

Abstract
Synchrotron radiation circular dichroism, Fourier transform infrared, and nuclear magnetic resonance spectroscopies, and small-angle x-ray scattering were used to monitor the reversible thermal unfolding of hen egg white lysozyme. The results were compared with crystal structures and high- and low-temperature structures derived from molecular-dynamics calculations. The results of both experimental and computational methods indicate that the unfolding process starts with the loss of β-structures followed by the reversible loss of helix content from ∼40% at 20°C to 27% at 70°C and ∼20% at 77°C, beyond which unfolding becomes irreversible. Concomitantly there is a reversible increase in the radius of gyration of the protein from 15 Å to 18 Å. The reversible decrease in forward x-ray scattering demonstrates a lack of aggregation upon unfolding, suggesting the change is due to a larger dilation of hydration water than of bulk water. Molecular-dynamics simulations suggest a similar sequence of events and are in good agreement with the 1HN chemical shift differences in nuclear magnetic resonance. This study demonstrates the power of complementary methods for elucidating unfolding/refolding processes and the nature of both the unfolded structure, for which there is no crystallographic data, and the partially unfolded forms of the protein that can lead to fibril formation and disease.
Introduction
Lysozyme is a protein that is an important factor in the innate immune response of higher organisms. It cleaves sugars of the outer cell walls of bacteria, breaking down their integrity and the bacteria's viability. Information on its thermal stability and folding intermediates may be medically relevant as the human form of the protein has been shown to be involved in the formation of amyloid fibers associated with familial amyloidosis, which has severe pathogenic consequences (1). Although a great deal is known from crystal structures at low or ambient temperatures, knowledge about the high-temperature structure and thermally induced unfolding intermediates and pathways is much more limited in the absence of crystallographic data.
Hen egg white lysozyme (HEWL) is a 129-residue protein that has been classified as “mostly α” in the CATH structure classification database (2) and as (α + β) protein by the SCOP classification system (3). One of its two domains consists of four α-helices and a 310 helix, and the other (residues 39–83) has a triple-stranded antiparallel β-sheet, an irregular loop, and a 310 helix (4) (see Fig. S1 A in the Supporting Material).
Previous studies have investigated the reversible and irreversible thermal denaturation processes of this structure using a variety of experimental techniques (e.g., small-angle x-ray scattering (SAXS) (5–8), quasielastic neutron scattering (9), differential scanning calorimetry (10,11), nuclear magnetic resonance (NMR) (12–14), Fourier transform infrared (FTIR) (15), circular dichroism (CD) (16), and Raman (11) spectroscopies). Despite all of these studies, however, the mechanism of this process and the forces that drive it remain unclear. In addition, the results of molecular-dynamics (MD) simulations have not always been consistent or in agreement with experimental observations (17–19). In this study, we used synchrotron radiation CD (SRCD) spectroscopy, which yields highly accurate values of the helical content of proteins, and NMR to determine the secondary structure, and SAXS measurements to examine the tertiary structure and the role of water (and in particular the hydration shell) in this transition. SAXS and FTIR spectroscopy were also used to verify the absence of aggregation.
These methods were combined to examine details of the unfolding/refolding processes and define the structural nature of the intermediates. The crystallographic structures at low temperature were compared with the quantitative SRCD, NMR, and MD results. They all yielded similar results, leading to confidence in the accuracy of the methods. In the absence of a crystallographic structure, the combined experimental and computational methods provide a view of the high-temperature form of the protein. Confidence in this view is reinforced by the correspondence between the experimental SRCD and NMR results and the MD calculations for the high-temperature structure.
Hence, despite the large differences in the sensitivities and physical bases of these techniques, the results of all of the methods and the computational studies are remarkably consistent.
Materials and Methods
Commercial samples of HEWL and standard experimental and computational procedures (described in the Supporting Material) were used throughout this study.
Results and Discussion
Far-ultraviolet SRCD spectroscopy
In addition to α-helices and β-sheet, HEWL contains a large amount of nonhelical, nonsheet structure in the turns and loops, as well as regions classified as “other” types of secondary structure, usually considered to be disordered. The CD spectrum (Fig. 1, top) is typical of a protein with a significant helical content but also some β-sheet structure. The peak at 224 nm is nearly entirely due to the helical features, whereas the one near 208 nm is due to a mixture of sheet and helix components. The positive peak near 190 nm has contributions from helix and sheet components and decreases as the unfolded component increases due to the negative contribution of disordered or other structures at this wavelength. By following the individual behaviors of these three peaks as a function of temperature, therefore, one can monitor the loss of helix, the loss of helix and sheet, and the gain in disordered structure, respectively.
Figure 1.
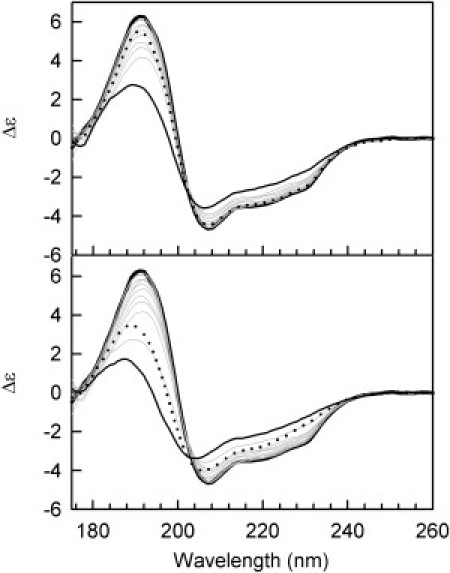
(Top) SRCD spectroscopy monitoring HEWL thermal denaturation between 25°C and 70°C in steps of 5°C. The largest curve at 191 nm (solid black) represents the spectrum obtained at 20°C, and the lowest (solid black) represents that obtained at 70°C. The spectra for the intermediate temperatures are displayed as solid gray curves. The dotted black curve represents the spectrum after cooling to 20°C and equilibration for 12 h. (Bottom) Thermal unfolding as in A, except that the highest temperature was 77°C.
The secondary structure of the protein calculated from the SRCD data at 20°C before heating is 40% helix, 12% sheet, plus 33% other secondary structure (Table 1), which corresponds well with the various crystallographic structures. Upon heating from 20°C to 70°C, the 224 nm peak decreases, resulting in a calculated reduction of more than one-third of the total helical content (Fig. 1, top; Table 1). However, the protein almost entirely regains its native helical content when cooled to 20°C (Table 1). The decrease relative to the initial spectrum is solely due to a small loss of protein as precipitate that occurs on cooling, which is reflected in the decrease in the associated high-tension signal, a measure of the pseudoabsorbance of the sample. The decrease in the 191 nm peak demonstrates that the unfolded helical fraction is mostly converted to other, or disordered, structure. The largest change in helical content occurs between 64°C and 70°C (Fig. 2, top), with a further fraction of helix being lost upon heating to 77°C (nearly 50% of the initial total). After heating to 77°C followed by cooling, only a fraction of the initial helical content is recovered (Fig. 1, bottom; Table 1). This suggests that an irreversible transition occurs between 70°C and 77°C, which results in only the refolding of the core helical residues, rather than complete refolding of all the native helical regions.
Table 1.
Calculated secondary structures (%) derived from analyses of SRCD data
| Analysis method |
||||||
|---|---|---|---|---|---|---|
| CONTINLL | SELCON | CDDSTR | AVG | SD | ||
| Folded 20°C | % Helix | 39 | 40 | 41 | 40 | 1 |
| % Other | 35 | 32 | 33 | 33 | 2 | |
| NRMSD | 0.023 | |||||
| Unfolded 70°C | % Helix | 27 | 27 | 24 | 26 | 2 |
| % Other | 39 | 36 | 38 | 38 | 2 | |
| NRMSD | 0.141 | |||||
| 20°C after 70°C | % Helix | 36 | 37 | 37 | 37 | 1 |
| % Other | 37 | 33 | 35 | 35 | 2 | |
| NRMSD | 0.026 | |||||
| Unfolded 77°C | % Helix | 20 | 19 | 19 | 19 | 0 |
| % Other | 43 | 41 | 40 | 41 | 2 | |
| NRMSD | 0.083 | |||||
| 20°C after 77°C | % Helix | 28 | 28 | 29 | 28 | 0 |
| % Other | 39 | 38 | 37 | 38 | 1 | |
| NRMSD | 0.156 | |||||
NRMSD is the goodness-of-fit parameter, AVG is the average value calculated by the different algorithms, and SD is the variation (±) between the secondary structures calculated by the different analysis algorithms.
Figure 2.
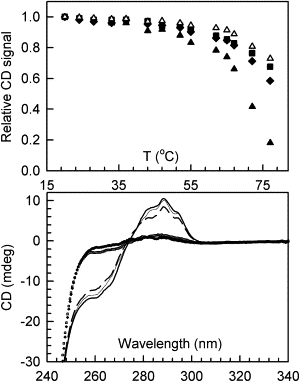
(Top) Changes in the SRCD signals as a function of temperature monitored at three wavelengths (191 nm (▴), 207 nm (■), and 222 nm (♦)), and the 191 nm signal scaled by a factor of 0.33 (▵). (Bottom) Near-UV CD spectra of HEWL at 20°C (thick solid line), 70°C (□), 20°C after cooling from 70°C (thin solid line), 77°C (♦), and 20°C after cooling from 77°C (dashed line).
The normalized root mean-square deviation (NRMSD) values for the folded and refolded from 70°C samples (Table 1) are very low, indicating that the spectra are well represented by the standard types of secondary structure. The high NRMSD values for both the unfolded samples and those refolded from 77°C are characteristic of the presence of additional nonstandard (disordered or aggregated) structures.
The spectra at all temperatures between 20°C and 64°C pass through an isosbestic point near 202 nm, suggesting that in this temperature range, unfolding is a two-state process and no significant intermediate structures are formed. However, the spectra at 70°C and 77°C do not pass through a common point, pointing to the presence of at least some proportion of a different structure at these temperatures. A singular value decomposition analysis of all spectra obtained from 20°C to 70°C (Fig. S2 A) indicates that they can be described by only two component curves, with the first curve mostly due to helical structure and the second curve representative of a classic disordered spectrum, which further suggests that there is no stable unfolding intermediate. This is further corroborated as the data can be fit with a second-order function, and in the plot of the magnitude of the SRCD change versus temperature (Fig. 2, top) the peaks at all wavelengths appear to decrease approximately in parallel. The 191 nm peak decreases to a greater extent as the changes at this wavelength are due to both a decrease in helix and a concomitant increase in other structure, both of which decrease the signal. When scaled to the same relative magnitude change, this curve virtually overlaps the others, also suggesting that it involves unfolding directly from helix to unordered in a concerted manner.
It is important to note that for these studies the use of SRCD as opposed to conventional CD was important, for several reasons: 1), the calculated secondary structure corresponds more closely to that determined by crystallography (40% for SRCD compared to 35% by conventional CD (data not shown)); 2), the small differences between the spectra at the intermediate temperatures meant that it was important to have the higher signal/noise ratio available in the SRCD spectra; and 3), it was only possible to accurately measure the data for the 191 nm peak using SRCD (rather than conventional CD spectroscopy (data not shown)), because the signal in that wavelength range from conventional CD instruments is too noisy to be reliable for subtle comparisons, especially at high temperatures.
Near-ultraviolet CD spectroscopy
The near-ultraviolet (UV) region of a CD spectrum (260–300 nm) arises from the aromatic amino acids in the protein, with tryptophan contributions dominating this spectral region. The chiral absorbances of the aromatic transitions are much less intense than those of the peptide backbone in the far-UV region. However, with longer-pathlength cells, they can be measured with conventional CD instruments.
HEWL contains six tryptophan residues (residues 28, 62, 63, 108, 111, and 123). Residues 62 and 63 are in a turn, and residue 123 is in a 310 helix. Residues 28 and 111 are located in helix 2 and helix 5, respectively, and residue 108 is buried in the center of the globular structure. In the crystal structures (20) residue 108 is located between a 310 helix [104–107] and helix 5 [109–115]. The first of these helices becomes irregular, and the second one is a 310 helix in the NMR structure at 35°C (21). Loss of the aromatic signal upon heating (Fig. 2, bottom) and its recovery upon cooling corroborate the far-UV data in that the latter residues appear to be located in regions that become unfolded (i.e., helices) and readopt a helical conformation upon refolding. The aromatic signal is also restored when the protein is cooled from 77°C, although it has a slightly lower magnitude, suggesting that only the core of the helical domain where the tryptophans are situated refolds effectively in this case.
FTIR spectroscopy
The effects of a temperature increase up to 80°C and subsequent cooling are shown in Fig. 3. The amide I band at 25°C is characterized by two distinct peaks at 1643 and 1652 cm−1, which have been assigned to 310-helix and α-helix structure, respectively (15). Note that the intensity ratio of these peaks corresponds to that previously reported at 52°C, i.e., after completion of the hydrogen-deuterium (H/D) exchange (15). As the temperature increases, the bands shift to higher wavenumbers without any significant change in spectral intensity, consistent with a weakening of H-bonds (Fig. 3). Note the absence of an H/D exchange-associated transition around 52°C, as the protein was already fully exchanged before the experiment. Only between 75°C and 80°C is a clear loss of intensity observed. It is accompanied by an increase in absorbance above 1665 cm−1 due to the formation of irregular structure. This is in agreement with previous work (15). However, the peaks arising from helical structures are still present at 80°C, suggesting that a significant fraction of the helical structure remains intact and the spectrum is clearly different from that reported for the unfolded protein (15). Upon cooling, the original spectrum is recovered at 25°C, although the ratio of the peak intensities at 1643 and 1652 cm−1 is somewhat different, with slightly more intensity at 1643 cm−1 (see Fig. S3 A). This indicates incomplete refolding, with more 310 helices or disordered structure, which also absorbs around 1640 cm−1, present after thermal unfolding. Of more importance, however, is the absence of any aggregation peaks that were previously shown to appear during cooling of fully unfolded HEWL (15).
Figure 3.
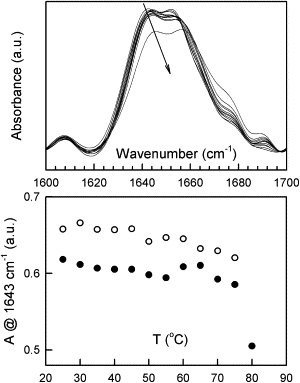
Temperature-induced changes in the IR spectrum (amide I band region) of HEWL between 20°C and 80°C. (Top) Deconvoluted spectra are shown with arrows indicating the direction of the spectral changes during heating. Bottom: Plot of the intensity change at 1643 cm−1 against temperature during heating (●) and cooling (□).
NMR spectroscopy
The temperature dependence of the one-dimensional 1H NMR spectra of HEWL (Fig. S4 A) indicates that the protein is unfolded at 80°C but still partially folded at 70°C. In general, thermal unfolding is fully reversible but becomes irreversible after a few hours at 80°C.
The amide proton and 15N chemical shifts at 37°C and 70°C and their differences (Δ1HN, Δ15N; Table S1), and the 15N-1H HSQC spectrum at 70°C (Fig. S5) are given in the Supporting Material. A superposition of the Δ1HN between 37°C and 70°C on the native NMR structure (PDB: 1E8L; Fig. 4) clearly illustrates that the α-domain, including the interdomain helix, has more native-like 1HN shifts than the β-domain at 70°C. NMR spectra obtained in a previous study suggested that in the unfolded protein all residues are in a solvent-accessible environment (13). Of the 45 residues for which protection factors have been determined at 69°C (12), 19 are associated with large Δ1HN-values (Table S1), and of these, nine correspond to hydrophobic residues. Eleven of the 26 protected residues that are not associated with large Δ1HN are hydrophobic. There is thus no simple correlation between presumed unfolding and protection against amide proton exchange.
Figure 4.
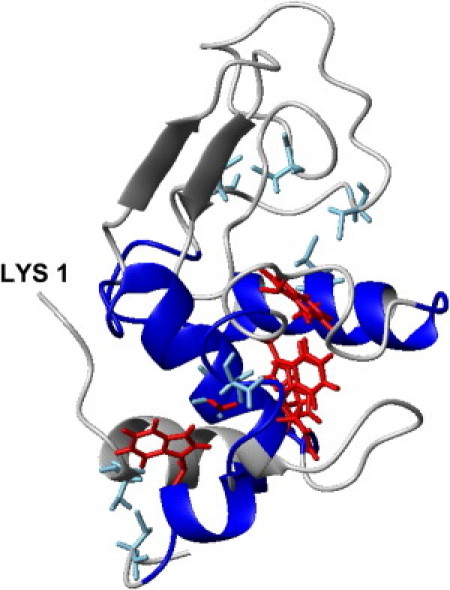
Differences in chemical shift between 37°C and 70°C plotted on the native NMR structure of HEWL (PDB entry 1E8L (36)). Blue: Stable parts of the structure where the 1HN chemical shift differences between 37°C and 70°C (Table S1) are small (i.e., > −0.07 ppm for three or more consecutive residues). Gray parts are largely unfolded at high temperature, as suggested by the larger random deviations. Red: Clearly observed native long-range NOEs. Light blue: Disulphide bonds.
All sheet structures except aa [51–53] are associated with large Δ1HN-values. Helix 1 [5–15] has a tendency to unfold at its N-terminal end, and there are also large Δ1HN-values in helix 3 [80–84], at both ends of helix 4 [88–101], and in helix 5 [109–115] (Table S1). Helix 2 [25–36] seems to be the most stable one, with only two N-terminal residues having a large Δ1HN. The [95–105] region, where a number of residues have large Δ1HN-values, is also one in which larger changes occur in crystals between –178°C and 22°C (22). The remaining total helical content at 70°C is 26% based on the average Δ1HN for three successive residues (Table S1), in good agreement with the SRCD results.
Not unexpectedly, the differences between the 1H chemical shifts of the cysteines (Table S1) tend to be smaller than those of their neighboring residues. The cysteines in the 6–127 bridge are both located in regions of large Δ1HN-values. In the 30–115 bridge, the first residue is located in the central part of helix 2 at the end of a region for which amide hydrogen-exchange protection factors at 69°C could be measured. Residue 115 is in a helix in the crystallographic models (20) but not in the NMR structure (21), and is also protected. The Δ1HN-values are small in both cases. In the 64–80 bridge, the first residue is located in a sheet and in a protected region, and residue 80 is part of a 310 helix. Both residues are flanked by aa with larger Δ1HN-values at 70°C. The situation is similar in the 76–94 bridge, where the two residues belong to protected regions and the Δ1HN-value for residue 94 is particularly small.
SAXS
The distance distributions calculated from the SAXS patterns at 20°C and 80°C, and the corresponding models (Fig. S6 and Fig. S7 A) clearly illustrate the expansion of the molecule at higher temperatures. The forward scattering I(0)/c, corrected for the change of density of the solution, decreases by ∼3% (Fig. 5). The glitch between 70°C and 78°C may be due to the influence of repulsive intermolecular forces on the values of I(0)/c for concentrated solutions. Similar deviations from ideal behavior in the region of the transition were also observed at lower concentration (M. Hirai, Gunma University, personal communication, 2009), and in a low-concentration SAXS study of thermal unfolding of the all-β protein neocarzinostatin, where a 9% increase in I(0)/c was found between 20°C and 80°C (23).
Figure 5.
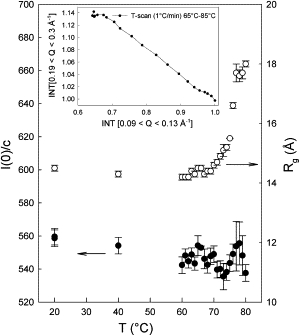
Forward scattering scaled to the effective concentration (I(0)/c) and Rg of HEWL solutions (50 mg mL−1) as a function of temperature. Note that the values at 20°C before and after unfolding are identical within experimental error. (Inset) The linearity of the correlation plot of the integrated intensity in the first maximum of the scattering pattern of HEWL and at the onset of the second maximum indicates that there are no detectable intermediates during unfolding.
The CRYSOL (24) fit between the SAXS data at 20°C and the crystallographic model (PDB entry: 6LYZ (4)) and other PDB entries for HEWL gives an average electron density of 0.4319 e/Å3 using the electron density of water (0.3340 e/Å3) for the solvent. Since the electron density of water drops from 0.3340 e/Å3 to 0.3241 e/Å3 between 20°C and 85°C, with everything else, including the hydration layer, being equal, this would lead to an increase of 20% of I(0)/c. Because the volume of the protein, or its specific volume, should not change by >∼1% (25), the lower I(0)/c must result mainly from a decrease in the difference in electron density between the hydration layer and the bulk solvent.
The radius of gyration, Rg, values increase slightly up to ∼72°C and then more rapidly up to a plateau at 80°C (Fig. 5). The limits of the transition are in agreement with thermodynamic data indicating that at 69°C and pH 3.8, 90% of the protein is still in the folded state (12). The linearity of the intensity correlation plot (Fig. 5, inset) strongly suggests that there are no intermediates in the transition between two states.
It was previously observed (5) that the change in Rg is independent of pH for 3.8 ≤ pH ≤ 7.2 but increases drastically at pH 2.2, indicating a different transition. The onset and midpoint of the transition monitored by differential scanning calorimetry are 70°C and 77°C at pH 4.8, and 66°C and 73°C at pH 7.2. The reversibility of the transition was found to be higher at pH 3.8 than at pH 7, as also illustrated here by the comparison of the SRCD results measured at pH 7 and those obtained by the other methods at pH 3.8. The Tm value in D2O buffers is generally 2–3°C higher than in equivalent H2O buffers (26). However, these differences should not affect the nature of the unfolded ensemble at the level of resolution achieved here.
Based on CRYSOL (24) fits of scattering patterns at different temperatures, it has been proposed that the decrease in I(0)/c and Rg during thermal unfolding of HEWL is accompanied by the collapse of the hydration shell (27). Of course, such fits are only legitimate as long as the structure does not significantly change with temperature (T< 55–60°C). An explanation of this effect must take into account that water in the vicinity of proteins differs from bulk water at ambient conditions. Although this is still a controversial subject (28–30), water molecules can have two environments—one akin to that of high-density amorphous ice, and one similar to that of low-density amorphous ice. The coexistence of these rapidly interconverting structures has been linked to heat, pressure, and cold denaturation (31). It can be assumed that the hydration layer, which has a higher density than bulk water at room temperature (32), has an H-bond distorted structure resembling high-density amorphous ice. Its preferential expansion is expected to reduce the contrast between the shell and bulk solvent. Rather than collapsing, the more fragile water network becomes a better solvent and the ratio of protein-water to protein-protein H-bonds increases. It has been proposed that, in contrast to the situation in cold denaturation, the water structure has an insignificant role in heat denaturation (31). This is in contradiction to the stabilization of the protein by TMAO, an osmolyte that does not interact with the protein but strengthens the water network (33). The midpoint of the heat denaturation transition of HEWL increases from 75°C in 0.1 M Tris-HCl to 80°C in buffer containing 1 M TMAO and 83°C at 2 M TMAO (34), but is insensitive to pH in the range of pH 5–7, over which the last may be expected to vary due to the temperature factor of Tris buffer. Unfolding of HEWL in very concentrated solution (∼200 mg mL−1) has been described as a two-stage process (11) whereby the breaking of the H-bond network of water above 47°C will induce a change in tertiary structure with enhanced solvent accessibility, but not in the secondary structure, which starts collapsing above 67°C. In hydrated HEWL powders, there is an abrupt change in hydration water dynamics at 67 ± 5°C coinciding with the onset of reversible denaturation (9). Some caution is required, however, in the interpretation of results obtained on systems in which crowding and confinement effects can alter the properties of the solvent.
The importance of solvent effects is also illustrated by thermal unfolding of HEWL in the presence of high concentrations of glycerol, where unfolding of the tertiary and secondary structures appears to be decoupled even though the protein is still hydrated. The influence of long-range effects due to changes in the water structure may also explain some of the difficulties involved in reproducing a sharp transition between two states in MD simulations.
MD simulations
Equilibration at 27°C of the initial crystallographic structure, which has a total helical content (α + 310) calculated by the STRIDE algorithm (35) of 45%, yields a structure with 37% helix. It should be taken into account, however, that different secondary structure calculation methods yield slightly different results, and different crystal structures of HEWL yield different values for the same algorithm. (For example, for a crystal structure of HEWL with 1.65 Å resolution (PDB code: 193l (20)) the STRIDE algorithm (35) gives 45% helix (33% α + 12% 310) and 7% sheet structure, whereas the DSSP method (36) yields 40% helix (α+310) and 6% sheet structure.) The STRIDE value for the NMR solution structure at 35°C, (PDB entry: 1E8L (21)) is 36% helix and 8.5% sheet.
After MD equilibration, the short 310 helices [20–22], [104–107], and [120–123] are lost, and the [80–84] region oscillates between a 310 and an α-helix. This is in agreement with the 100-fold lower protection factors of the 310 helix 3 [80–83] compared to other helices in NMR amide hydrogen-exchange experiments (12), and the absence of helices [20–22] and [104–107] in the NMR solution structure at 35°C (21). Identical results were found after independent 2 ns equilibrations at 32°C and 37°C, but not at 29°C. The scattering curve of the initial MD model fits the experimental SAXS data at 20°C and yields an Rg-value of 15.0 Å (Fig. S8 A), which is close to that of the crystallographic models (15.2(2) Å), the average Rg calculated from three NMR models (14.95(5) Å), and the value obtained by SAXS after extrapolation to zero concentration (15.4(2) Å) (32). In addition, the secondary structure content of the MD model is close to that obtained from SRCD (Table 1) and the crystallographic models. The initial MD is thus in good agreement with the solution structures at both the secondary and tertiary levels of organization.
To simulate the thermal unfolding process, the initial structure was equilibrated at 227°C and nine different trajectories were followed. The root mean-square deviation (RMSD) relative to the initial structure was similar for all trajectories up to 2 ns (Fig. S9 A), but the later unfolding differed in each case. The Rg-values of the structures at the end of 10 ns, calculated using CRYSOL (24) with a hydration shell density equal to that of bulk solvent (except for the initial structure), are listed in Table 2. The RMS average Rg for the nine runs is 16.3 Å. A description of the time course of the secondary structure in different trajectories, which is not accessible experimentally, is given in the Supporting Material.
Table 2.
Helical content and Rg of the initial structure and nine trajectories after 10 ns
| Structure | % Helix (α + 310) | Rg (Å) |
|---|---|---|
| Initial | 37.2 | 15.0 |
| Run1 | 23.4 | 15.6 |
| Run 2 | 14.9 | 18.0 |
| Run 3 | 26.0 | 15.2 |
| Run 4 | 27.1 | 15.4 |
| Run 5 | 32.3 | 15.4 |
| Run 6 | 24.9 | 16.8 |
| Run 7 | 27.2 | 17.3 |
| Run 8 | 24.6 | 16.2 |
| Run 9 | 16.7 | 16.3 |
The helical content of the individual α-helices calculated as averages over the last 2 ns of the trajectories is given in Table 3, and Fig. 6 illustrates the fate of the secondary structural elements in the initial structure (Fig. S1) in the final MD structures at the end of 10 ns at 227°C in simulation runs 2 and 7. The correlation map between distances in the final structures in runs 2 and 7 and in the initial structure (Fig. S10) illustrates that the difference between the initial and final structures is much larger in run 2 than in run 7, where the most important modification concerns the interaction between β-strands and the [83–95] region containing helix 4.
Table 3.
Average local helical content during the last 2 ns in MD
| Helix | Residues | % Helix | Notes |
|---|---|---|---|
| 1 (A) | 5–15 | 70 | N-terminal end is less stable |
| 2 (B) | 25–35 | 67 | Center is stable, may open at either end or both ends |
| 3 (C) | 80–84 | 80 | Fluctuates between 310 and α-helix |
| 4 (D) | 88–101 | 71 | Center is stable, may open at either end or both ends |
| 5 | 109–115 | 56 | Often completely lost, but is regained |
| Average | 66 |
Figure 6.
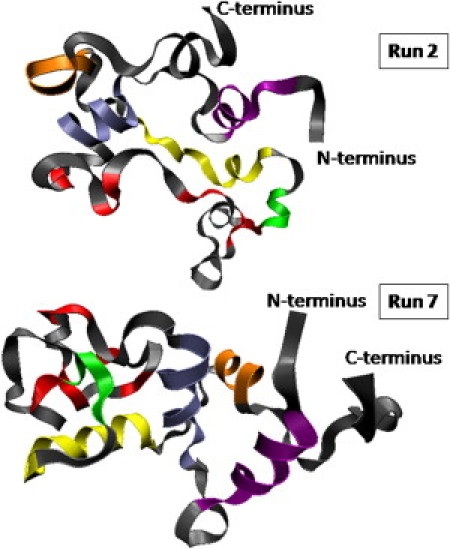
Models of two of the MD structures obtained after 10 ns at 227°C. The scattering pattern of run 7 (bottom) gives the best agreement with the experimental data at 80°C, as illustrated in Fig. S11. The color-coding corresponds to the different structural elements in the initial structure in Fig. S1. Run 2 is the only trajectory in which helix 4 is completely unfolded.
Nearly a third of the helices are lost on average after 10 ns, leaving an average helical content of 25% for the nine trajectories in Table 2, which is very similar to the 27% found by SRCD and NMR. The loss of helical content is accompanied by larger Rg-values that vary between 15 Å and 18 Å, but only those of runs 2 and 7 are close to the experimental one at 80°C, whereas in three cases (runs 3, 4, and 5) the Rg remains close to that of the folded structure even though the β-structures are already lost, as corroborated by the increase in RMSD (Fig. S8). Note that the fit to the experimental data at 80°C of run 7 (Fig. S11 A), which corresponds to the helical content found by SRCD (27%), is better than that of run 2 (14% helix), where the side maximum between 0.25 < Q < 0.5 Å−1 in the scattering pattern of the initial structure is replaced by a feature at lower angles (Fig. S11). The maximum dimension of the distance distribution function is 55 Å in run 2 and 66 Å in run 7, as opposed to an experimental value of 55–60 Å at 80°C.
In a previous study (17), Rg-values (16.8 Å) significantly below those observed experimentally at 80°C were also obtained when unfolding of helices was facilitated by insertion of water molecules into the cavities produced during MD simulations. The authors predicted that the β-domain would be stable throughout the unfolding process, and reported a lower helical content in the unfolded state (17–22%) than found in our simulations or SRCD and NMR studies. Another MD study of HEWL unfolding found that the β-domain unfolded very early on in different trajectories, whereas the α-domain remained structured (19). The unfolded state (Rg = 17.4 Å) had a total helix content of 34%, which is significantly higher than the value found by SRCD or NMR (27%).
A simulation at 27°C using an RMSD constraint algorithm reached maximum Rg-values of 16.7 Å (18) and indicated that the unfolding process involves partial unfolding of the three-stranded β-sheet to a two-stranded one during a phase in which helices 1–3 remain to a large extent native. Complete unfolding of the β-domain was accompanied by partial unfolding of the α-helices, with helices 1 and 2 unfolding through progressive hydration by insertion of water molecules into their H-bond network.
Taken together, the more-recent MD simulations suggest that the β-domain unfolds before the α-domain and with progressive hydration of the helices. The relative stability of the α- and β-domains may also be related to their hydropathy (Fig. S12 A). The average hydropathy index (38) of HEWL is −0.47 (−0.54, 0.32, 2.22, 0.46, −0.70 for helices 1–5, respectively, i.e., 0.35 on average) and −1.08 for the region [37–79], which corresponds to the nonhelical part of the β-domain (Fig. 6). Helix 3, which is very stable in the simulations, has the highest index (2.22), and helix 5, which has a tendency to disappear, has the lowest (−0.70).
Conclusions
The reversible thermal unfolding of HEWL is a continuous transition that is not associated with any latent heat (10). The experiments above confirm that the secondary and tertiary structures are highly stable against thermal unfolding, with very little change between 20°C and 64°C.
When the temperature is increased to 70°C, the sheet structures in the β-domain and some of the helical structure are lost, with a concomitant gain in other or disordered structure, but the protein can refold. Further unfolding of the helical structure above 70°C leads to a large increase in the Rg of the molecule. Only when the temperature is equilibrated above 80°C does unfolding become irreversible. The fact that the β-structures are the first to disappear in these equilibrium unfolding experiments suggests that the α-helices are both thermodynamically and kinetically more stable, since upon refolding the α-domain forms first (39).
Previous hydrogen-exchange experiments indicated that during refolding after guanidinium chloride denaturation the α-domain refolds before the β-domain (40), but that during reversible thermal denaturation the two domains unfold cooperatively (12), and it was estimated from thermodynamic data that, at 69°C, 90% of the molecules are still in the folded state. This implies that at this temperature, large-scale cooperative structural fluctuations must take place, not unlike those predicted by the MD calculations.
The spectroscopic experiments examined the ensemble of molecules and cannot distinguish whether after “irreversible unfolding” all molecules refold to a nearly native secondary structure, or whether some fully refold and others remain unfolded (and aggregated). The latter case is most likely, given the change in absorbance between the initial far-UV SRCD spectrum and the refolded one at 20°C, and because the refolded spectrum in the near-UV is essentially the same as the initial spectrum, albeit slightly smaller in magnitude. This phenomenon is probably due to the fact that at pH 5.9 (conditions under which the SRCD measurements were made), the intermolecular interactions are less repulsive, which furthers aggregation. This was also previously observed by differential scanning calorimetry with HEWL solutions of different pH values (5). Whereas the optical measurements yield number-averaged signals, SAXS gives weight average values of I(0)/c and Rg2, and is thus more sensitive to aggregation. The fact that the forward scattering returns to the same value upon cooling (Fig. 5) indicates that at pH 3.8 there is no loss of material (e.g., through formation of large aggregates, no longer detectable in SAXS). Note that due to the disulphide bonds, the number of different conformations accessible to the molecule remains very limited in comparison with a free 129-residue excluded volume chain of Cα-carbons (Fig. S13 A).
Based on the data obtained by the different methods, it can be concluded that in aqueous solution, unfolding initially occurs by the loss of sheet structures in the β-domain and a small part of the helical structures in the α-domain below 70°C, followed by the concerted unfolding of ultimately nearly 50% of the helical structure and the tertiary structures at higher temperatures. The fact that cosolutes such as TMAO and glycerol, which are preferentially excluded from the protein surface (41), have a strong influence on the reversible thermal unfolding suggests that the roles of the structure and dynamics of the solvent in this process have hitherto been underestimated. Since an increase in temperature is expected to diminish the solubility of apolar groups, it seems somewhat contradictory to regard hydrophobic effects as the main driving force in protein (un)folding. That other factors are dominant is also suggested by the fact that the solubility of hydrophobic residues increases with temperature (42). Based on the transfer model, it was recently convincingly argued that folding and unfolding depend mainly on the balance of solvent-backbone and backbone-backbone H-bonds (43), which is certainly also influenced by solvent-solvent and solvent-cosolute H-bonding. The success of this approach suggests that a better understanding of thermal denaturation, and possibly also cold denaturation (31), could be obtained by extending the transfer model to the temperature extrema of the stability range of proteins.
In summary, in this study we combined several experimental methods and compared the results with crystallographic structures at low temperatures and the results of MD simulations. The experimental and computational methods provide information on the high-temperature structure, for which there are no crystallographic data, as well as on the unfolding/refolding process and the secondary and tertiary structures of the folding intermediates.
Acknowledgments
C.A. and M.K. thank Ayse Ozlem Aykut and Çetin Baloğlu for their help with the MD runs, and Alpay Taralp for useful discussions. R.B. thanks Prof. Dr. H. R. Kalbitzer for granting access to the 500 MHz NMR spectrometer and Auremol software package.
The SRCD work was supported by project grants from the Biotechnology and Biological Sciences Research Council (UK) to B.A.W. and by synchrotron beam time grants to B.A.W. Access to the CD1 beam line at the Institute for Storage Ring Facilities was provided under the European Union Integrated Infrastructure Initiative (I3) Integrated Activity on Synchrotron and Free Electron Laser Science (IA-SFS), contract No. RII3-CT-2004-506008. F.M. is a postdoctoral fellow of the Research Foundation Flanders. A.M. was supported in part by grants from the Fonds de la Recherche Fondamentale et Collective (contracts 2.4550.05 and 2.4530.09) and by the Belgian Program of Interuniversity Attraction Poles initiated by the Federal Office for Scientific Technical and Cultural Affairs (PAI nr. P6/19). The authors declare no conflict of interest.
Contributor Information
Filip Meersman, Email: Filip.Meersman@chem.kuleuven.be.
B.A. Wallace, Email: b.wallace@mail.cryst.bbk.ac.uk.
Supporting Material
References
- 1.Frare E., Mossuto M.F., Fontana A. Identification of the core structure of lysozyme amyloid fibrils by proteolysis. J. Mol. Biol. 2006;361:551–561. doi: 10.1016/j.jmb.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 2.Orengo C.A., Michie A.D., Thornton J.M. CATH—a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 3.Murzin A.G., Brenner S.E., Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 4.Diamond R. Real-space refinement of the structure of hen egg-white lysozyme. J. Mol. Biol. 1974;82:371–391. doi: 10.1016/0022-2836(74)90598-1. [DOI] [PubMed] [Google Scholar]
- 5.Arai S., Hirai M. Reversibility and hierarchy of thermal transition of hen egg-white lysozyme studied by small-angle x-ray scattering. Biophys. J. 1999;76:2192–2197. doi: 10.1016/S0006-3495(99)77374-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirai M., Arai S., Iwase H. Complementary analysis of thermal transition multiplicity of hen egg-white lysozyme at low pH using X-ray scattering and scanning calorimetry. J. Phys. Chem. B. 1999;103:549–556. [Google Scholar]
- 7.Shang, W. 2003. Design of a time-resolved data acquisition system for x-ray scattering and application to proteins in solution and semi-crystalline polymers. Master's thesis. University of Siegen, Siegen, Germany.
- 8.Hirai M., Koizumi M., Inoue K. Hierarchical map of protein unfolding and refolding at thermal equilibrium revealed by wide-angle X-ray scattering. Biochemistry. 2004;43:9036–9049. doi: 10.1021/bi0499664. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y., Lagi M., Chen S.H. Observation of high-temperature dynamic crossover in protein hydration water and its relation to reversible denaturation of lysozyme. J. Chem. Phys. 2009;130:135101. doi: 10.1063/1.3081137. [DOI] [PubMed] [Google Scholar]
- 10.Hirai M., Arai S., Takizawa T. Small-angle X-ray scattering and calorimetric studies of thermal conformational change of lysozyme depending on pH. J. Phys. Chem. B. 1998;102:1308–1313. [Google Scholar]
- 11.Hédoux A., Ionov R., Danéde F. Evidence of a two-stage thermal denaturation process in lysozyme: a Raman scattering and differential scanning calorimetry investigation. J. Chem. Phys. 2006;124:14703. doi: 10.1063/1.2139087. [DOI] [PubMed] [Google Scholar]
- 12.Radford S.E., Buck M., Evans P.A. Hydrogen exchange in native and denatured states of hen egg-white lysozyme. Proteins. 1992;14:237–248. doi: 10.1002/prot.340140210. [DOI] [PubMed] [Google Scholar]
- 13.McDonald C.C., Phillips W.D., Glickson J.D. Nuclear magnetic resonance study of the mechanism of reversible denaturation of lysozyme. J. Am. Chem. Soc. 1971;93:235–246. doi: 10.1021/ja00730a039. [DOI] [PubMed] [Google Scholar]
- 14.Evans P.A., Topping K.D., Dobson C.M. Hydrophobic clustering in nonnative states of a protein: interpretation of chemical shifts in NMR spectra of denatured states of lysozyme. Proteins. 1991;9:248–266. doi: 10.1002/prot.340090404. [DOI] [PubMed] [Google Scholar]
- 15.Meersman F., Heremans K. Temperature-induced dissociation of protein aggregates: accessing the denatured state. Biochemistry. 2003;42:14234–14241. doi: 10.1021/bi035623e. [DOI] [PubMed] [Google Scholar]
- 16.Goux W.J., Hooker T.M., Jr. The chiroptical properties of proteins. II. Near-ultraviolet circular dichroism of lysozyme. Biopolymers. 1980;19:2191–2208. doi: 10.1002/bip.1980.360191205. [DOI] [PubMed] [Google Scholar]
- 17.Williams M.A., Thornton J.M., Goodfellow J.M. Modelling protein unfolding: hen egg-white lysozyme. Protein Eng. 1997;10:895–903. doi: 10.1093/protein/10.8.895. [DOI] [PubMed] [Google Scholar]
- 18.Gilquin B., Guilbert C., Perahia D. Unfolding of hen egg lysozyme by molecular dynamics simulations at 300K: insight into the role of the interdomain interface. Proteins. 2000;41:58–74. [PubMed] [Google Scholar]
- 19.Kazmirski S.L., Daggett V. Non-native interactions in protein folding intermediates: molecular dynamics simulations of hen lysozyme. J. Mol. Biol. 1998;284:793–806. doi: 10.1006/jmbi.1998.2192. [DOI] [PubMed] [Google Scholar]
- 20.Vaney M.C., Maignan S., Ducriux A. High-resolution structure (1.33 A) of a HEW lysozyme tetragonal crystal grown in the APCF apparatus. Data and structural comparison with a crystal grown under microgravity from SpaceHab-01 mission. Acta Crystallogr. D Biol. Crystallogr. 1996;52:505–517. doi: 10.1107/S090744499501674X. [DOI] [PubMed] [Google Scholar]
- 21.Schwalbe H., Grimshaw S.B., Smith L.J. A refined solution structure of hen lysozyme determined using residual dipolar coupling data. Protein Sci. 2001;10:677–688. doi: 10.1110/ps.43301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurinov I.V., Harrison R.W. The influence of temperature on lysozyme crystals. Structure and dynamics of protein and water. Acta Crystallogr. D Biol. Crystallogr. 1995;51:98–109. doi: 10.1107/S0907444994009261. [DOI] [PubMed] [Google Scholar]
- 23.Pérez J., Vachette P., Durand D. Heat-induced unfolding of neocarzinostatin, a small all-β protein investigated by small-angle X-ray scattering. J. Mol. Biol. 2001;308:721–743. doi: 10.1006/jmbi.2001.4611. [DOI] [PubMed] [Google Scholar]
- 24.Svergun D., Barberato C., Koch M.H.J. CRYSOL - a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 1995;28:768–773. [Google Scholar]
- 25.Chalikian T.V., Bresiauer K.J. On volume changes accompanying conformational transitions of biopolymers. Biopolymers. 1996;39:619–626. doi: 10.1002/(sici)1097-0282(199611)39:5<619::aid-bip1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 26.Efimova Y.M., Haemers S., van Well A.A. Stability of globular proteins in H2O and D2O. Biopolymers. 2007;85:264–273. doi: 10.1002/bip.20645. [DOI] [PubMed] [Google Scholar]
- 27.Koizumi M., Hirai H., Hirai M. Collapse of the hydration shell of a protein prior to thermal unfolding. J. Appl. Cryst. 2007;40:s175–s178. [Google Scholar]
- 28.Huang C., Wikfeldt K.T., Nilsson A. Reply to Soper et al.: Fluctuations in water around a bimodal distribution of local hydrogen-bonded structural motifs. Proc. Natl. Acad. Sci. USA. 2010;107:E45. [Google Scholar]
- 29.Huang C., Wikfeldt K.T., Nilsson A. The inhomogeneous structure of water at ambient conditions. Proc. Natl. Acad. Sci. USA. 2009;106:15214–15218. doi: 10.1073/pnas.0904743106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soper A.K., Teixeira J., Head-Gordon T. Is ambient water inhomogeneous on the nanometer-length scale? Proc. Natl. Acad. Sci. USA. 2010;107:E44. doi: 10.1073/pnas.0912158107. author reply E45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsai C.-J., Maizel J.V., Jr., Nussinov R. The hydrophobic effect: a new insight from cold denaturation and a two-state water structure. Crit. Rev. Biochem. Mol. Biol. 2002;37:55–69. doi: 10.1080/10409230290771456. [DOI] [PubMed] [Google Scholar]
- 32.Svergun D.I., Richard S., Zaccai G. Protein hydration in solution: experimental observation by x-ray and neutron scattering. Proc. Natl. Acad. Sci. USA. 1998;95:2267–2272. doi: 10.1073/pnas.95.5.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meersman F., Bowron D., Koch M.H. Counteraction of urea by trimethylamine N-oxide is due to direct interaction. Biophys. J. 2009;97:2559–2566. doi: 10.1016/j.bpj.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nandi P.K., Bera A., Sizaret P.-Y. Osmolyte trimethylamine N-oxide converts recombinant α-helical prion protein to its soluble β-structured form at high temperature. J. Mol. Biol. 2006;362:810–820. doi: 10.1016/j.jmb.2006.07.060. [DOI] [PubMed] [Google Scholar]
- 35.Frishman D., Argos P. Knowledge-based protein secondary structure assignment. Proteins. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 36.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 37.Reference deleted in proof.
- 38.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 39.Radford S.E., Dobson C.M., Evans P.A. The folding of hen lysozyme involves partially structured intermediates and multiple pathways. Nature. 1992;358:302–307. doi: 10.1038/358302a0. [DOI] [PubMed] [Google Scholar]
- 40.Miranker A., Radford S.E., Dobson C.M. Demonstration by NMR of folding domains in lysozyme. Nature. 1991;349:633–636. doi: 10.1038/349633a0. [DOI] [PubMed] [Google Scholar]
- 41.Timasheff S.N. Protein-solvent preferential interactions, protein hydration, and the modulation of biochemical reactions by solvent components. Proc. Natl. Acad. Sci. USA. 2002;99:9721–9726. doi: 10.1073/pnas.122225399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ji P., Feng W. Solubility of amino acids in water and aqueous solutions by the statistical associating fluid theory. Ind. Eng. Chem. Res. 2008;47:6275–6279. [Google Scholar]
- 43.Bolen D.W., Rose G.D. Structure and energetics of the hydrogen-bonded backbone in protein folding. Annu. Rev. Biochem. 2008;77:339–362. doi: 10.1146/annurev.biochem.77.061306.131357. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.