG/POPC | 128 ± 11 | 10,350 ± 890 | −5.39 ± 0.05 |
| POPC | 1690 ± 210 | 785 ± 99 | −3.89 ± 0.07 |
| DPPG/DPPC | 48 ± 12 | 31,100 ± 8200 | −6.03 ± 0.15 |
| DPPC | 241 ± 137 | 6,000 ± 3400 | −5.07 ± 0.33 |
| (B) Diameter (nm) | |||
| 46 | 5.7 ± 1.3 | 234,000 ± 53,000 | −7.21 ± 0.13 |
| 77 | 75 ± 14 | 17,500 ± 3300 | −5.70 ± 0.11 |
| 93 | 100 ± 5 | 13,200 ± 630 | −5.53 ± 0.03 |
| 116 | 169 ± 6 | 7,800 ± 280 | −5.23 ± 0.02 |
| 162 | 136 ±39 | 9,700 ± 2800 | −5.35 ± 0.17 |
| 46 (DPPC)∗ | 46 ± 10 | 31,000 ± 7000 | −6.04 ± 0.12 |
| (C) Mutants | |||
| 93 nm vesicles | |||
| WT | 100 ± 5 | 13,200 ± 630 | −5.53 ± 0.03 |
| A30P | 282 ± 12 | 4,690 ± 190 | −4.93 ± 0.02 |
| A53T | 102 ± 9 | 12,900 ± 1100 | −5.52 ± 0.05 |
| E46K | 46.6 ± 4.6 | 28,400 ± 2800 | −5.98 ± 0.06 |
| 46 nm vesicles | |||
| WT | 5.7 ± 1.3 | 234,000 ± 53,000 | −7.21 ± 0.13 |
| A30P | 8.9 ± 2.2 | 149,000 ± 36,000 | −6.95 ± 0.14 |
| A53T | 7.0 ± 1.1 | 189,000 ± 31,000 | −7.09 ± 0.10 |
| E46K | 1.6 ± 0.4 | 804,000 ± 207,000 | −7.93 ± 0.15 |
(A) WT αS and 93 nm diameter vesicles with varying anionic headgroups or chain saturation. (B) WT αS and 1:1 POPS/POPC (∗or DPPC) vesicles of different curvatures. (C) αS mutants and 1:1 POPS/POPC vesicles of either 93 nm or 46 nm diameter. Kp and ΔG of partitioning were calculated as described in the text.
Many previous studies of the effects of various anionic lipids on αS binding indicated that the presence of a negatively charged lipid is required for binding of αS (13,21–23,40,41), and thus the very low affinity of αS for POPC was unsurprising (Fig. 3 A and Table 1). However, the free energy of partitioning for POPC is still a significant −3.89 kcal/mol, suggesting that electrostatic interactions are not the only important factor for binding. To further explore the role of other bilayer properties, we made 93 nm vesicles composed of 1:1 DPPG/DPPC. The lipid tails of POPG and POPC are asymmetric in length and contain one unsaturated chain; in contrast, DPPG and DPPC tails are symmetric and saturated. Furthermore, whereas POPG and POPC form disordered fluid phases at room temperature, DPPG and DPPC form gel phases (55). We measured the binding of αS to DPPG/DPPC vesicles and found that Kp was increased threefold as compared to fluid-phase POPG/POPC (Fig. 3 A and Table 1). A larger effect was seen with the zwitterionic lipids, where we observed an almost 10-fold enhancement in Kp of αS for DPPC relative to POPC (Fig. 3 A and Table 1). Our findings are in contrast to previous work that suggested that αS bound zwitterionic gel-phase SUVs but not LUVs or giant unilamellar vesicles (GUVs) (18–20,41). Moreover, the Kp for DPPC vesicles is only approximately a factor of 2 less than that for disordered fluid-phase vesicles containing 50% anionic PG or PS (Table 1), suggesting a specific, strong interaction with these lipids.
Curvature and PD mutants
We studied the effect of vesicle diameter on αS binding using POPS/POPC vesicles ranging from 46 nm to 182 nm in diameter (Fig. 3 B and Table 1). We found that αS binds more strongly to vesicles with a smaller diameter, in agreement with some previous reports (19,23,35,39), but that highly curved SUVs are not required for binding either anionic or zwitterionic membranes. The difference in binding 77–182 nm vesicles is moderate and the Kp values change by just over a factor of 2 for these vesicles, corresponding to a difference in ΔG of <0.5 kcal/mol. However, there is a strong increase in binding 46 nm vesicles: for 1:1 POPS/POPC vesicles, Kp is >10 times greater for 46 nm vesicles compared to 77 nm. A similarly dramatic increase in affinity was observed for SUVs made of 100% DPPC (∼5 times greater; Table 1), as well as 100% POPS and 100% POPC (data not shown).
The effect of αS pathological mutants on interactions between αS and lipid membranes is of interest because it has been suggested that perturbation of normal membrane interactions may be important for the role αS plays in PD (56). Given our observation that WT αS binds with a significantly higher affinity to SUVs, which are similar in size to synaptic vesicles, we measured the affinity of WT αS and the PD mutants for both 93 nm LUVs and 46 nm SUVs composed of 1:1 POPS/POPC. For both sizes of vesicles, we find that the relative binding strengths of these mutants are E46K>WT=A53T>A30P (Fig. 3 C and Table 1), in support of the previously observed trend (13,20,22,41,57,58). The ΔG of partitioning ranges from −4.93 to −5.98 kcal/mol for 93 nm vesicles, and from −6.95 to −7.93 kcal/mol for 46 nm vesicles. However, these ΔG calculations show that there is only an ∼1 kcal/mol difference in free energies between the strongest (E46K) and weakest (A30P) interactions for both LUVs and SUVs.
To determine whether the large difference in binding affinity between LUVs and SUVs could be explained by an increase in the amount of αS bound per vesicle, we performed experiments under saturating conditions. In contrast to our previous measurements, for which we added vesicles to a fixed concentration of protein, we used a fixed concentration of lipid and varied the amount of protein until the amount of bound protein reached saturation. A plot of the molar ratio of lipids to bound αS as a function of total αS concentration plateaus at a value that corresponds to the number of lipids in the binding site (Fig. 4). These experiments were carried out at pH 5.0 where stronger binding allowed for a more accurate calculation of saturation, using vesicles composed of 1:1 POPS/POPC. The plateau value was ∼20 lipids in the outer leaflet per αS for 93 nm and 46 nm vesicles (∼40 in the bilayer), an estimate of the number of lipids in an αS binding site. These experiments were repeated using 93 nm 100% POPS vesicles with the same results (data not shown).
Figure 4.
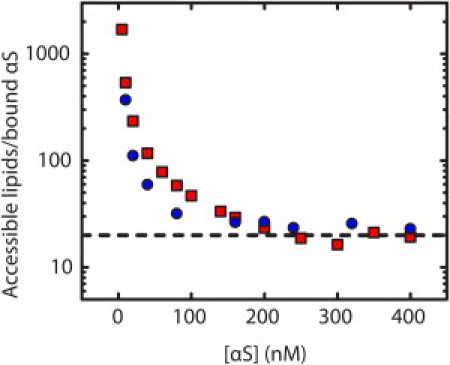
Determination of the number of lipids in a binding site. αS was added to 93 nm (squares) or 46 nm (circles) 1:1 POPS/POPC vesicles at pH 5. The curve saturates at an accessible lipid/bound protein molar ratio of ∼20:1 (dashed line).
Discussion
Strength of binding interaction
A rigorous treatment of membrane partitioning includes corrections for electrostatic effects that depend on the charge of the protein, the charge density of the membrane, and the buffer conditions (59,60). We chose not to correct for these factors because the presence of specific anionic lipids may contribute to in vivo membrane affinities. Therefore, the Kp-values reported here are not due to purely hydrophobic partitioning; rather, they reflect both hydrophobic and electrostatic contributions. Although exact comparisons with other published thermodynamic parameters for αS-membrane binding are not possible due to differences in experimental conditions (e.g., vesicle compositions and diameters, temperatures, and buffers), we will consider our own measurements in the context of these published values as closely as possible. For simplicity, we have calculated the affinity coefficients as defined in each study from our measurements.
Stöckl et al. (41) reported a Ka,app-value of ∼2 μM−1 for αS binding 1:1 DOPG/DOPC GUVs, which is significantly lower than our Ka,app of ∼8 mM−1 for 1:1 POPG/POPC 93 nm LUVs. Although our data suggest that increasing vesicle diameter will have a minimal effect on Ka.app for vesicles larger than ∼77 nm diameter, the higher affinity we measure may be due to our use of LUVs instead of GUVs, or our use of an assay that does not rely on a secondary change in fluorescence to detect binding. Kamp and Beyer (19) and Nuscher et al. (20) used fluorescence anisotropy and isothermal titration calorimetry to measure ΔG values ranging from −10.1 to −10.7 kcal/mol for DPPC and POPG/POPC SUVs < 40 nm in diameter. We calculate ΔG = −7.05 kcal/mol and ΔG = −6.04 kcal/mol for 46 nm 1:1 POPS/POPC and DPPC vesicles, respectively, which should correspond closely to their results. Given the dramatic increase in affinity that we measure between 77 nm and 46 nm vesicles, it is possible that the remaining ∼3–4 kcal/mol discrepancy may be due to the slight difference in SUV diameters used. However, it is also possible that other experimental factors played a role, as the authors did not observe binding to DPPC or POPG/POPC LUVs, in contrast to our observations.
Another fluorescence study found Kd,app-values of 1.556 μM and 1.863 μM for WT and A53T binding POPS LUVs, respectively, and 1.645 μM for WT binding ∼100 nm POPC LUVs (39). These values are lower than the Kd,app-values we measure for ∼93 nm LUVs: 100 μM and 102 μM for WT and A53T binding 1:1 POPS/POPC, respectively, and 1.7 mM for WT binding POPC. The POPS results can be partially explained by the molar fraction of POPS used, since our Kd values would be expected to be ∼10-fold larger than for 100% POPS LUVs. There is qualitative agreement between our results and those of Narayanan and Scarlata (39), both of which reveal similar binding affinities for WT and A53T. However, they found little difference between binding to POPC as compared to POPS vesicles, in conflict with our observations.
Also of interest are other amphipathic membrane-binding proteins, as exemplified by a recent study of a helix from an N-BAR domain, which plays a role in membrane remodeling in endocytosis (61). Kp-values of N-BAR range from 2 × 104 to 5.5 × 104 for POPG and POPS LUVs extruded through 30 nm, 100 nm, and 400 nm pores (61), and these values are in very good agreement with our measured values for αS under similar conditions (Table 1). Both αS and N-BAR show a Kp orders of magnitude weaker for POPC than anionic lipids, suggesting that anionic lipids mediate binding for both proteins. In light of a recent study showing that αS is capable of tubulating and otherwise rearranging lipid bilayers (30) and long-standing observations that αS can disrupt lipid bilayers (21,22,30–37), the comparable affinities of the two proteins suggest that αS may have a similar functional role in rearranging or maintaining bilayer structures.
For the PD mutants, our quantitative results can be used to provide insight into the source of altered binding in the A30P and E46K mutations. The E46K mutation exchanges a residue with −1 charge for one with +1 charge, causing a net charge difference of +2. This is expected to enhance the affinity of αS for negatively charged membranes due to electrostatic attraction. Model polylysine peptides have been used to calculate that partitioning of one lysine residue into a negatively charged membrane under conditions similar to ours should result in ∼1 kcal/mol enhancement in binding (62). Because this lysine replaces a negatively charged glutamic acid, we might expect twice that effect, or ∼2 kcal/mol. For comparison, when ΔGassociation is calculated from the apparent association constant, Ka,app, the difference between WT and E46K is 0.45 kcal/mol for 93 nm vesicles and 0.72 kcal/mol for 46 nm vesicles, much less than expected. To understand this discrepancy, we considered a helical wheel model of the N-terminal α-helix of αS, which suggests that E46 is on the solvent-exposed face of the helix (63). Thus the interaction of E46K with negatively charged lipid headgroups is likely to be limited, and increased binding is not due to lysine partitioning into the membrane. The added positive charge may play a role in attracting αS to the membrane from bulk solution, thus increasing the concentration of αS near the membrane and the measured Kp.
The A30P mutation inserts a proline into the region of the protein that is α-helical when bound to the lipid bilayer. Proline is known to introduce a kink into helices, which has been suggested as the reason for reduced A30P binding. An additional possibility concerns the A30 position as a lipid-exposed residue (27,28,63). Alanine is a more hydrophobic residue than proline, and inserting proline in place of alanine in the lipid interface is likely to reduce beneficial hydrophobic interactions (64,65). The affinity of A30P is ∼30 times greater for the 46 nm SUVs than for 93 nM LUVs, as compared to WT, where the affinity is ∼18 times greater (Table 1). Thus the relative difference in binding affinities is much smaller between A30P and WT for SUVs than LUVs, suggesting that the mechanism of disruption is less important for highly curved bilayers, such as the synaptic vesicles relevant to the native function of αS. The relatively minor differences in the binding affinities of WT, A30P, and A53T suggest that a perturbation of functional binding interactions is not necessarily a mechanistic link to disease.
Effects of composition
One finding of significant interest from our studies is that Kp of αS is >60-fold greater for PA than for PS or PG (Table 1). The higher charge on the PA headgroup at pH 7.4 (−1.2) compared to PS and PG (−1) offers a partial explanation. We expect both hydrophobic and electrostatic effects to contribute to the free energy of binding (as indicated by binding to zwitterionic POPC; Table 1), and if we reasonably assume that the hydrophobic contributions should be similar for all lipid compositions compared, we can expect log(Kp) to scale linearly with the charge density of the membrane (66,67). The Kp derived from our data is much higher than this, suggesting that other properties of PA must play a role in αS binding. It has been suggested that the smaller PA headgroup results in additional space for a protein to bind without rearranging lipids in the membrane, which is consistent with our results (38). Single-molecule fluorescence resonance energy transfer measurements of αS bound to POPA and POPS vesicles show similar structures, indicating that there is no significant structural difference (data not shown) (25). Binding affinity also does not scale with the chain packing density (PA>PS>PG) (68), in agreement with models showing that αS interacts mainly with lipid headgroups (69).
The affinity of αS for specific anionic lipids can be examined with respect to their physiological concentrations. It has been shown in aging mice that amounts of PA in the brain increase while PG, PS, and PC decrease (70). In mouse models transgenic for αS, the amount of PA decreases, PG and PS increase, and PC is unaffected, implying that the presence of αS alters lipid metabolism. Since we have shown that αS can bind PA much more tightly than PS and PG, it is possible that membrane interactions of αS play a role in this process (71–74). Depending on the exact relative amounts of PA, PS, and PG in the relevant membrane and changes in these amounts due to aging, the effect of aging on lipid composition could significantly enhance or decrease αS binding.
Curvature and binding density
The number of lipids in the αS binding site gives insight into the geometry of the interaction, and reports of this number in the literature are varied. Values range from 3 to 377 lipids in the bilayer, although these numbers should be taken as an upper limit because they were not necessarily measured under saturating conditions (18,19,21,30). Measurements taken under saturating conditions have yielded values of at least 36 lipids from electron spin resonance (40), fewer than three lipids by nuclear magnetic resonance (30), and 85 lipids by FCS (38). Our results suggest that 20 surface-accessible lipids make up a binding site, corresponding to ∼40 total lipids, in excellent agreement with ESR measurements (40). The theoretical minimum number of accessible lipids needed to bind αS as an extended helix has been estimated to be 12–30 (30,38,40), and our measured value falls in the middle of this range. Although these calculations vary considerably with the area per lipid headgroup and number of helix-forming residues used, our measurements are likely consistent with αS forming relatively tightly packed, extended helices.
Because we measure the same number of lipids in a binding site for 93 nm vesicles composed of POPS or 1:1 POPS/POPC as well as for 46 nm 1:1 POPS/POPC vesicles (Fig. 4), it is likely that the membrane surface area—not the lipid charge or number of defects—limits the density of αS at saturation. This is in contrast to a recent study that examined binding of the amphipathic helix from endophilin A1 (75) using a novel fluorescence method that allows the size of individual vesicles within a heterogeneously sized population to be characterized. The authors found that the binding affinity changed by a factor of ∼2 for vesicles spanning a range from 50 nm to 200 nm, whereas the density of protein bound was dramatically higher for smaller vesicles. They concluded that the increase in binding was due to an increased density of defects in the more highly curved surfaces rather than an increased affinity for the curved surfaces themselves.
Our own results suggest the opposite conclusion for αS, namely, that it has an increased affinity for highly curved bilayers, with the attraction for binding at sites of defects playing a more minor role. This idea is supported by our observations of a moderate increase in binding affinity for gel-phase vesicles over disordered fluid-phase vesicles as compared to the dramatic increase observed for SUVs over LUVs composed of 1:1 POPS/POPC and DPPC (Table 1), as well as POPS and POPC. Gel-phase membranes contain more defects compared to fluid-phase membranes, and this has specifically been shown for DPPC as compared to POPC (76–78). The smaller difference between DPPG/DPPC and POPG/POPC is likely due to the strength of the electrostatic interaction, which dominates when anionic lipids are present. Nonelectrostatic contributions to binding must dominate the interaction of αS with zwitterionic lipids, and may increase the stability of αS interacting with membranes with a significant proportion of zwitterionic lipids, such as those found in vivo.
The mean curvature of the lipid bilayer, 1/R, changes by a factor of ∼4 between the 182 nm and 46 nm vesicles. In similarity to our observations for αS, a sharp binding affinity transition at these curvatures has been observed for proteins containing amphipathic lipid-packing sensor motifs, which have been suggested to be important for controlling or sensing membrane curvature (79). The native function of αS likely involves interactions with synaptic vesicles, and our findings in a model system suggest that the curvature of the membrane may drive this interaction, perhaps by closely matching the ideal curvature of the N-terminal helix. Twisting of the helix is necessary to attain the observed 11/3 periodicity (80), which may provide the necessary ideal curvature for binding SUVs or synaptic vesicles (27).
Conclusions
Although the physiological relevance of interactions of αS with lipid membranes is widely recognized, many of the factors that affect these interactions are poorly understood. Our results provide quantitative measurements of the contributions from disease-associated protein mutations, as well as the effects of membrane curvature, lipid headgroup, and lipid phase state. They suggest a model in which strong binding is driven by electrostatic attraction between the negatively charged bilayer and positively charged N-terminus of αS, and an intrinsic affinity for highly curved lipid surfaces, which may be modulated by specific lipid components and the presence of bilayer defects. These findings demonstrate that αS may interact with a variety of cellular membranes in vivo and provide insight into the role of αS in disease.
Acknowledgments
We thank Adam Trexler for purifying the αS and, along with Drs. Abhinav Nath and Eva Sevcsik, for insightful discussions and critical reading of and comments on the manuscript. We thank Professor David Eliezer for the gift of the αS plasmid, and Professor Mary Roberts for advice on making SUVs.
This work was supported by grants from the Ellison Medical Foundation (to E.R.), the Raymond and Beverly Sackler Institute for Biological, Physical and Engineering Sciences (to E.R.), and the National Institutes of Health (training grant GM08203 to E.R.M.).
Supporting Material
References
- 1.Krüger R., Kuhn W., Riess O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat. Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 2.Polymeropoulos M.H., Lavedan C., Nussbaum R.L. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 3.Zarranz J.J., Alegre J., de Yebenes J.G. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 4.Bradbury J. α-Synuclein gene triplication discovered in Parkinson's disease. Lancet Neurol. 2003;2:715. doi: 10.1016/s1474-4422(03)00601-x. [DOI] [PubMed] [Google Scholar]
- 5.Singleton A.B., Farrer M., Gwinn-Hardy K. α-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 6.Eliezer D., Kutluay E., Browne G. Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 7.Iwai A., Masliah E., Saitoh T. The precursor protein of non-A β component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- 8.Abeliovich A., Schmitz Y., Rosenthal A. Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 9.Liu S., Ninan I., Arancio O. α-Synuclein produces a long-lasting increase in neurotransmitter release. EMBO J. 2004;23:4506–4516. doi: 10.1038/sj.emboj.7600451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yavich L., Tanila H., Jäkälä P. Role of α-synuclein in presynaptic dopamine recruitment. J. Neurosci. 2004;24:11165–11170. doi: 10.1523/JNEUROSCI.2559-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murphy D.D., Rueter S.M., Lee V.M. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cabin D.E., Shimazu K., Nussbaum R.L. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking α-synuclein. J. Neurosci. 2002;22:8797–8807. doi: 10.1523/JNEUROSCI.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perrin R.J., Woods W.S., George J.M. Interaction of human α-synuclein and Parkinson's disease variants with phospholipids. Structural analysis using site-directed mutagenesis. J. Biol. Chem. 2000;275:34393–34398. doi: 10.1074/jbc.M004851200. [DOI] [PubMed] [Google Scholar]
- 14.Jo E.J., Fuller N., Fraser P.E. Defective membrane interactions of familial Parkinson's disease mutant A30P α-synuclein. J. Mol. Biol. 2002;315:799–807. doi: 10.1006/jmbi.2001.5269. [DOI] [PubMed] [Google Scholar]
- 15.Li J., Uversky V.N., Fink A.L. Effect of familial Parkinson's disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochemistry. 2001;40:11604–11613. doi: 10.1021/bi010616g. [DOI] [PubMed] [Google Scholar]
- 16.Choi W., Zibaee S., Goedert M. Mutation E46K increases phospholipid binding and assembly into filaments of human α-synuclein. FEBS Lett. 2004;576:363–368. doi: 10.1016/j.febslet.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 17.Ding T.T., Lee S.-J., Lansbury P.T., Jr. Annular α-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry. 2002;41:10209–10217. doi: 10.1021/bi020139h. [DOI] [PubMed] [Google Scholar]
- 18.Kjaer L., Giehm L., Otzen D. The influence of vesicle size and composition on α-synuclein structure and stability. Biophys. J. 2009;96:2857–2870. doi: 10.1016/j.bpj.2008.12.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamp F., Beyer K. Binding of α-synuclein affects the lipid packing in bilayers of small vesicles. J. Biol. Chem. 2006;281:9251–9259. doi: 10.1074/jbc.M512292200. [DOI] [PubMed] [Google Scholar]
- 20.Nuscher B., Kamp F., Beyer K. α-Synuclein has a high affinity for packing defects in a bilayer membrane: a thermodynamics study. J. Biol. Chem. 2004;279:21966–21975. doi: 10.1074/jbc.M401076200. [DOI] [PubMed] [Google Scholar]
- 21.Jo E., McLaurin J., Fraser P.E. α-Synuclein membrane interactions and lipid specificity. J. Biol. Chem. 2000;275:34328–34334. doi: 10.1074/jbc.M004345200. [DOI] [PubMed] [Google Scholar]
- 22.Zhu M., Li J., Fink A.L. The association of α-synuclein with membranes affects bilayer structure, stability, and fibril formation. J. Biol. Chem. 2003;278:40186–40197. doi: 10.1074/jbc.M305326200. [DOI] [PubMed] [Google Scholar]
- 23.Davidson W.S., Jonas A., George J.M. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 24.Ulmer T.S., Bax A., Nussbaum R.L. Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem. 2005;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- 25.Trexler A.J., Rhoades E. α-Synuclein binds large unilamellar vesicles as an extended helix. Biochemistry. 2009;48:2304–2306. doi: 10.1021/bi900114z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drescher M., Veldhuis G., Huber M. Antiparallel arrangement of the helices of vesicle-bound α-synuclein. J. Am. Chem. Soc. 2008;130:7796–7797. doi: 10.1021/ja801594s. [DOI] [PubMed] [Google Scholar]
- 27.Jao C.C., Hegde B.G., Langen R. Structure of membrane-bound α-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. USA. 2008;105:19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Georgieva E.R., Ramlall T.F., Eliezer D. Membrane-bound α-synuclein forms an extended helix: long-distance pulsed ESR measurements using vesicles, bicelles, and rodlike micelles. J. Am. Chem. Soc. 2008;130:12856–12857. doi: 10.1021/ja804517m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bussell R., Jr., Eliezer D. Effects of Parkinson's disease-linked mutations on the structure of lipid-associated α-synuclein. Biochemistry. 2004;43:4810–4818. doi: 10.1021/bi036135+. [DOI] [PubMed] [Google Scholar]
- 30.Bodner C.R., Dobson C.M., Bax A. Multiple tight phospholipid-binding modes of α-synuclein revealed by solution NMR spectroscopy. J. Mol. Biol. 2009;390:775–790. doi: 10.1016/j.jmb.2009.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drescher M., van Rooijen B.D., Huber M. A stable lipid-induced aggregate of α-synuclein. J. Am. Chem. Soc. 2010;132:4080–4082. doi: 10.1021/ja909247j. [DOI] [PubMed] [Google Scholar]
- 32.Volles M.J., Lansbury P.T., Jr. Vesicle permeabilization by protofibrillar α-synuclein is sensitive to Parkinson's disease-linked mutations and occurs by a pore-like mechanism. Biochemistry. 2002;41:4595–4602. doi: 10.1021/bi0121353. [DOI] [PubMed] [Google Scholar]
- 33.Giannakis E., Pacífico J., Barnham K.J. Dimeric structures of α-synuclein bind preferentially to lipid membranes. Biochim. Biophys. Acta. 2008;1778:1112–1119. doi: 10.1016/j.bbamem.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 34.Madine J., Hughes E., Middleton D.A. The effects of α-synuclein on phospholipid vesicle integrity: a study using 31P NMR and electron microscopy. Mol. Membr. Biol. 2008;25:518–527. doi: 10.1080/09687680802467977. [DOI] [PubMed] [Google Scholar]
- 35.Zhu M., Fink A.L. Lipid binding inhibits α-synuclein fibril formation. J. Biol. Chem. 2003;278:16873–16877. doi: 10.1074/jbc.M210136200. [DOI] [PubMed] [Google Scholar]
- 36.van Rooijen B.D., Claessens M.M., Subramaniam V. Membrane binding of oligomeric α-synuclein depends on bilayer charge and packing. FEBS Lett. 2008;582:3788–3792. doi: 10.1016/j.febslet.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 37.van Rooijen B.D., Claessens M., Subramaniam V. Lipid bilayer disruption by oligomeric α-synuclein depends on bilayer charge and accessibility of the hydrophobic core. Biochim. Biophys. Acta. 2009;1788:1271–1278. doi: 10.1016/j.bbamem.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 38.Rhoades E., Ramlall T.F., Eliezer D. Quantification of α-synuclein binding to lipid vesicles using fluorescence correlation spectroscopy. Biophys. J. 2006;90:4692–4700. doi: 10.1529/biophysj.105.079251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Narayanan V., Scarlata S. Membrane binding and self-association of α-synucleins. Biochemistry. 2001;40:9927–9934. doi: 10.1021/bi002952n. [DOI] [PubMed] [Google Scholar]
- 40.Ramakrishnan M., Jensen P.H., Marsh D. α-Synuclein association with phosphatidylglycerol probed by lipid spin labels. Biochemistry. 2003;42:12919–12926. doi: 10.1021/bi035048e. [DOI] [PubMed] [Google Scholar]
- 41.Stöckl M., Fischer P., Herrmann A. α-Synuclein selectively binds to anionic phospholipids embedded in liquid-disordered domains. J. Mol. Biol. 2008;375:1394–1404. doi: 10.1016/j.jmb.2007.11.051. [DOI] [PubMed] [Google Scholar]
- 42.Haque F., Pandey A.P., Hovis J.S. Adsorption of α-synuclein on lipid bilayers: modulating the structure and stability of protein assemblies. J. Phys. Chem. B. 2010;114:4070–4081. doi: 10.1021/jp1006704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pandey A.P., Haque F., Hovis J.S. Clustering of α-synuclein on supported lipid bilayers: role of anionic lipid, protein, and divalent ion concentration. Biophys. J. 2009;96:540–551. doi: 10.1016/j.bpj.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen P.S., Toribara T.Y., Warner H. Microdetermination of phosphorus. Anal. Chem. 1956;28:1756–1758. [Google Scholar]
- 45.Fiske C.H., Subbarow Y. The colorimetric determination of phosphorus. J. Biol. Chem. 1925;66:375–400. [Google Scholar]
- 46.Elbaum-Garfinkle S., Ramlall T., Rhoades E. The role of the lipid bilayer in tau aggregation. Biophys. J. 2010;98:2722–2730. doi: 10.1016/j.bpj.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ries J., Schwille P. New concepts for fluorescence correlation spectroscopy on membranes. Phys. Chem. Chem. Phys. 2008;10:3487–3497. doi: 10.1039/b718132a. [DOI] [PubMed] [Google Scholar]
- 48.Thompson N.L. Fluorescence correlation spectroscopy. In: Lakowicz J., editor. Topics in Fluorescence Microscopy. Plenum Press; New York: 1991. pp. 337–378. [Google Scholar]
- 49.Rigler R., Mets U., Kask P. Fluorescence correlation spectroscopy with high count rate and low-background—analysis of translational diffusion. Eur. Biophys. J. 1993;22:169–175. [Google Scholar]
- 50.Pu M.M., Fang X.M., Roberts M.F. Correlation of vesicle binding and phospholipid dynamics with phospholipase C activity: insights into phosphatidylcholine activation and surface dilution inhibition. J. Biol. Chem. 2009;284:16099–16107. doi: 10.1074/jbc.M809600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.White S.H., Wimley W.C. Membrane protein folding and stability: physical principles. Annu. Rev. Biophys. Biomol. Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 52.MacDonald R.C., MacDonald R.I., Hu L.-R. Small-volume extrusion apparatus for preparation of large, unilamellar vesicles. Biochim. Biophys. Acta. 1991;1061:297–303. doi: 10.1016/0005-2736(91)90295-j. [DOI] [PubMed] [Google Scholar]
- 53.Takamori S., Holt M., Jahn R. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 54.Zhou W., Freed C.R. Tyrosine-to-cysteine modification of human α-synuclein enhances protein aggregation and cellular toxicity. J. Biol. Chem. 2004;279:10128–10135. doi: 10.1074/jbc.M307563200. [DOI] [PubMed] [Google Scholar]
- 55.Jacobson K., Papahadjopoulos D. Phase transitions and phase separations in phospholipid membranes induced by changes in temperature, pH, and concentration of bivalent cations. Biochemistry. 1975;14:152–161. doi: 10.1021/bi00672a026. [DOI] [PubMed] [Google Scholar]
- 56.Auluck P.K., Caraveo G., Lindquist S. α-Synuclein: membrane interactions and toxicity in Parkinson's disease. Ann. Rev. Cell Dev. Biol. 2010 doi: 10.1146/annurev.cellbio.042308.113313. May 25 (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 57.Karpinar D.P., Balija M.B.G., Zweckstetter M. Pre-fibrillar α-synuclein variants with impaired β-structure increase neurotoxicity in Parkinson's disease models. EMBO J. 2009;28:3256–3268. doi: 10.1038/emboj.2009.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bodner C.R., Maltsev A.S., Bax A. Differential phospholipid binding of α-synuclein variants implicated in Parkinson's disease revealed by solution NMR spectroscopy. Biochemistry. 2010;49:862–871. doi: 10.1021/bi901723p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seelig J. Titration calorimetry of lipid-peptide interactions. Biochim. Biophys. Acta. 1997;1331:103–116. doi: 10.1016/s0304-4157(97)00002-6. [DOI] [PubMed] [Google Scholar]
- 60.McLaughlin S. The electrostatic properties of membranes. Annu. Rev. Biophys. Biophys. Chem. 1989;18:113–136. doi: 10.1146/annurev.bb.18.060189.000553. [DOI] [PubMed] [Google Scholar]
- 61.Fernandes F., Loura L.M.S., Prieto M. Role of helix 0 of the N-BAR domain in membrane curvature generation. Biophys. J. 2008;94:3065–3073. doi: 10.1529/biophysj.107.113118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ben-Tal N., Honig B., McLaughlin S. Binding of small basic peptides to membranes containing acidic lipids: theoretical models and experimental results. Biophys. J. 1996;71:561–575. doi: 10.1016/S0006-3495(96)79280-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jao C.C., Der-Sarkissian A., Langen R. Structure of membrane-bound α-synuclein studied by site-directed spin labeling. Proc. Natl. Acad. Sci. USA. 2004;101:8331–8336. doi: 10.1073/pnas.0400553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 65.Eisenberg D. Three-dimensional structure of membrane and surface proteins. Annu. Rev. Biochem. 1984;53:595–623. doi: 10.1146/annurev.bi.53.070184.003115. [DOI] [PubMed] [Google Scholar]
- 66.Rusu L., Gambhir A., Rädler J. Fluorescence correlation spectroscopy studies of peptide and protein binding to phospholipid vesicles. Biophys. J. 2004;87:1044–1053. doi: 10.1529/biophysj.104.039958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murray D., Arbuzova A., McLaughlin S. Electrostatic properties of membranes containing acidic lipids and adsorbed basic peptides: theory and experiment. Biophys. J. 1999;77:3176–3188. doi: 10.1016/S0006-3495(99)77148-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marsh D. Structural and thermodynamic determinants of chain-melting transition temperatures for phospholipid and glycolipids membranes. Biochim. Biophys. Acta. 2010;1798:40–51. doi: 10.1016/j.bbamem.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 69.Perlmutter J.D., Braun A.R., Sachs J.N. Curvature dynamics of α-synuclein familial Parkinson disease mutants: molecular simulations of the micelle- and bilayer-bound forms. J. Biol. Chem. 2009;284:7177–7189. doi: 10.1074/jbc.M808895200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rappley I., Myers D.S., Selkoe D.J. Lipidomic profiling in mouse brain reveals differences between ages and genders, with smaller changes associated with α-synuclein genotype. J. Neurochem. 2009;111:15–25. doi: 10.1111/j.1471-4159.2009.06290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sharon R., Bar-Joseph I., Selkoe D.J. The formation of highly soluble oligomers of α-synuclein is regulated by fatty acids and enhanced in Parkinson's disease. Neuron. 2003;37:583–595. doi: 10.1016/s0896-6273(03)00024-2. [DOI] [PubMed] [Google Scholar]
- 72.Golovko M.Y., Faergeman N.J., Murphy E.J. α-Synuclein gene deletion decreases brain palmitate uptake and alters the palmitate metabolism in the absence of α-synuclein palmitate binding. Biochemistry. 2005;44:8251–8259. doi: 10.1021/bi0502137. [DOI] [PubMed] [Google Scholar]
- 73.Assayag K., Yakunin E., Sharon R. Polyunsaturated fatty acids induce α-synuclein-related pathogenic changes in neuronal cells. Am. J. Pathol. 2007;171:2000–2011. doi: 10.2353/ajpath.2007.070373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barceló-Coblijn G., Golovko M.Y., Murphy E.J. Brain neutral lipids mass is increased in α-synuclein gene-ablated mice. J. Neurochem. 2007;101:132–141. doi: 10.1111/j.1471-4159.2006.04348.x. [DOI] [PubMed] [Google Scholar]
- 75.Hatzakis N.S., Bhatia V.K., Stamou D. How curved membranes recruit amphipathic helices and protein anchoring motifs. Nat. Chem. Biol. 2009;5:835–841. doi: 10.1038/nchembio.213. [DOI] [PubMed] [Google Scholar]
- 76.Rinia H.A., de Kruijff B. Imaging domains in model membranes with atomic force microscopy. FEBS Lett. 2001;504:194–199. doi: 10.1016/s0014-5793(01)02704-1. [DOI] [PubMed] [Google Scholar]
- 77.Mou J., Yang J., Shao Z. Atomic force microscopy of cholera toxin B-oligomers bound to bilayers of biologically relevant lipids. J. Mol. Biol. 1995;248:507–512. doi: 10.1006/jmbi.1995.0238. [DOI] [PubMed] [Google Scholar]
- 78.Bailey A.L., Cullis P.R. Membrane fusion with cationic liposomes: effects of target membrane lipid composition. Biochemistry. 1997;36:1628–1634. doi: 10.1021/bi961173x. [DOI] [PubMed] [Google Scholar]
- 79.Drin G., Antonny B. Amphipathic helices and membrane curvature. FEBS Lett. 2010;584:1840–1847. doi: 10.1016/j.febslet.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 80.Bussell R., Jr., Eliezer D. A structural and functional role for 11-mer repeats in α-synuclein and other exchangeable lipid binding proteins. J. Mol. Biol. 2003;329:763–778. doi: 10.1016/s0022-2836(03)00520-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.