

Abstract
Alpha-synuclein (αS) is a 140-amino-acid protein that is involved in a number of neurodegenerative diseases. In Parkinson's disease, the protein is typically encountered in intracellular, high-molecular-weight aggregates. Although αS is abundant in the presynaptic terminals of the central nervous system, its physiological function is still unknown. There is strong evidence for the membrane affinity of the protein. One hypothesis is that lipid-induced binding and helix folding may modulate the fusion of synaptic vesicles with the presynaptic membrane and the ensuing transmitter release. Here we show that membrane recognition of the N-terminus is essential for the cooperative formation of helical domains in the protein. We used circular dichroism spectroscopy and isothermal titration calorimetry to investigate synthetic peptide fragments from different domains of the full-length αS protein. Site-specific truncation and partial cleavage of the full-length protein were employed to further characterize the structural motifs responsible for helix formation and lipid-protein interaction. Unilamellar vesicles of varying net charge and lipid compositions undergoing lateral phase separation or chain melting phase transitions in the vicinity of physiological temperatures served as model membranes. The results suggest that the membrane-induced helical folding of the first 25 residues may be driven simultaneously by electrostatic attraction and by a change in lipid ordering. Our findings highlight the significance of the αS N-terminus for folding nucleation, and provide a framework for elucidating the role of lipid-induced conformational transitions of the protein within its intracellular milieu.
Introduction
The protein alpha-synuclein (αS) is known to be implicated in both idiopathic and inherited forms of Parkinson's disease (PD) as well as in a number of other neurodegenerative disorders (1–6). This protein was first recognized as the major component of disease-associated intracytoplasmic fibrillar deposits in specific types of neurons (Lewy bodies) considered to be the pathological hallmark of sporadic PD (7,8). In dilute solution, the intrinsically disordered state of monomeric αS is characterized by a dynamic ensemble of transiently interconverting conformations involving a broad energy landscape (9). Molecular crowding in the native intracellular environment may shift this dynamic equilibrium in favor of a particular folded state. At higher concentrations, there is a striking aggregation propensity and—upon long-term incubation—fibrillar structures eventually form that bear remarkable resemblance to the intracellular amyloid deposits encountered in PD. Three single-site mutations of the αS protein are known to be associated with inherited early-onset variants of PD (10–12). These mutant proteins undergo accelerated oligomer formation, and two of them rapidly form large fibrillar structures (13). A hydrophobic sequence domain, referred to as non-amyloid-β component (NAC) (residues 61–95), has a critical role for nucleation of the aggregation process (14). In addition, the amino-acid sequence of αS is characterized by seven imperfect repeats containing a KTKEGV consensus motif (Fig. 1). Similar motifs are found in apolipoprotein A-I, which forms amphipathic helices upon lipid-binding (15–17). This is in line with the observation that the intrinsically disordered αS monomer undergoes a coil→helix transition upon binding to small unilamellar vesicles (SUVs) that feature a negatively charged lipid-water interface (18) or interfacial packing defects (19). The amphipathic nature of the αS N-terminus may play a role in membrane interaction similar to rat endophilin N-BAR domain (20,21), which may allow for curvature sensing (22). Notable contributions for our recent understanding of membrane interactions and of the conditions that promote the misfolding and aggregation of αS have been provided by Anthony Fink (23).
Figure 1.

Amino-acid sequence of α-synuclein. The N-terminal region is blue, the aggregation-prone (NAC) domain is in green, and the C-terminal region is in red. The seven imperfect repeats are indicated in bold type. Protein fragments are schematically shown as bars above (synthetic small peptides) or below (chemically truncated protein). For clarity, the N-terminal truncated mutants are omitted.
A variety of spectroscopic techniques have been employed to obtain high-resolution structural information on the interface-associated state of the protein (24–29). Pulsed electron spin resonance spectroscopy has provided evidence for a continuous single helix when the protein is associated with a flat lipid interface (26–28), in conformity with recent computational work (30). Moreover, truncation of the C-terminus results in accelerated aggregation of the protein (31), indicating that the flexibility of the C-terminus may result in stabilization of rapidly interconverting structures in solution, thereby inhibiting aggregation (32). The C-terminus has also been associated with age-related, calcium-induced membrane interaction and subsequent protein aggregation (33). The N-terminus confers toxicity to the protein, while deletion of an N-terminal peptide not only alleviates toxicity but also decreases the helix propensity (34). Yet there are still unresolved questions regarding the lipid selectivity of interfacial αS binding. The affinity of the monomeric protein for packing defects in a phospholipid bilayer (19), and the observation that membrane-assisted binding and folding occurs in the presence of vesicles containing sphingomyelin and cholesterol, with a known tendency to undergo lateral phase separation (35–37), underscores the need for additional research. Sphingomyelin and cholesterol are abundant in synaptic vesicle membranes (38) and have also been found in Lewy bodies (39), suggesting that lipid domains (rafts) could provide a platform for αS lipid binding (40). Here we use a combination of circular dichroism (CD) spectroscopy and isothermal titration calorimetry (ITC) to establish the role of the N-terminus for membrane-assisted binding and folding of the monomeric protein. Membrane-assisted folding an intrinsic property of αS; notably the high folding propensity of the short N-terminal sequence comprising residues 1–25 suggests that nucleation of the cooperative coil→helix transition of the adjacent large protein domains emerges from the amino-terminus of the protein.
Methods
Circular dichroism spectra were recorded with a model No. 810 spectropolarimeter (JASCO, Gross Umstadt, Germany). The experiments were carried out using a quartz cell with 0.2-cm pathlength. Far-ultraviolet spectra for the 25-amino-acid fragments were recorded from 260–200 nm, and the signal/noise ratio was improved by accumulating and averaging four scans. Spectra for the 1–20 peptide were collected on a DSM 20 double-beam spectropolarimeter (Olis, Bogart, GA) with the same pathlength cell from 300 to 190 nm for both low and high salt conditions, at a rate of 0.25 nm s−2 for three scans. The temperature on each spectrometer was controlled within an accuracy of ±0.5°C. For the spectra of the peptides bound to vesicles, the lineshapes of the vesicles alone in buffer were acquired at each temperature and then subtracted. Calculation of the helicity was performed as described previously (19). Briefly, where % helicity = 100 fhelix. The mean-residue ellipticities at 222 nm for the completely unfolded and completely folded peptides were obtained from and where n equals the number of amino acids in the polypeptide (41).
Isothermal titration calorimetry measurements were performed with a VP-ITC instrument (MicroCal, Northampton, MA). Typically, 40 injections of 7 μL each were performed into the calorimeter chamber containing 1.41 mL of the protein or peptide solution. The data were processed using the software ORIGIN (MicroCal), and thermodynamic parameters were determined from the sigmoidal titration curves, assuming independent saturable binding sites in the outer vesicle interface. First, the ligand concentration was equated with the total lipid concentration in the syringe, neglecting the aggregation state of the lipids. Second, N independent lipid binding sites on the protein were assumed (19). The fractional occupancy of lipid-binding sites on the protein can be obtained from the total heat released, which is a function of N, the microscopic binding constant Kb, and the change in enthalpy. The number N can be identified with the total lipids associated with one protein molecule. This yields the enthalpy per mole of protein, and a single macroscopic binding constant Kb. Details of the calculation are described elsewhere (19).
Results
Membrane-assisted folding of N-terminal α-synuclein peptides is revealed by circular dichroism spectroscopy
Small peptides and larger truncated protein variants were selected from the αS primary sequence to investigate the initiation of the structural transition from an unfolded into a membrane-associated helical state (Fig. 1). The fragments were selected so as to cover sequence domains that have been previously shown to be critical for aggregation of the protein (42), or for the coil/helix transition induced upon contact with membranes or with detergent micelles (25,43–46). Fig. 1 summarizes the peptides and the truncated proteins used in this study and their correlation with previously identified sequence domains.
Circular dichroism spectroscopy, using the ellipticity at 222 nm, reliably detects the transition from a random coil to a helical peptide conformation as long as no other conformations are involved. Here we assessed interactions of the αS-derived peptides with SUVs containing lipids with acidic headgroups, lipids with zwitterionic headgroups below the chain melting phase transition temperature (Tm) of the acyl chains, and lipid mixtures—with and without net surface charges—that undergo phase separation. The first peptide to be studied was the N-terminal amino-acid sequence 1–20. This αS fragment has no net charge at neutral pH and comprises one of the seven repeat motifs (KTKEGV; see Fig. 1). The peptide was mixed in the cuvette of the CD spectrometer with SUVs consisting of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) at a phospholipid/peptide molar ratio of RL/P = 167. These vesicles alone undergo a broad thermal transition centered at 39°C due to packing stress in the curved membrane, rather than 41°C as found for multilamellar dispersions. An affinity of full-length αS for packing defects in such vesicles has recently been discovered by combining CD spectroscopy and isothermal titration calorimetry (19,35).
Far-ultraviolet CD spectra (47) were recorded over a temperature range from 45 to 20°C, either with 10 mM phosphate buffer alone (pH 7.0), or with the same buffer in the presence of 140 mM KCl (Fig. 2, A and B, respectively). These temperature downscans reveal a change in ellipticity that is most prominent at 222 nm, indicating that there is, to some extent, a transition from a random-coil to a helical conformation. Notably, the conformational transition occurs over the temperature interval from 32 to 20°C (i.e., it commences below the phase transition of the vesicles). This may be contrasted to the more-abrupt conformational change that occurs close to the lipid phase-transition temperature when monomeric full-length αS interacts with DPPC vesicles (19). Isodichroic points at 202 nm can be recognized, both in the presence of low and high ionic strength, suggesting that there are just two interconverting peptide conformers in the temperature range of the conformational transition of the 1–20 peptide.
Figure 2.
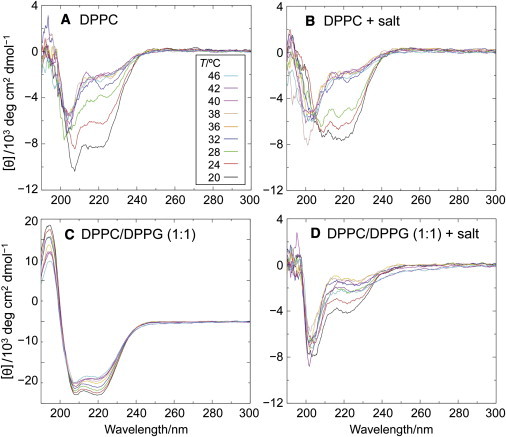
Temperature dependence of circular dichroism spectra reveals membrane binding and coil/helix transition of N-terminal peptide 1–20. Vesicle compositions: (A) DPPC; (B) DPPC, 140 mM NaCl; (C) DPPC/DPPG (1:1); and (D) DPPC/DPPG (1:1), 140 mM NaCl. All samples contained 10 mM Na phosphate, pH 7.0. Total phospholipid concentration was 5 mM and the peptide concentration was 30 μM in a cuvette of 0.2-cm pathlength. CD spectra in terms of decreasing ellipticity at 222 nm were recorded at temperatures of 46, 42, 40, 38, 36, 32, 28, 24, and 20°C. Control spectra acquired without peptide, but with the same buffer and phospholipid concentration, were subtracted for each temperature.
Next, we studied the interaction of the 1–20 peptide with SUVs comprising an equimolar mixture of DPPC and 1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol (DPPG). These lipids mix almost ideally in the liquid-crystalline state of the vesicles, according to the narrow heat capacity peak at 39°C. Hence, there are two contributions to the interfacial lipid-peptide interaction below the phase transition temperature. These are the negative surface charges provided by the phosphoglycerol headgroups, and the interfacial stress due to lipid freezing in the highly curved vesicle membrane (19). The peptide indeed undergoes a coil→helix transition in the presence of these vesicles (Fig. 2, C and D). At low ionic strength the electrostatic interaction seems to prevail, because there is no sudden change of the mean-residue ellipticity [Θ]222 that can be correlated with lipid chain melting (Fig. 2 C). Instead, the spectra change continuously with decreasing temperature, and a well-defined isodichroic point is obtained.
Increasing the ionic strength has a strong effect on the lipid interaction of the peptide (Fig. 2 D); i.e., the CD spectra now reflect the lipid phase transition, similar to the result obtained with DPPC alone, as shown in Fig. 2, A and B. At 20°C, the ellipticity minimum at 222 nm and a prominent minimum at 205 nm indicate that, upon electrostatic shielding, there is even less helical folding than in the presence of DPPC vesicles (Fig. 2 B). Considering that the 1–20 sequence has no net charge at neutral pH, it must be concluded that the positive and negative amino-acid charges (K6, K10, K12, and D2, E13, E20, respectively) are asymmetrically distributed, so as to enable an optimal interaction of the positive lysine side chains with the negative lipid headgroups. A helical-wheel representation indeed shows the uneven distribution of charged and uncharged amino acids (Fig. 3). As a result, we propose that the nonpolar side of the helix immerses into the lipid interface with the lysines being in close contact with the PG headgroups and the aspartate and glutamate side chains facing the aqueous phase. It follows that the axis of this sided-helix can be assumed to run parallel with the membrane surface. This is in full agreement with the structure elucidated in the simulations of Perlmutter et al. (30).
Figure 3.
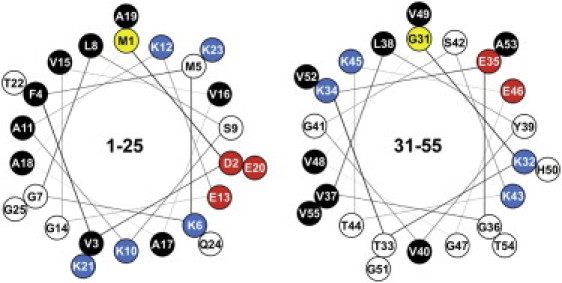
Helical-wheel representation of 1–25 and 31–55 peptides. View is from the N-terminus down the helix axis. Adjacent amino acids are offset by an angle of 100 deg. Residue designations are (yellow) first amino acid in the sequence; (red) negative amino acid charges; (blue) positive amino acid charges; (white) slightly polar, neutral amino acids; and (black) nonpolar, hydrophobic amino acids. Note the opposing lysine clusters, separated by 200 deg (K10, K21, and K12, K23 for the 1–25 peptide, and K32, K43 and K34, K45 for the 31–55 peptide, respectively).
The mean-residue ellipticity obtained for the 1–20 peptide in the presence of DPPC/DPPG vesicles at 20°C and low ionic strength (see Fig. 2 C) yields a helicity of 69% (see Methods). This may be due either to an imperfect folding in the bound state, assuming that all of the peptide is bound, or to the presence of unbound peptide in equilibrium with a bound portion. To address this problem, isothermal titration calorimetry was used (Fig. 4 A) which yielded N = 78 lipid molecules associated with one peptide molecule. A single macroscopic binding constant Kb = 1.02 × 106 M–1 is obtained, which characterizes the association of the N lipids with the peptide molecule, provided that the peptide-lipid binding equilibrium can be described by P + Lc = PLc, where P is the peptide and Lc the virtual cluster of N lipids associated with the protein (see Methods for details) (19). The percentage of vesicle-associated peptide, PLc, can be calculated from this binding equilibrium (48). For the experiment shown in Fig. 2 C (total protein and lipid concentrations, 30 μM and 5 mM, respectively), it turns out that the bound portion of the peptide amounts to 97.3%. Considering the helicity of 69% this indicates that a considerable portion of the 1–20 peptide remains disordered, even when almost all of the peptide is lipid-associated. Similarly, ITC yielded 99.9% binding for the full-length protein at the same temperature and total lipid/protein ratio (Fig. 4 C), although the helicity was only 48% (see Fig. 5) (19).
Figure 4.
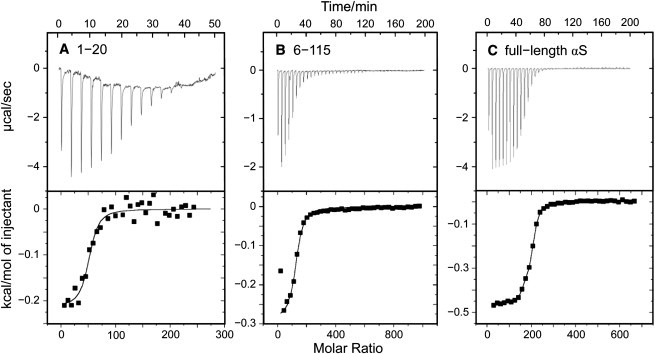
Isothermal titration calorimetry yields percentage of vesicle-bound peptide or protein. SUV suspensions were titrated at 20°C into a 1.4-mL calorimeter cell containing the peptide or protein solutions. Single 7-μL injection volumes were typically employed. (A) Here, 30 μM of peptide 1–20 and (B) 8.5 μM peptide 6–115 in phosphate buffer, pH 7.0, titrated with DPPC/DPPG vesicles; molar ratio of the lipids 1:1, total lipid concentration in the syringe, 40 mM. (C) Here, 8.7 mM αS titrated with 45 mM DPPC/DPPG vesicles; other conditions as in panels A and B. The number of associated lipids and the binding constants Kb were obtained using the binding model for N independent sites provided with the software ORIGIN (MicroCal). (For details, see (19).)
Figure 5.

Circular dichroism spectroscopy manifests helical folding due to local sequence properties and lipid composition of vesicle membranes. The helicity was obtained from mean-residue ellipticity values at 222 nm. Small unilamellar vesicles (phospholipid concentration 5 mM) and αS or αS-derived peptides in 100 mM KCl, 20 mM Na phosphate, pH 7.4, were mixed in a 0.2-cm pathlength CD cuvette, and incubated for 5 min to allow for temperature equilibration. The molar ratio of phospholipids in the binary mixtures was 1:1 and the lipid/protein (peptide) molar ratio was 167:1. Temperature, 20°C; typical errors ±6%. (Methods).
Peptide fragments reveal the modular organization of full-length α-synuclein
Overlapping αS fragments, ranging from 25 to 28 residues, were studied next, allowing us to scan the αS sequence for membrane-mediated helix folding. Peptides were selected so as to cover the αS sequence up to residue 85. Experimental conditions (100 mM KCl, pH 7.4) were chosen to be close to those of the intracellular milieu. Here we show the helicity (Fig. 5) as obtained from the mean-residue ellipticity values at 222 nm at 20°C (see Methods). This simplifying approach seems justified, as deconvolution of the spectra (49) typically yielded <15% of contributions from structures other than helix or coil. We cannot rule out that a larger fraction of extended β-structure contributes to the CD spectra in cases where deconvolution is not feasible due to poor signal/noise. However, we believe that the temperature dependence of the spectra reflects an increasing contribution of helical folding with decreasing temperature, particularly if the temperature scans involve a lipid chain melting phase transition. There is a distinct propensity for helix formation of the full-length protein and the 1–25 fragment, triggered by contact with any of the vesicular membranes shown in Fig. 5. In contrast, the 1–20 peptide does not fold in the presence of the liquid-crystalline 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC)/1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG) vesicles, but does so, although to a lesser extent, in the presence of DPPC vesicles below the chain melting phase transition temperature. The 31–55 peptide has even less helix propensity in the presence of vesicles comprising POPC/POPG or DPPC alone, though it exhibits a surprising affinity for the DPPC/DPPG interface. This peptide incorporates three positive net charges (including H50) and two KTKEGV motifs. The particular arrangement of these residues along the helical circumference allows for strong simultaneous charge and hydrophobic interactions in the disordered defect zones of the DPPC/DPPG vesicle interface below the transition temperature.
These results clearly show that the N-terminal peptides not only respond to electrostatic forces, but also to packing defects in the vesicular membrane. Separation of these contributions can be achieved by monitoring the temperature dependence of the coil→helix transition in the presence of the different vesicles (Fig. 6). For comparison, the temperature dependence of the mean-residue ellipticity of the full-length αS protein is included (Fig. 6 A). In the presence of vesicles consisting of DPPC alone, the full-length protein undergoes helix folding in the temperature range of the chain melting transition of the vesicles, in accord with previous findings (19). By contrast, the negative surface charge of POPC/POPG vesicles results in a partially folded protein over the entire temperature range (18). In DPPC/DPPG vesicles, the negative headgroup charges give rise to mean-residue ellipticities similar to those obtained in the presence of the POPC/POPG mixture. A further decrease to more negative ellipticity values occurs at the transition temperature, which is in line with a cumulative effect of electrostatic interactions and interfacial packing defects.
Figure 6.
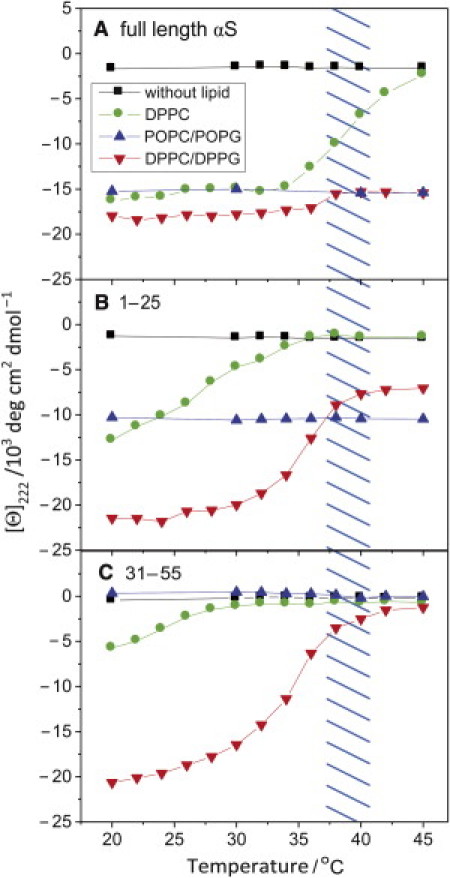
Temperature dependence of mean-residue ellipticity at 222 nm reveals varying affinities of N-terminal sequence domains to surface charges and membrane defects. (A) Full-length α-synuclein, (B) 1-25 peptide, and (C) 31-35 peptide. Small unilamellar vesicles (phospholipid concentration 5 mM) were mixed with 6 μM αS or 30 μM of the peptides in 100 mM KCl, 20 mM Na phosphate, pH 7.4, corresponding to phospholipid/protein molar ratios of 833:1 and 167:1. The molar ratio of phospholipids in the binary mixtures was 1:1. The temperature range of the phase transition for DPPC and DPPC/DPPG vesicles is indicated by the hatched area. Note the additive effect of electrostatic interaction and chain melting phase transition on the mean-residue ellipticity of the full-length protein and the 1–25 peptide.
The temperature dependence of the membrane-induced coil→helix transition of the 1–25 peptide is similar to the thermal behavior of the full-length protein, but surprisingly different from the 31–55 peptide (Fig. 6, B and C). Peptide 1–25, similar to αS itself, assumes a helix (∼30% helicity) when mixed with POPC/POPG vesicles in a temperature-independent manner, while the strong temperature dependence in the presence of DPPC/DPPG vesicles is clearly related to the chain melting phase transition. The phase transition in the presence of DPPC vesicles alone induces a similar conformational change, but over a much broader temperature range than for the full-length protein. In contrast, the 31–55 fragment does not undergo helix folding in the presence of the liquid-crystalline POPC/POPG vesicles, and DPPC vesicles are only able to elicit a small effect at temperatures far from the lipid phase-transition temperature (Fig. 6 C). The interaction with DPPC/DPPG vesicles, however, is surprisingly strong for this peptide, and the coil/helix transition commences in the temperature range where lipid chain melting occurs. The mean-residue ellipticity of –21,000 deg cm2 dmol–1 obtained at 20°C during a temperature downscan (which translates into 61% helicity) clearly exceeds the values reached by the other peptides, except for the 1–25 sequence. Evidently, there is a nearly perfect match when the 31–55 peptide helix dips into the molecularly rough interface of the curved gel-state DPPC/DPPG vesicles, which arises from the dislocation stress field. However, there may be no such opportunity for the POPC/POPG vesicles with a liquid-crystalline interface relatively free of defects. Specifically, the lysine residues within the KTK motifs are offset by ∼200 deg, which puts them on opposite sides of the α-helix as depicted by the helical wheel in Fig. 3. This spatial arrangement may allow for proper positioning of the helix due to interaction of the positive amino-acid charges with the negative surface potential of the membrane, thereby dragging the nonpolar residues of the sided helix deeper into the membrane interface.
The N-terminus triggers interfacial binding and helix folding of full-length α-synuclein
Next, we constructed a number of truncated αS variants by stepwise removal of residues from the N-terminus, as well as two fragments that lacked the acidic C-terminal tail of the protein (Fig. 7). Various lipid compositions were studied, including a mixture of neutral lipids that undergoes lateral phase separation (36). There was always a stoichiometric excess of the vesicles, i.e., the total lipid/protein molar ratios were RL/P = 588 (for the αS deletion mutants) or RL/P = 833 (for the chemically truncated or for the full-length protein), which is more than twice the saturation ratio as determined by CD or ITC titration (19). The largest absolute mean-residue ellipticities were obtained with the 1–115 protein variant for all lipid mixtures shown in Fig. 7. Perhaps this is not surprising if one considers that the C-terminal sequence domain does not take part in the helix folding of the full-length protein (50). Assuming that the truncated sequence reaches the same extent of helix folding as the full-length protein, one may expect that the mean-residue ellipticity increases by ∼18%. However, the ellipticity was only ∼5% larger than the value obtained for full-length αS, probably due to some disorder in the new C-terminus after removal of 25 residues. In addition to these end effects, there may be a loss of stabilization of the protein structure, owing to an interaction between the C-terminus of αS and the central hydrophobic domain of the protein (51) (see Discussion).
Figure 7.
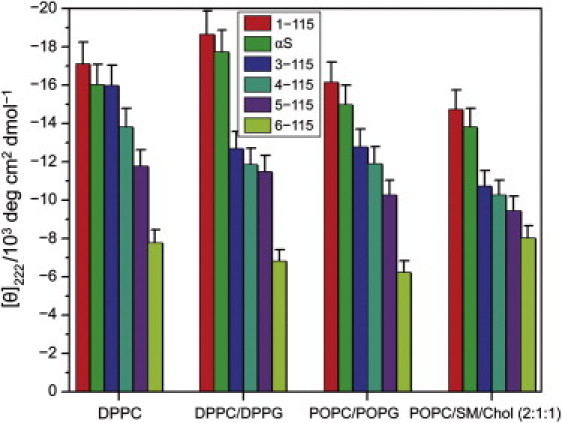
Stepwise removal of N-terminal amino acids from α-synuclein yields a progressive decrease of membrane-induced helical folding. Mean-residue ellipticities of SUVs were determined in 20 mM sodium phosphate, pH 7.4, and 100 mM KCl. Total SUV lipid concentration was 5 mM; protein concentrations were 8.5 μM and 6 μM for the deletion and truncation mutants, corresponding to lipid/protein molar ratios of 588 and 833, respectively. The molar ratio of phospholipids in the binary mixtures was 1:1. Temperature, 20°C.
The salient feature of Fig. 7 is the systematic reduction of the membrane-induced mean-residue ellipticity upon stepwise truncation of the N-terminus. This suggests that the N-terminal amino acids are essential for membrane recognition and for the initiation of the folding of the entire protein. Even removal of the first two N-terminal amino acids (methionine and aspartate) has a remarkably strong effect in membranes with a mixed lipid composition. These results indicate that the initiation of the αS coil→helix transition is an intrinsic feature of the N-terminal protein sequence, independent of the particular properties of the membrane interface. The low mean-residue ellipticity of the 6–115 fragment obtained with any of the vesicle membranes is also in excellent agreement with the notion that the N-terminus determines the folding of the full-length protein. Isothermal titration calorimetry at 20°C yielded N = 123 for the lipids associated with one protein molecule and a binding constant Kb = 2.2 × 106 M–1 (see Fig. 4 B), which gives 99% binding when allowance is made for the protein and lipid concentrations pertinent to the CD experiment. The helicity of 19% obtained under the same conditions (see Fig. 7) suggests that there is even more disorder in the membrane-bound state of this truncated version of αS than in the bound full-length protein. Comparing this with the result obtained for the 1–115 fragment (helicity 51%), it is obvious that the five N-terminal amino acids are critical for the folding of the protein, regardless of the high folding propensity of the downstream peptide 31–55 (see Figs. 6 and 7).
Discussion
Membrane affinity and helix formation is intrinsic to the N-terminus of α-synuclein
The results presented here afford new insights into the structural properties of the various αS sequence motifs. The conformational transition of αS is triggered by the N-terminus, as shown by the membrane affinity and helix propensity of the 1–25 peptide fragment, rather than by being a function of cooperative all-or-none folding of the entire protein. Moreover, our data suggest that the 1–25 peptide features a similar selectivity for different lipid compositions as the full-length protein. Notably, the adjacent 9–36 sequence that is part of the NMR-derived N-terminal helix formed in the presence of SDS micelles (25) is much less prone to undergo a spontaneous conformational transition at a membrane interface. Membrane-induced helix folding is negligible in the presence of POPC/POPG vesicles for all peptides shown in Fig. 5, except for the 1–25 fragment. Only the 1–25 and 1–20 peptides respond to the electroneutral defect interface of gel-state DPPC vesicles. Hence, the N-terminus triggers the coil/helix transition of the protein in different membranes.
The low affinity of the 1–20 peptide for the POPC/POPG interface is not surprising, because this peptide has no net charge. The 1–20 peptide was chosen to demonstrate that even weak interaction with a defective membrane interface, in the absence of strong electrostatic forces, leads to partial helix folding. There are, however, positive side-chain charges in excess over negative ones for the 9–36, 31–55, and 41–65 peptides (two, two, and one, respectively). Hence, the mere charge excess is not sufficient for membrane recognition and helix folding of these fragments. Rather, it must be assumed that there is a slight preponderance of partially folded structures in the ensemble of rapidly interconverting conformations, which makes the 1–25 peptide more adapted to recognize the lipid interface than the C-terminal segments. This peptide selectivity is in qualitative agreement with a recent NMR study, which demonstrated preferential lipid binding and folding for the N-terminal sequence of the full-length protein (52).
Packing defects and membrane surface potential are determinants of α-synuclein binding
Notably, there is a high proclivity of the peptides for the folded state in the presence of the DPPC/DPPG vesicles. This lipid mixture provides two favorable properties simultaneously, i.e., packing defects and negative surface charges. The accessibility of the hydrophobic membrane core is evidently crucial for both hydrophobic and electrostatic contact of the extended full-length protein helix (27). The detrimental effect of αS oligomers on bilayer integrity indeed conforms with this paradigm (53). In contrast to αS oligomers, however, binding of the monomeric protein results in stabilization of a defective interface (19,35). Further support comes from the observation that the full-length protein and peptide 1–25 are able to reduce the calcium or detergent-induced fusion of small unilamellar vesicles (see Fig. S1 in the Supporting Material). A recent study, demonstrating that overexpression of αS in a primary hippocampal cell culture results in considerable inhibition of neurotransmitter release (54), is also in line with a retarding effect of αS upon vesicle fusion. The reduced clustering of synaptic vesicles near the active zone observed by these authors may indeed be related to the observed inhibition of in vitro vesicle fusion as αS stabilizes the synaptic vesicle reserve pool (1,19).
The strong interaction of the 31–55 peptide with gel-state DPPC/DPPG vesicles seems surprising, considering the broken-helix model that has emerged from previous high-resolution NMR studies. In the presence of SDS micelles, the protein forms two antiparallel helices connected by a hinge region that comprises most of the residues of this peptide (24,25,46,55). However, a contiguous helix—comprising at least residues 3–90—exists in a vesicular membrane interface with a much larger radius of curvature than a micelle (27). Evidently, the membrane-stabilizing effect of αS requires that almost two-thirds of the primary sequence of the protein be in a helical state to warrant the optimal interfacial contact, as well as to avoid aggregation of the most unfolded and hydrophobic central part of the αS sequence (52). Thus, the hinge may be required for the protein to adapt to membrane curvature after attachment and folding of the N-terminal 25 amino acid residues. The strong tendency for helix formation of the hinge region—and likewise of peptides 9–36 and 41–65—may come into play only after nucleation by the N-terminal sequence domain.
The gradual decrease of the mean-residue ellipticities upon removal of amino acids from the N-terminus reflects an intrinsic protein property, because it is alike for different lipid compositions. This holds true also for vesicles composed of POPC, sphingomyelin, and cholesterol (molar ratio 2:2:1), strongly suggesting that the αS binding mode is the same for this electroneutral interface as for membranes with net interfacial charges or for vesicles in the gel state. This lipid mixture is close to the classic (i.e., equimolar) raft mixture of the three lipids (36), which suggests that the favorable interaction between cholesterol and sphingomyelin results in partial phase separation and microdomain formation.
Mechanistic aspects and biomedical implications
Finally, a tentative model of αS binding and membrane interaction can be derived from our results. We hypothesize that the N-terminus of αS serves as a nucleation center for membrane binding of the full-length protein and helix folding up to approximately residue 100. A two-stage model considers binding of residues 1–25 as the initial step, and subsequent cooperative binding and folding of residues 26–100 as the second step (52,56). The first state leaves the protein prone to aggregation due to solvent exposure and disordering of the most hydrophobic central part of the protein (52). In the second state, the long helix fold protects from aggregation, and allows for the full membrane-stabilizing capacity of the protein (35). Early aggregates, usually referred to as oligomers, are believed to represent the real toxic species of the protein. Toxic oligomer formation in a membrane interface may be particularly deleterious for cell viability when it affects intracellular storage vesicles (34). This attains even greater importance when one considers that the overwhelming number of synuclein-related disease cases is idiopathic. The culprit of the early aggregation then may be an alteration of membrane properties, e.g., the lipid composition of synaptic vesicles, rather than a defective protein structure. A subtle change in the interfacial properties may result in an extension of the time interval between the hypothetical binding modes of the protein, which may enhance the chance for hydrophobic protein-protein contact. Our approach, using peptide fragments of an intrinsically unstructured protein as employed here for αS, not only simplifies the elucidation of the initial events that lead to protein aggregation or folding in a membrane interface, but also may be useful for rational drug design and for the design of rapid drug screening methodology.
Acknowledgments
We thank Ulf Dettmer and Lucas Martin for technical advice in preparing the deletion mutants and John Fitch and Chad Park for helpful discussions regarding CD spectroscopy and ITC.
This work was supported by the Arizona Biomedical Research Foundation, the Deutsche Forschungsgemeinschaft (under grant No. SFB 596, Teilprojekt No. B10), and the National Institutes of Health, Bethesda, MD.
Footnotes
Tim Bartels' present address is Center for Neurologic Diseases, Brigham & Women's Hospital, Harvard Medical School, Boston, MA 02115.
Supporting Material
References
- 1.Beyer K. Mechanistic aspects of Parkinson's disease: α-synuclein and the biomembrane. Cell Biochem. Biophys. 2007;47:285–299. doi: 10.1007/s12013-007-0014-9. [DOI] [PubMed] [Google Scholar]
- 2.Haass C., Selkoe D.J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 3.Dev K.K., Hofele K., van der Putten H. Part II: α-synuclein and its molecular pathophysiological role in neurodegenerative disease. Neuropharmacology. 2003;45:14–44. doi: 10.1016/s0028-3908(03)00140-0. [DOI] [PubMed] [Google Scholar]
- 4.Moore D.J., West A.B., Dawson T.M. Molecular pathophysiology of Parkinson's disease. Annu. Rev. Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- 5.Wakabayashi K., Yoshimoto M., Takahashi H. α-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci. Lett. 1998;249:180–182. doi: 10.1016/s0304-3940(98)00407-8. [DOI] [PubMed] [Google Scholar]
- 6.Gai W.P., Power J.H.T., Blessing W.W. Multiple-system atrophy: a new α-synuclein disease? Lancet. 1998;352:547–548. doi: 10.1016/s0140-6736(05)79256-4. [DOI] [PubMed] [Google Scholar]
- 7.Spillantini M.G., Schmidt M.L., Goedert M. α-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 8.Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- 9.Dedmon M.M., Lindorff-Larsen K., Dobson C.M. Mapping long-range interactions in α-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc. 2005;127:476–477. doi: 10.1021/ja044834j. [DOI] [PubMed] [Google Scholar]
- 10.Polymeropoulos M.H., Lavedan C., Nussbaum R.L. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 11.Krüger R., Kuhn W., Riess O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat. Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 12.Zarranz J.J., Alegre J., de Yebenes J.G. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 13.Conway K.A., Lee S.-J., Lansbury P.T., Jr. Acceleration of oligomerization, not fibrilization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han H., Weinreb P.H., Lansbury P.T., Jr. The core Alzheimer's peptide NAC forms amyloid fibrils which seed and are seeded by β-amyloid: is NAC a common trigger or target in neurodegenerative disease? Chem. Biol. 1995;2:163–169. doi: 10.1016/1074-5521(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 15.Surewicz W.K., Epand R.M., Hui S.W. Human apolipoprotein A-I forms thermally stable complexes with anionic but not with zwitterionic phospholipids. J. Biol. Chem. 1986;261:16191–16197. [PubMed] [Google Scholar]
- 16.Segrest J.P., Jones M.K., Anantharamaiah G.M. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J. Lipid Res. 1992;33:141–166. [PubMed] [Google Scholar]
- 17.Ajees A.A., Anantharamaiah G.M., Murthy H.M. Crystal structure of human apolipoprotein A-I: insights into its protective effect against cardiovascular diseases. Proc. Natl. Acad. Sci. USA. 2006;103:2126–2131. doi: 10.1073/pnas.0506877103. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Davidson W.S., Jonas A., George J.M. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 19.Nuscher B., Kamp F., Beyer K. α-synuclein has a high affinity for packing defects in a bilayer membrane: a thermodynamics study. J. Biol. Chem. 2004;279:21966–21975. doi: 10.1074/jbc.M401076200. [DOI] [PubMed] [Google Scholar]
- 20.Gallop J.L., Jao C.C., McMahon H.T. Mechanism of endophilin N-BAR domain-mediated membrane curvature. EMBO J. 2006;25:2898–2910. doi: 10.1038/sj.emboj.7601174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Itoh T., Takenawa T. Mechanisms of membrane deformation by lipid-binding domains. Prog. Lipid Res. 2009;48:298–305. doi: 10.1016/j.plipres.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 22.Botelho A.V., Huber T., Brown M.F. Curvature and hydrophobic forces drive oligomerization and modulate activity of rhodopsin in membranes. Biophys. J. 2006;91:4464–4477. doi: 10.1529/biophysj.106.082776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fink A.L. The aggregation and fibrillation of α-synuclein. Acc. Chem. Res. 2006;39:628–634. doi: 10.1021/ar050073t. [DOI] [PubMed] [Google Scholar]
- 24.Chandra S., Chen X., Südhof T.C. A broken α-helix in folded α-synuclein. J. Biol. Chem. 2003;278:15313–15318. doi: 10.1074/jbc.M213128200. [DOI] [PubMed] [Google Scholar]
- 25.Ulmer T.S., Bax A., Nussbaum R.L. Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem. 2005;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- 26.Georgieva E.R., Ramlall T.F., Eliezer D. Membrane-bound α-synuclein forms an extended helix: long-distance pulsed ESR measurements using vesicles, bicelles, and rodlike micelles. J. Am. Chem. Soc. 2008;130:12856–12857. doi: 10.1021/ja804517m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jao C.C., Hegde B.G., Langen R. Structure of membrane-bound α-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. USA. 2008;105:19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drescher M., Veldhuis G., Huber M. Antiparallel arrangement of the helices of vesicle-bound α-synuclein. J. Am. Chem. Soc. 2008;130:7796–7797. doi: 10.1021/ja801594s. [DOI] [PubMed] [Google Scholar]
- 29.Bodner C.R., Dobson C.M., Bax A. Multiple tight phospholipid-binding modes of α-synuclein revealed by solution NMR spectroscopy. J. Mol. Biol. 2009;390:775–790. doi: 10.1016/j.jmb.2009.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perlmutter J.D., Braun A.R., Sachs J.N. Curvature dynamics of α-synuclein familial Parkinson disease mutants: molecular simulations of the micelle- and bilayer-bound forms. J. Biol. Chem. 2009;284:7177–7189. doi: 10.1074/jbc.M808895200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu C.W., Giasson B.I., Thomas P.J. A precipitating role for truncated α-synuclein and the proteasome in α-synuclein aggregation: implications for pathogenesis of Parkinson disease. J. Biol. Chem. 2005;280:22670–22678. doi: 10.1074/jbc.M501508200. [DOI] [PubMed] [Google Scholar]
- 32.Bertoncini C.W., Jung Y.S., Zweckstetter M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured α-synuclein. Proc. Natl. Acad. Sci. USA. 2005;102:1430–1435. doi: 10.1073/pnas.0407146102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamamizu-Kato S., Kosaraju M.G., Narayanaswami V. Calcium-triggered membrane interaction of the α-synuclein acidic tail. Biochemistry. 2006;45:10947–10956. doi: 10.1021/bi060939i. [DOI] [PubMed] [Google Scholar]
- 34.Vamvaca K., Volles M.J., Lansbury P.T., Jr. The first N-terminal amino acids of α-synuclein are essential for α-helical structure formation in vitro and membrane binding in yeast. J. Mol. Biol. 2009;389:413–424. doi: 10.1016/j.jmb.2009.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kamp F., Beyer K. Binding of α-synuclein affects the lipid packing in bilayers of small vesicles. J. Biol. Chem. 2006;281:9251–9259. doi: 10.1074/jbc.M512292200. [DOI] [PubMed] [Google Scholar]
- 36.Bartels T., Lankalapalli R.S., Brown M.F. Raftlike mixtures of sphingomyelin and cholesterol investigated by solid-state 2H NMR spectroscopy. J. Am. Chem. Soc. 2008;130:14521–14532. doi: 10.1021/ja801789t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bunge A., Müller P., Huster D. Characterization of the ternary mixture of sphingomyelin, POPC, and cholesterol: support for an inhomogeneous lipid distribution at high temperatures. Biophys. J. 2008;94:2680–2690. doi: 10.1529/biophysj.107.112904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takamori S., Holt M., Jahn R. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 39.Gai W.P., Yuan H.X., Jensen P.H. In situ and in vitro study of colocalization and segregation of α-synuclein, ubiquitin, and lipids in Lewy bodies. Exp. Neurol. 2000;166:324–333. doi: 10.1006/exnr.2000.7527. [DOI] [PubMed] [Google Scholar]
- 40.Cooper A.A., Gitler A.D., Lindquist S. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scholtz J.M., Qian H., Baldwin R.L. Parameters of helix-coil transition theory for alanine-based peptides of varying chain lengths in water. Biopolymers. 1991;31:1463–1470. doi: 10.1002/bip.360311304. [DOI] [PubMed] [Google Scholar]
- 42.Uversky V.N., Fink A.L. Amino acid determinants of α-synuclein aggregation: putting together pieces of the puzzle. FEBS Lett. 2002;522:9–13. doi: 10.1016/s0014-5793(02)02883-1. [DOI] [PubMed] [Google Scholar]
- 43.Eliezer D., Kutluay E., Browne G. Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 44.Bussell R., Jr., Eliezer D. A structural and functional role for 11-mer repeats in α-synuclein and other exchangeable lipid binding proteins. J. Mol. Biol. 2003;329:763–778. doi: 10.1016/s0022-2836(03)00520-5. [DOI] [PubMed] [Google Scholar]
- 45.Bisaglia M., Tessari I., Mammi S. A topological model of the interaction between α-synuclein and sodium dodecyl sulfate micelles. Biochemistry. 2005;44:329–339. doi: 10.1021/bi048448q. [DOI] [PubMed] [Google Scholar]
- 46.Ulmer T.S., Bax A. Comparison of structure and dynamics of micelle-bound human α-synuclein and Parkinson disease variants. J. Biol. Chem. 2005;280:43179–43187. doi: 10.1074/jbc.M507624200. [DOI] [PubMed] [Google Scholar]
- 47.Brown M.F., Schleich T. Circular dichroism and gel filtration behavior of subtilisin enzymes in concentrated solutions of guanidine hydrochloride. Biochemistry. 1975;14:3069–3074. doi: 10.1021/bi00685a005. [DOI] [PubMed] [Google Scholar]
- 48.Jia Z., Ramstad T., Zhong M. Determination of protein-drug binding constants by pressure-assisted capillary electrophoresis (PACE)/frontal analysis (FA) J. Pharm. Biomed. Anal. 2002;30:405–413. doi: 10.1016/s0731-7085(02)00223-6. [DOI] [PubMed] [Google Scholar]
- 49.Provencher S.W., Glöckner J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry. 1981;20:33–37. doi: 10.1021/bi00504a006. [DOI] [PubMed] [Google Scholar]
- 50.Der-Sarkissian A., Jao C.C., Langer R. Structural organization of α-synculein fibrils by site-directed spin labeling. J. Biol. Chem. 2003;278:37530–37535. doi: 10.1074/jbc.M305266200. [DOI] [PubMed] [Google Scholar]
- 51.McClendon S., Rospigliosi C.C., Eliezer D. Charge neutralization and collapse of the C-terminal tail of α-synuclein at low pH. Protein Sci. 2009;18:1531–1540. doi: 10.1002/pro.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bodner C.R., Maltsev A.S., Bax A. Differential phospholipid binding of α-synuclein variants implicated in Parkinson's disease revealed by solution NMR spectroscopy. Biochemistry. 2010;49:862–871. doi: 10.1021/bi901723p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Rooijen B.D., Claessens M.M., Subramaniam V. Lipid bilayer disruption by oligomeric α-synuclein depends on bilayer charge and accessibility of the hydrophobic core. Biochim. Biophys. Acta. 2009;1788:1271–1278. doi: 10.1016/j.bbamem.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 54.Nemani V.M., Lu W., Edwards R.H. Increased expression of α-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bortolus M., Tombolato F., Maniero A.L. Broken helix in vesicle and micelle-bound α-synuclein: insights from site-directed spin labeling-EPR experiments and MD simulations. J. Am. Chem. Soc. 2008;130:6690–6691. doi: 10.1021/ja8010429. [DOI] [PubMed] [Google Scholar]
- 56.Ferreon A.C.M., Gambin Y., Deniz A.A. Interplay of α-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc. Natl. Acad. Sci. USA. 2009;106:5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.