

Abstract
Cardiac excitation-contraction coupling is a highly coordinated process that is controlled by protein kinase signaling pathways, including Ca2+/calmodulin-dependent protein kinase II (CaMKII) and protein kinase A (PKA). Increased CaMKII expression and activity (as occurs during heart failure) destabilizes EC coupling and may lead to sudden cardiac death. To better understand mechanisms of cardiac CaMKII function, we integrated dynamic CaMKII-dependent regulation of key Ca2+ handling targets with previously validated models of cardiac EC coupling, Ca2+/calmodulin-dependent activation of CaMKII, and β-adrenergic activation of PKA. Model predictions are validated against CaMKII-overexpression data from rabbit ventricular myocytes. The model demonstrates how overall changes to Ca2+ handling during CaMKII overexpression are explained by interactions between individual CaMKII substrates. CaMKII and PKA activities during β-adrenergic stimulation may synergistically facilitate inotropic responses and contribute to a CaMKII-Ca2+-CaMKII feedback loop. CaMKII regulated early frequency-dependent acceleration of relaxation and EC coupling gain (which was highly sarcoplasmic reticulum Ca2+ load-dependent). Additionally, the model identifies CaMKII-dependent ryanodine receptor hyperphosphorylation as a proarrhythmogenic trigger. In summary, we developed a detailed computational model of CaMKII and PKA signaling in cardiac myocytes that provides unique insights into their regulation of normal and pathological Ca2+ handling.
Introduction
Ca2+/Calmodulin-dependent protein kinase II (CaMKII) is a key regulator of cardiac excitation-contraction coupling (ECC) that has emerged as a potential therapeutic target for heart failure and arrhythmia (1). The activation of CaMKII by Ca2+-bound calmodulin (CaM) affords it the ability to decode both the frequency and location of Ca2+ signals (2,3). CaMKII expression is increased in failing human myocardium (4) and hyperactivity has been linked to the induction of hypertrophy and arrhythmias (5,6). Chronic (7) and acute (8) CaMKII overexpression studies have demonstrated that this kinase regulates myocyte Ca2+ handling, though the specific mechanisms by which these changes occur are unclear. Both CaMKII and protein kinase A (PKA) phosphorylate a number of Ca2+ handling targets with similar functional consequences (including L-type Ca2+ channels (LCCs), ryanodine receptors (RyRs), and phospholamban (PLB)), though how these two pathways communicate with one another remains unresolved.
To examine how the known molecular mechanisms of CaMKII and PKA interact to coordinate ECC and trigger arrhythmia, we developed an integrated computational model of CaMKII and PKA signaling in the rabbit ventricular myocyte. The model includes dynamic phosphorylation of LCCs, RyRs, and PLB by CaMKII and PKA, as well as functional regulation of additional targets distinct to each kinase. These interactions were integrated with previously validated models of Ca2+/CaM-dependent activation of CaMKII (3), β-adrenergic activation of PKA (9), and cardiac ECC in the rabbit ventricular myocyte (10). With this framework, we examined a number of unanswered questions in cardiac CaMKII/PKA signaling:
How do the individual actions of CaMKII substrates work together to modulate overall Ca2+ handling?
Can CaMKII enhance its own activity via a CaMKII-Ca2+-CaMKII positive feedback loop?
Does β-adrenergic stimulation increase dyadic CaMKII activity via enhanced Ca2+ fluxes and, if so, does this additional activity contribute to inotropic responses?
Finally, under what conditions can CaMKII activity lead to the development of cellular arrhythmias?
The model shows how synergy between individual CaMKII activities modulates overall myocyte Ca2+ handling, that CaMKII-PKA synergy may contribute to full adrenergic responses, and that CaMKII-dependent modulation of RyR function can trigger arrhythmias.
Methods
Dynamic CaMKII and PKA-dependent phosphorylation were modeled using mass action and Michaelis-Menten kinetics (11). PKA phosphorylation modules are essentially the same as those in our previous model (9). Model parameters were obtained from the biochemical literature or estimated by nonlinear least-squares fitting to cellular data as described in the Supporting Material.
Structurally, the model consists of three main modules:
-
1.
Activation of CaMKII and phosphorylation of ECC targets,
-
2.
β-adrenergic activation of PKA and phosphorylation, and
-
3.
The cardiac ECC model of the rabbit ventricular myocyte from Shannon et al. (10), including detailed cellular electrophysiology, Ca2+ diffusion between distinct compartments, and graded Ca2+ release from the sarcoplasmic reticulum (SR), in which regulation by modules 1 and 2 was included.
To model the effects of CaMKII and PKA on LCC modal gating (12,13), we adapted an established Markovian formulation for the LCC (14) (see Fig. S1 in the Supporting Material). All simulations were performed in MATLAB (The MathWorks, Natick, MA) using the stiff ODE solver ode15s.
Phosphorylation modules
LCC
CaMKII phosphorylation rapidly facilitates LCC current (ICa), seen as a stepwise increase in peak ICa with repeated stimulation (Fig. S2) (13,15,16). CaMKII alters LCC gating by increasing the fraction of channels operating in mode 2, characterized by high Po (13). Facilitation is enhanced during CaMKII overexpression (8) and eliminated during CaMKII knockout. Additional properties of CaMKII-dependent changes to ICa are provided in Fig. S3. PKA increases LCC Po and availability via phosphorylation of α1C and β2a sites, respectively (9).
RyR
CaMKII phosphorylation of RyRs (mainly at Ser2815 in rabbit) has been reported to increase channel Ca2+ sensitivity, opening probability, and leak (17–20). Basal phosphorylation and biphasic dephosphorylation by PP1 and PP2A at Ser2815 were included by fitting to experimental data of increasing phosphatase inhibition by okadaic acid (Fig. S4). PKA also enhances RyR Ca2+ sensitivity during adrenergic stimulation.
PLB, INa, and Ito
CaMKII phosphorylation of PLB (at Thr17) may be <5% in normal conditions (21). In our model, cytosolic CaMKII is not sufficient to regulate PLB (3). To achieve appropriate frequency-dependent Thr17 phosphorylation (22), we modeled this using dyadic cleft CaMKII signals (see the Supporting Material). PLB phosphorylation by both CaMKII and PKA (at Ser16) relieves SERCA inhibition and roughly doubles the pump's Ca2+ sensitivity (22–24). Also, PKA activation of inhibitor-1 blocks PP1 near PLB, thereby increasing Thr17 phosphorylation during adrenergic stimulation (Fig. S6). Lastly, CaMKII overexpression in rabbit myocytes delays INa recovery from inactivation, enhances INa,L magnitude (Fig. S7 A), hastens Ito recovery from inactivation (Fig. S8 A), and increases Ito magnitude (Fig. S8 B) (25,26).
Others
PKA-specific regulation of the slow delayed rectifier K+ current (IKs), troponin I, and the cystic fibrosis transmembrane conductance regulator Cl− current were included. An overall model schematic is provided in Fig. 1, with further details of model equations, parameters, and justification provided in Table S1 in the Supporting Material.
Figure 1.
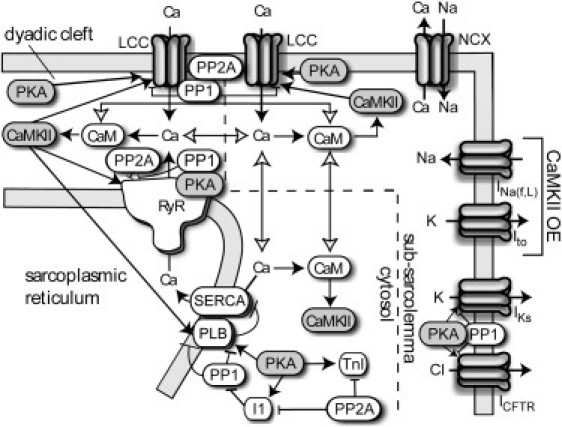
Model schematic. CaMKII is activated by Ca2+/CaM binding in the dyadic cleft, subsarcolemma, and cytosolic compartments. Active CaMKII phosphorylates LCCs, RyRs, and PLB. During overexpression simulations, CaMKII-dependent alterations to INa(f,L) and Ito were included. PKA phosphorylates LCCs, RyRs, PLB, Inhibitor-1, troponin I (TnI), IKs, and cystic fibrosis transmembrane conductance regulator (CFTR) and phosphatases 1 and 2A oppose phosphorylation by either kinase.
Results
Frequency-dependence of CaMKII-mediated phosphorylation
Phosphorylation of all CaMKII substrates is predicted to exhibit positive frequency dependence, though phosphorylation levels and kinetics are quantitatively quite distinct (Fig. 2). The model predicts highly dynamic CaMKII activity and oscillatory phosphorylation kinetics, which adapt to changes in pacing with distinct kinetics (Fig. 2 A). LCCs exhibit moderate to high phosphorylation during control scenarios (Fig. 2 B). LCC phosphorylation reaches steady state within ∼5 beats when the myocyte is paced from rest, consistent with ICa facilitation kinetics in Fig. S2. With CaMKII overexpression (CaMKII-OE, 6× increase), LCC phosphorylation saturates at lower frequencies. In contrast, RyR and PLB phosphorylation exhibit moderate positive frequency-dependence in control conditions. RyR phosphorylation increases from ∼15% at rest to ∼29% at 3 Hz pacing (Fig. 2 C), while PLB phosphorylation is <10% at all frequencies (Fig. 2 D). During CaMKII-OE, however, RyR and PLB phosphorylation is amplified. The predicted increase in RyR phosphorylation with CaMKII-OE at 1 Hz is similar to that seen experimentally (8) (Fig. S5). Together, these predictions indicate that the frequency-dependence of CaMKII substrates may vary considerably even when regulated by the same Ca2+ and CaMKII signals.
Figure 2.

Time-course and frequency-dependence of CaMKII phosphorylation. (A) Dyadic CaMKII activity and phosphorylation profiles during a CaMKII-OE simulation at low (0.5 Hz) and higher (2 Hz) frequency pacing. Note the differences between phosphorylation kinetics, which are very fast at the LCC, slower at the RyR, and very slow at PLB. (B–D) Time-averaged steady-state phosphorylation levels at (B) LCC, (C) RyR, and (D) PLB are frequency-dependent and sensitive to the amount of active CaMKII (control versus OE).
CaMKII-dependent changes to Ca2+ handling and electrophysiology
At 1 Hz pacing, Ca2+ transient magnitude (Δ[Ca2+]i) was 402 nM in control simulations, though CaMKII-OE slightly reduced Δ[Ca2+]i to 366 nM (∼91% of control) (Fig. 3 A). Experimental Ca2+ transients showed a similar, though statistically insignificant, decrease in Δ[Ca2+]i with acute CaMKII-OE (72 ± 8% of control) (Fig. 3 C, left columns). With CaMKII-KO, the model predicted a decrease in Δ[Ca2+]i to 281 nM (Fig. 3 A, dashed line). Both CaMKII-OE and CaMKII-KO reduced action potential duration (APD), consistent with the experimentally measured APD for CaMKII-OE but somewhat overestimating the amount of APD shortening due to CaMKII inhibition in rabbit myocytes (26) (Fig. 3 B).
Figure 3.

Effects of CaMKII on myocyte Ca2+-handling and electrophysiology. (A) At 1 Hz pacing, both CaMKII-OE and CaMKII-KO depress Δ[Ca2+]i compared to control. Diastolic [Ca2+]i for control, OE, and KO simulations was 92.6, 69.1, and 95.8 nM, respectively. (B) APD is shorter during CaMKII-OE (194 ms) and CaMKII-KO (199 ms) simulations compared to control (213 ms). (C) Comparison of Δ[Ca2+]i and [Ca2+]SRT during CaMKII-OE in model (gray bars) and experimental data (black bars, data from (8), n.s. = not significant). (D) CaMKII overexpression increases fractional release of Ca2+ from the SR (data from (8)). (E and F) Frequency-dependence of Δ[Ca2+]i and [Ca2+]SRT. (E) Model predicts slightly smaller Δ[Ca2+]i at larger frequencies (≥1 Hz) with CaMKII-OE and depressed Δ[Ca2+]i at all frequencies during CaMKII-KO. (F) CaMKII-OE dramatically decreases SR Ca2+ across all frequencies, while CaMKII-KO allows greater SR filling at higher frequencies.
Total SR Ca2+ content ([Ca2+]SRT) was reduced in response to CaMKII-OE in both model predictions (∼23%) and published experiments (∼40 ± 15%) (Fig. 3 C, right). Additionally, fractional SR Ca2+ release was enhanced in both model predictions (127% of control) and experimental data (158 ± 23% of control). Absolute fractional release values at 1 Hz pacing are also in agreement with experimental data (Fig. 3 D). The model exhibits the positive frequency dependence of Δ[Ca2+]i (Fig. 3 E) and [Ca2+]SRT (Fig. 3 F) observed in rabbit ventricular myocytes (though SR content changes only slightly beyond 1 Hz) (10). During CaMKII-OE, however, Δ[Ca2+]i and [Ca2+]SRT progressively deviate from control values. At low frequencies (0.25 and 0.5 Hz), Δ[Ca2+]i slightly rises in response to CaMKII-OE, though the relationship flips at frequencies ≥1 Hz. These predictions can be explained by the quantitative differences between phosphorylation levels at low and high frequencies (Fig. 2). Also, CaMKII-KO reduced Δ[Ca2+]i despite a higher SR Ca2+ content, suggesting the potential role of CaMKII in the positive frequency dependence of Δ[Ca2+]i in normal myocytes.
As additional validations, postrest decay was enhanced by CaMKII-OE (as seen in similar experiments (8), Fig. S10) and [Na]i was slightly greater at all tested frequencies during CaMKII-OE (though larger increases have been observed experimentally (25), perhaps due to additional mechanisms; see Fig. S7, B and C). The included effects on INa(f,L) and Ito in combination contributed to slightly larger [Ca2+]i transients during pacing (Fig. S9). These results demonstrate that our model is capable of predicting key changes to myocyte Ca2+ handling and electrophysiology that have been shown experimentally with CaMKII-OE.
Role of CaMKII in FDAR and ECC gain
CaMKII is believed to participate in frequency-dependent acceleration of relaxation (FDAR), though the mechanisms for such control are unclear and may not involve PLB (21,27,28) or SERCA (29). Between 0.5 and 3 Hz, we found that steady-state FDAR (quantified as the t50 of Δ[Ca2+]i relaxation) was only modestly CaMKII-dependent (CaMKII-OE and KO made the slope of FDAR slightly more and less negative, respectively; see Fig. S11). As an additional test for CaMKII-dependence, we monitored the time course of FDAR development during a rapid switch from 0.25 to 1 Hz pacing (see Fig. 4 A and Fig. S12). As expected, our control model rapidly adapted to faster pacing. However, the early phase of FDAR was blunted in CaMKII-KO simulations, suggesting CaMKII contributes to the early phase of FDAR. Additional simulations were performed with CaMKII-dependent LCC or RyR phosphorylation eliminated (Fig. S12, right panels), showing that LCC phosphorylation is necessary for this early phase. Steady-state FDAR was enhanced by elimination of RyR phosphorylation but reduced by elimination of LCC phosphorylation (Fig. S12 F). Thus, overall FDAR appears to depend on a balance of LCC and RyR phosphorylation by CaMKII.
Figure 4.
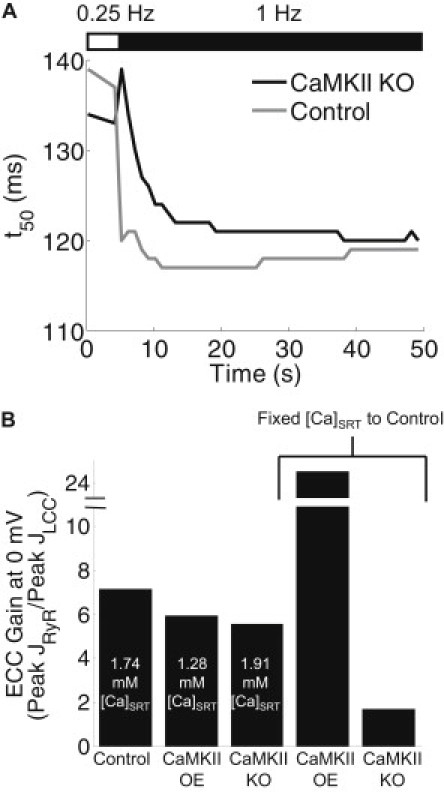
CaMKII-dependent enhancement of FDAR via LCC phosphorylation and effects on ECC gain. (A) Time course of Δ[Ca2+]it50 values in response to increased pacing frequency (0.25–1 Hz at 4 s). Control simulations showed rapid FDAR adaptation, though CaMKII-KO slowed early FDAR. (B) ECC gain assessed by voltage-clamp (−80 mV holding potential, 0 mV test for 200 ms, 500 ms basic cycle length) until steady state was reached. ECC gain is reported as peak steady-state JRyR over peak steady-state JLCC. ECC gain was reduced by both CaMKII-OE and CaMKII-KO simulations, though SR loads varied considerably between conditions. The last two bars are from simulations with SR content fixed to the control value. In this case, CaMKII-OE dramatically increased ECC gain at 0 mV (from 7.2 to 25), while CaMKII-KO decreased ECC gain (to 1.7) compared to control.
ECC gain, defined by the ratio of peak RyR Ca2+ flux to peak trigger LCC Ca2+ flux, is a central element of ECC. To determine whether CaMKII modulates ECC gain, we paced the model myocyte under voltage-clamp to steady state (−80 mV holding potential, 0 mV test, 2 Hz) and quantified peak LCC and RyR fluxes (JLCC and JRyR, respectively). Surprisingly, the control model exhibited the highest ECC gain, despite increased ICa amplitude and RyR Po in the CaMKII-OE model (Fig. 4 B, left columns). However, [Ca2+]SRT was not equal across the three tested conditions (see labels in Fig. 4 B). To eliminate the influence of SR loading on these results, we ran additional simulations for CaMKII-OE and KO at fixed [Ca2+]SRT (1.74 mM, the control level) and found that, in these conditions, CaMKII-OE dramatically enhanced ECC gain, while gain in the KO condition was depressed substantially (Fig. 4 B, right columns).
Comparative analysis of CaMKII target effects
We used the model to further examine how individual CaMKII substrates produce the combined effects discussed earlier. Individual substrate contributions to the overall CaMKII-OE response at 1 Hz were isolated by overexpressing CaMKII and allowing CaMKII-dependent phosphorylation of a single substrate; the results are summarized in Table 1. CaMKII enhancement of ICa in isolation mainly enhanced Ca2+ transients (∼31%). On the other hand, RyR effects substantially decreased Δ[Ca2+]i (∼24%) and [Ca2+]SRT (∼23%), while PLB phosphorylation alone modestly affected all parameters, perhaps due to the low quantitative phosphorylation levels at PLB-Thr17. Net Ito and INa effects increased Δ[Ca2+]i (∼7%) and decreased APD (∼12%). Thus, the overall decreased Δ[Ca2+]i and [Ca2+]SRT seen with CaMKII-OE may be primarily explained by increased phosphorylation of RyRs, although LCC phosphorylation largely compensates for the decreased Δ[Ca2+]i and, to a lesser extent, [Ca2+]SRT. Interestingly, all targets positively influenced fractional release to a small extent, but the overall 27% increase is only seen when combining all effects. This indicates that global changes to ECC are dictated by the relative strengths of several CaMKII interactions working in synergy. Results from 1 Hz CaMKII-KO simulations are also shown for comparison.
Table 1.
Comparative analysis of individual CaMKII interactions
| Control |
CaMKII overexpression |
KO |
|||||
|---|---|---|---|---|---|---|---|
| Parameter | All targets | All targets | LCC effect only | RyR effect only | PLB effect only | Ito/INa effects only | All targets |
| Δ[Ca]i (nM) | 402 | 365 | 525 | 306 | 414 | 431 | 281 |
| [Ca]SRT (mM) | 1.87 | 1.44 | 1.91 | 1.44 | 1.88 | 1.86 | 1.93 |
| Fractional release | 0.34 | 0.43 | 0.39 | 0.39 | 0.35 | 0.35 | 0.26 |
| APD90 (ms) | 213 | 194 | 220 | 213 | 210 | 190 | 199 |
Effects of overexpressing CaMKII for individual targets (LCC, RyR, PLB, and Ito/INa) on overall Δ[Ca2+]i, [Ca2+]SRT, fractional release, and APD during 1 Hz pacing. RyR phosphorylation is the only effect responsible for decreasing Δ[Ca2+]i and [Ca2+]SRT, while no one effect dominates the overall increase in fractional release. The net effect of Ito and INa regulation mostly contributes to APD shortening. The effects of global CaMKII inhibition are provided in the last column.
CaMKII-Ca2+-CaMKII positive feedback during normal ECC and β-adrenergic stimulation
We hypothesized that positive feedback from CaMKII to Ca2+ to CaMKII (via enhanced Ca2+ fluxes) may be a prominent feedback loop. To test this in the model, we analyzed dyadic CaMKII activity during control and CaMKII-OE simulations (Fig. 5 A). At 2 Hz pacing, CaMKII-OE increased average CaMKII activity (59.5% vs. 53.8% for control) and t50% (222 vs. 210 ms). To test whether this increased activity is the result of feedback as opposed to the fact that overall CaMKII availability is enhanced, additional CaMKII-OE simulations were run with phosphorylation levels at each CaMKII substrate fixed to their steady-state diastolic levels (CaMKII-OE + Target KO). In this case, CaMKII average activity and t50% were 57.5% and 216 ms, respectively, suggesting that overexpression of CaMKII alone enhances activity, though the subsequent CaMKII-dependent enhancement of Ca2+ fluxes feeds back to increase activity further.
Figure 5.
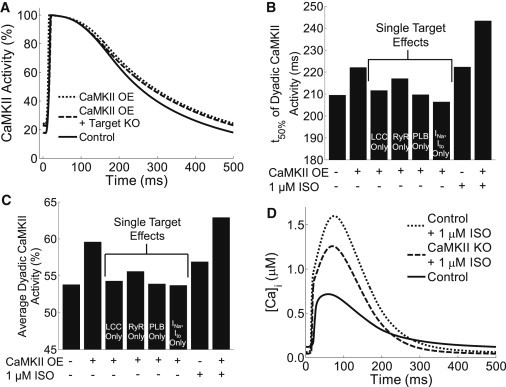
CaMKII-Ca2+-CaMKII feedback and β-adrenergic response. (A) Single 2 Hz dyadic cleft CaMKII transients from control, CaMKII-OE, and CaMKII-OE + Target KO (i.e., all target phosphorylation levels fixed to control values) simulations. Feedback is evident because t50 and average CaMKII activity increase with overexpression alone, though CaMKII-OE with all functional targets present increases these parameters further. (B and C) Analysis of individual target contributions to enhanced CaMKII activity. ISO also increased t50 and average activity of CaMKII. (D) ISO substantially increases [Ca2+]i transients during 2 Hz pacing, though the full response depends on synergy between CaMKII and PKA substrates.
We next evaluated how much each individual CaMKII substrate contributed to overall CaMKII-Ca2+-CaMKII feedback. Interestingly, most CaMKII substrates contributed to the overall feedback (except Ito and INa), with RyR phosphorylation producing the strongest individual effect. However, all targets working together produced the strongest response (Fig. 5, B and C, middle columns). Increases in Ca2+ fluxes by other kinases may enhance CaMKII activity in a similar manner. To test whether β-adrenergic signaling through PKA enhances CaMKII activity, we ran additional simulations with 1 μM isoproterenol (ISO) (see Fig. S13 for sample outputs from the β-adrenergic model). Through PKA enhancement of Ca2+ fluxes, CaMKII activity and t50% were enhanced, which was further potentiated by CaMKII-OE (Fig. 5, B and C, right columns).
We next tested whether this feedback loop contributes to the increased Ca2+ transients during β-adrenergic stimulation. As expected for the control model, ISO significantly enhanced Ca2+ transients (∼2.6-fold) (Fig. 5 D). But when CaMKII was eliminated (CaMKII-KO), the ISO response was partially diminished. We found that the change in Ca2+ transients from CaMKII-KO to “control + ISO” models (Δ[Ca2+]i of 1181 nM) was larger than the sum of the Ca2+ transient increases from CaMKII-KO to control (Δ[Ca2+]i = 239 nM) and “CaMKII-KO + ISO” (Δ[Ca2+]i = 860 nM), indicating synergy between PKA and CaMKII in the overall β-adrenergic response. Thus, this feedback loop appears to be both stimulated by and partially contribute to the response of Ca2+ transients to β-adrenergic stimulation.
Arrhythmogenesis
Abnormal CaMKII signaling is suggested to be proarrhythmogenic (5,6,30), so the model was used to test whether the included mechanisms could predict such scenarios. The model was stimulated to steady state with 2 Hz pacing and spontaneous activity was monitored after abrupt stimulus removal. Control simulations with 1 μM ISO show [Ca2+]i (Fig. 6 A) and membrane potential (Fig. 6 B) profiles that return to rest after stimulus removal. With CaMKII-OE, however, peak [Ca2+]i is reduced by ∼36% and delayed after-depolarizations (DADs) are seen after stimulus removal. Typically, DADs occur after repolarization of the membrane has completed and are triggered by aberrant release of Ca2+ from the SR (30). In the model, enhanced RyR Ca2+ sensitivity together with rising [Ca2+]SR increases RyR leak. Initially, RyR leak slowly increases dyadic [Ca2+] and begins to decrease [Ca2+]SR once Jleak > JSERCA. But once dyadic [Ca2+] rises sufficiently, it triggers RyR opening, causing sharp increases in dyadic Ca2+ and then subsarcolemmal [Ca2+]. Increased subsarcolemmal [Ca2+] stimulates inward Na+/Ca2+ exchange (INCX), resulting in membrane depolarization (DADs).
Figure 6.

CaMKII overexpression induces DADs. (A and B) After abrupt removal of a >60 s, 2 Hz stimulus (see arrows labeled stimuli), DADs are observed in CaMKII-OE simulations with ISO. Note that CaMKII-OE also reduces Δ[Ca2+]i in response to ISO (A). SR Ca2+ release during each DAD was assessed by integrating total RyR flux during afterdepolarizations. The smaller 65-μM release event produced a subthreshold DAD, while the larger 86-μM release induced a full AP (B). Both values fall on either side of our model's threshold of ∼70 μM for generation of a full AP, which is quantitatively consistent with an experimentally measured value of ∼64 μM (31).
To validate the predicted relationship between SR Ca2+ release and DAD generation, we induced graded SR Ca2+ release events after 2-Hz pacing during CaMKII-OE + ISO and measured subsequent changes in membrane potential. We found that our model has a full DAD threshold of ∼70 μmol/L cytosol Ca2+, which is quantitatively consistent with an experimentally measured threshold of ∼64 μmol/L cytosol (31) (see Fig. S14 for details of this analysis).
Interestingly, CaMKII-OE + ISO produced DADs only at frequencies ≥2 Hz. During CaMKII-OE alone at 2 Hz, diastolic RyR leak (Jleak) increased from ∼5 to 12 μmol/L cytosol/s, though [Ca2+]SRT was insufficient for spontaneous release (Fig. 3 F). However, the addition of ISO during CaMKII-OE increased [Ca2+]SRT. This effect, coupled with CaMKII-induced increases in RyR Ca2+ sensitivity and Jleak (∼15 μM/s in this case), induced spontaneous RyR opening that triggered the predicted DADs. At 3 Hz, increased RyR phosphorylation further lowered the threshold for SR release, triggering a subthreshold DAD even in the absence of ISO (data not shown). With ISO alone in the control model, Jleak increased only slightly (from ∼5 to ∼7 μM/s) and no DADs were produced. If CaMKII phosphorylation of RyRs is specifically eliminated during CaMKII-OE + ISO, no DADs are seen (Fig. S15). In contrast, if PKA-mediated phosphorylation of RyRs is eliminated, DADs are still observed.
Discussion
We incorporated CaMKII and PKA-dependent regulation of cardiac myocyte Ca2+ handling into a computational model of the rabbit ventricular myocyte. From this study, we were able to obtain new mechanistic insights regarding how these kinases coordinate control of excitation-contraction coupling. We also explored scenarios where CaMKII-induced dysregulation of ECC promoted cellular arrhythmias.
Several groups have developed models of CaMKII-dependent regulation of ECC. Hund and Rudy (32) found that ECC gain increases with frequency due to CaMKII, leading to a positive Ca2+ transient-frequency relationship. Grandi et al. (33) showed that static CaMKII-dependent modulations to ICa, Ito, and INa have distinct functional consequences on APD, though their collective activity promotes AP shortening. Hund et al. (34) showed that CaMKII hyperactivity and effects on INa contribute to abnormal Ca2+ homeostasis and AP upstroke velocity in infarct border zones. Hashambhoy et al. (35) developed a detailed, stochastic model of CaMKII-LCC interactions, finding that CaMKII-induced redistribution of LCC gating modes explains ICa facilitation.
Our model builds on this past work, providing a number of methodological advances that led to new mechanistic insights. Methodological advances include:
-
1.
Development of biochemically detailed phosphorylation models of LCCs, RyRs, and PLB regulated by Ca2+/CaM-activated CaMKII.
-
2.
Integration of these phosphorylation modules with well-validated models of β-adrenergic signaling and ECC.
-
3.Successful validation of our model against a number of experimental readouts, including CaMKII effects on
- i. LCC facilitation and recovery from inactivation;
- ii. RyR and PLB phosphorylation;
- iii. SR Ca2+ content;
- iv. fractional release;
- v. Ca2+ transient amplitude;
- vi. APD;
- vii. FDAR; and
- viii. postrest decay.
Using this quantitative framework, model analysis provided a number of new mechanistic insights, showing how:
-
1.
CaMKII substrates are differentially phosphorylated in a context-dependent manner.
-
2.
Interactions among CaMKII substrates determine overall consequences to myocyte Ca2+ handling and APD.
-
3.
CaMKII participates in a CaMKII-Ca2+-CaMKII positive feedback loop that is potentiated by enhanced Ca2+ fluxes through PKA signaling.
-
4.
Synergy between PKA and CaMKII, including effects on inhibitor-1, facilitates full responses to β-adrenergic stimulation.
-
5.
CaMKII is responsible for early adaptation of FDAR via LCC phosphorylation.
-
6.
CaMKII modulates ECC gain, though this influence is highly dependent on SR Ca2+.
-
7.
CaMKII hyperphosphorylation of RyRs is arrhythmogenic and further potentiated by adrenergic stimulation.
CaMKII substrate phosphorylation and regulation of Ca2+ handling
While there is limited kinetic data regarding CaMKII phosphorylation at the modeled targets, our quantitative steady-state levels are generally consistent with available experimental measurements (see the Supporting Material) (20,21). At the LCC, we assumed quick kinetics to mimic rapid CaMKII-dependent ICa facilitation (13,15,16). As suggested by a number of experiments (21,29), we predicted slower kinetics at PLB that resulted in quantitatively small phosphorylation levels. While we predicted somewhat slow RyR kinetics, some data suggests that CaMKII-dependent RyR phosphorylation occurs on the order of ∼3 min (21). Such slow kinetics are difficult to reconcile with the much faster functional consequences of ICa facilitation (13,15,16), given that RyRs are concentrated near LCCs in dyads. Further experimental work is needed to clarify these issues. Also, data regarding CaMKII-dependent RyR regulation is somewhat conflicting (36,37), though our results indicate that positive regulation is critical for predictions of reduced SR Ca2+ content and enhanced leak during CaMKII-OE (as in experiments (11)).
Our model reproduced a range of data from rabbit myocytes acutely overexpressing CaMKII, including Ca2+ transient amplitudes, [Ca2+]SRT, fractional release, APD, changes in [Na]i, postrest decay, and FDAR (8,25,26). In transgenic mouse models, similar results are observed (in terms of Ca2+ sparks and [Ca2+]SRT) though these animals show dramatically depressed Ca2+ transients and longer APD. In addition to species differences, these myocytes exhibit compensatory changes in protein expression including decreased RyR, PLB, and SERCA with increased Na+/Ca2+ exchanger, that are not observed in the rabbit experiments. These alterations may partially explain phenotypic differences between these animal models. Koivumäki et al. (38) recently modeled CaMKII-OE and found that inclusion of these secondary expression changes was critical for reproducing the mouse phenotype. Also, our predicted changes in [Na]i are qualitatively similar but smaller than the levels measured experimentally (25), suggesting that CaMKII may regulate [Na]i through additional mechanisms.
We predicted that CaMKII-KO decreases steady-state Δ[Ca2+]i and APD compared with control conditions. But in CaMKII-KO or CaMKII inhibited mice, Δ[Ca2+]i is normal (39,40), though secondary compensatory mechanisms play a role in maintaining Ca2+ handling (40). In Wagner et al. (26), the CaMKII inhibitor AIP somewhat reduced APD in normal myocytes but reversed the AP shortening effect of CaMKII-OE. The subtle model-experiment differences here may be due to partial CaMKII inhibition by AIP but complete CaMKII elimination in our model.
We provided additional evidence for the role of CaMKII in early FDAR and ECC gain. Several experiments have indicated that FDAR adaptation and PLB-Thr17 phosphorylation are temporally dissimilar (21,29). Our model agrees with these findings, where FDAR is rapid and PLB phosphorylation is slow and quantitatively small in control scenarios. However, we found that CaMKII-KO slowed FDAR and that LCC phosphorylation appeared to facilitate this quick adaptation. In addition, CaMKII-OE made the slope of steady-state FDAR more negative (consistent with experiments (7)), while the converse was true for CaMKII-KO. CaMKII inhibition either strongly inhibits (27), partially inhibits (8), or fails to affect (29) steady-state FDAR in experiments; our included mechanisms support findings of only slight CaMKII-dependence on steady-state FDAR. We also showed that the presence of CaMKII enhances ECC gain, though gain was smaller during CaMKII-OE. However, gain is highly SR-load-dependent (10), so we tested our model in scenarios of matched SR load. We found that CaMKII-OE dramatically enhanced gain, while CaMKII-KO produced the opposite effect. Thus, CaMKII can enhance gain, though this effect may be masked in conditions of low SR Ca2+.
Role of CaMKII-Ca2+-CaMKII feedback
The model predicted that the three dynamic CaMKII substrates contribute to CaMKII-Ca2+-CaMKII feedback, though RyR phosphorylation was most prominent. CaMKII activity was also enhanced in simulations with the β-adrenergic agonist ISO due to enhanced Ca2+ fluxes via PKA phosphorylation. We found that CaMKII helped facilitate overall β-adrenergic enhancement of Ca2+ transients via synergy between PKA and CaMKII-dependent phosphorylation. Specifically, it appeared that PKA-dependent increases in LCC and SERCA flux, combined with CaMKII-dependent RyR fluxes, were a major source of crosstalk. PKA activation of inhibitor-1, another route of crosstalk between these pathways, substantially increased Thr17 PLB phosphorylation independent of enhanced Ca2+ signals.
Interestingly, Curran et al. (19) found that the CaMKII inhibitor KN-93 eliminated ISO-induced enhancement of peak [Ca2+]i in rabbit myocytes, in accordance with our results. However, KN-93 can inhibit ICa in a CaMKII-independent manner which complicates these findings (41). In another study, transgenic mice expressing a CaMKII inhibitor (AC3-I) produced full adrenergic responses, though these results are complicated by secondary PKA-dependent increases in ICa (40). Our results thus suggest that, excluding secondary compensatory mechanisms, CaMKII can play a role in facilitating inotropic responses to acute adrenergic stimulation.
Arrhythmogenesis
Hyperactive CaMKII is suspected to be proarrhythmogenic, due mainly to its ability to enhance LCC gating and RyR sensitivity (5,30). We found that CaMKII-OE + ISO with ≥2 Hz pacing or CaMKII-OE alone with ≥3 Hz pacing produced DADs after abrupt stimulus removal. Use of a well-validated model of the RyR (in terms of open probability, leak, fractional release, and response to agonists (e.g., caffeine)) and a Na+/Ca2+ exchanger model that responds to subsarcolemmal Ca2+ signals was critical for our analysis (10). Our model demonstrates that CaMKII-dependent increases in RyR Ca2+ sensitivity and leak can lower the threshold for spontaneous SR Ca2+ release, while ISO-induced enhancement of [Ca2+]SRT can potentiate these effects. We also showed that the threshold amount of Ca2+ release required to trigger a full DAD is consistent with experimental measurements (31). In our simulations, elimination of CaMKII's ability to hyperphosphorylate the RyR (i.e., fixing phosphorylation to control levels) eliminated DADs, with less influence from PKA-dependent RyR phosphorylation. Sag et al. (42) demonstrated that β-adrenergic stimulation of CaMKII-OE mouse myocytes increased the number of DADs, consistent with our model predictions. While DADs involve the complex interplay of several mechanisms, the model illustrates a quantitatively significant contribution from CaMKII-dependent hyperphosphorylation of RyRs.
While early after-depolarizations (EADs) have been linked to abnormal CaMKII signaling (5,6,42), EADs were not observed in our model, even during adrenergic stimulation. CaMKII is believed to induce EADs through its interactions with LCCs (5). Though LCC-mediated EADs can be generated using deterministic computational models (43), the CaMKII-dependent LCC alterations included here, with and without adrenergic stimulation, were insufficient. Stochastic models of LCC modal gating can trigger EADs (44), which may suggest that EAD formation via CaMKII is sensitive to stochasticity. CaMKII-RyR interactions may also be sensitive to stochasticity. Also, the mechanism of DAD formation could be sensitive to model formulation (e.g., a single dyad versus multiple, independent dyads). Given that our CaMKII-RyR model is sufficient to predict global changes in [Ca2+]SRT and SR leak seen experimentally and the much larger computational burden for stochastic simulations, our deterministic model appears to be an appropriate first step for analyzing changes in Ca2+ handling that may lead to arrhythmia.
Limitations
While we were careful to focus on well-validated interactions constrained by quantitative measurements, uncertainties in the exact mechanisms underlying the functional consequences of these interactions are unavoidable. In our CaMKII model, basal kinase activity is low (<1%) due to limited Ca2+/CaM binding at diastolic Ca2+ levels and high [PP1] in the dyadic cleft. However, increased Ca2+ spark frequency during CaMKII-OE indirectly suggests that CaMKII is partially active during rest (7,8,45). This discrepancy could be due to a change in the balance of kinase and phosphatase activities (3), but additional experimental data and analysis are needed.
While we included several CaMKII activation mechanisms (e.g., Ca2+/CaM-dependent, autonomous, ECC feedback, and ISO), there are still other Ca2+-independent mechanisms that control CaMKII activity. Erickson et al. (46) demonstrated that CaMKII can be oxidized at methionine residues (281/282), conferring additional memory to CaMKII. Christensen et al. (47) modeled CaMKII oxidation, showing increased activity in infarct border zones that was predicted to reduce conduction velocity and increase susceptibility to conduction block. CaMKII can also autophosphorylate at Thr306 after Thr287 phosphorylation, but Thr287 autophosphorylation is not prominent in our simulations. In addition, CaMKII can be activated during β1-specific adrenergic stimulation via formation of a β-arrestin-CaMKII-Epac1 complex, which may confer additional signaling potential to CaMKII during adrenergic stimulation (48). These additional activation mechanisms may be examined in subsequent studies.
Acknowledgments
This work was supported by grant No. HL094476 from the National Institutes of Health and grant No. 0830470N from the American Heart Association.
Supporting Material
References
- 1.Couchonnal L.F., Anderson M.E. The role of calmodulin kinase II in myocardial physiology and disease. Physiology (Bethesda) 2008;23:151–159. doi: 10.1152/physiol.00043.2007. [DOI] [PubMed] [Google Scholar]
- 2.Maier L.S., Bers D.M. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc. Res. 2007;73:631–640. doi: 10.1016/j.cardiores.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Saucerman J.J., Bers D.M. Calmodulin mediates differential sensitivity of CaMKII and calcineurin to local Ca2+ in cardiac myocytes. Biophys. J. 2008;95:4597–4612. doi: 10.1529/biophysj.108.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoch B., Meyer R., Karczewski P. Identification and expression of δ-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ. Res. 1999;84:713–721. doi: 10.1161/01.res.84.6.713. [DOI] [PubMed] [Google Scholar]
- 5.Anderson M.E., Braun A.P., Sung R.J. KN-93, an inhibitor of multifunctional Ca2+/calmodulin-dependent protein kinase, decreases early afterdepolarizations in rabbit heart. J. Pharmacol. Exp. Ther. 1998;287:996–1006. [PubMed] [Google Scholar]
- 6.Wu Y., Temple J., Anderson M.E. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106:1288–1293. doi: 10.1161/01.cir.0000027583.73268.e7. [DOI] [PubMed] [Google Scholar]
- 7.Maier L.S., Zhang T., Bers D.M. Transgenic CaMKIIδC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ. Res. 2003;92:904–911. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- 8.Kohlhaas M., Zhang T., Maier L.S. Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circ. Res. 2006;98:235–244. doi: 10.1161/01.RES.0000200739.90811.9f. [DOI] [PubMed] [Google Scholar]
- 9.Saucerman J.J., Healy S.N., McCulloch A.D. Proarrhythmic consequences of a KCNQ1 AKAP-binding domain mutation: computational models of whole cells and heterogeneous tissue. Circ. Res. 2004;95:1216–1224. doi: 10.1161/01.RES.0000150055.06226.4e. [DOI] [PubMed] [Google Scholar]
- 10.Shannon T.R., Wang F., Bers D.M. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys. J. 2004;87:3351–3371. doi: 10.1529/biophysj.104.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saucerman J.J., McCulloch A.D. Mechanistic systems models of cell signaling networks: a case study of myocyte adrenergic regulation. Prog. Biophys. Mol. Biol. 2004;85:261–278. doi: 10.1016/j.pbiomolbio.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Yue D.T., Herzig S., Marban E. β-adrenergic stimulation of calcium channels occurs by potentiation of high-activity gating modes. Proc. Natl. Acad. Sci. USA. 1990;87:753–757. doi: 10.1073/pnas.87.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dzhura I., Wu Y., Anderson M.E. Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat. Cell Biol. 2000;2:173–177. doi: 10.1038/35004052. [DOI] [PubMed] [Google Scholar]
- 14.Mahajan A., Shiferaw Y., Weiss J.N. A rabbit ventricular action potential model replicating cardiac dynamics at rapid heart rates. Biophys. J. 2008;94:392–410. doi: 10.1529/biophysj.106.98160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grueter C.E., Abiria S.A., Colbran R.J. L-type Ca2+ channel facilitation mediated by phosphorylation of the β-subunit by CaMKII. Mol. Cell. 2006;23:641–650. doi: 10.1016/j.molcel.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 16.Yuan W., Bers D.M. Ca-dependent facilitation of cardiac Ca current is due to Ca-calmodulin-dependent protein kinase. Am. J. Physiol. 1994;267:H982–H993. doi: 10.1152/ajpheart.1994.267.3.H982. [DOI] [PubMed] [Google Scholar]
- 17.Wehrens X.H., Lehnart S.E., Marks A.R. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 2004;94:e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 18.Ai X., Curran J.W., Pogwizd S.M. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 19.Curran J., Hinton M.J., Shannon T.R. β-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ. Res. 2007;100:391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- 20.Huke S., Bers D.M. Ryanodine receptor phosphorylation at Serine 2030, 2808, and 2814 in rat cardiomyocytes. Biochem. Biophys. Res. Commun. 2008;376:80–85. doi: 10.1016/j.bbrc.2008.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huke S., Bers D.M. Temporal dissociation of frequency-dependent acceleration of relaxation and protein phosphorylation by CaMKII. J. Mol. Cell Cardiol. 2007;42:590–599. doi: 10.1016/j.yjmcc.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hagemann D., Kuschel M., Xiao R.P. Frequency-encoding Thr17 phospholamban phosphorylation is independent of Ser16 phosphorylation in cardiac myocytes. J. Biol. Chem. 2000;275:22532–22536. doi: 10.1074/jbc.C000253200. [DOI] [PubMed] [Google Scholar]
- 23.Kranias E.G. Regulation of Ca2+ transport by cyclic 3′,5′-AMP-dependent and calcium-calmodulin-dependent phosphorylation of cardiac sarcoplasmic reticulum. Biochim. Biophys. Acta. 1985;844:193–199. doi: 10.1016/0167-4889(85)90090-4. [DOI] [PubMed] [Google Scholar]
- 24.Odermatt A., Kurzydlowski K., MacLennan D.H. The Vmax of the Ca2+-ATPase of cardiac sarcoplasmic reticulum (SERCA2a) is not altered by Ca2+/calmodulin-dependent phosphorylation or by interaction with phospholamban. J. Biol. Chem. 1996;271:14206–14213. doi: 10.1074/jbc.271.24.14206. [DOI] [PubMed] [Google Scholar]
- 25.Wagner S., Dybkova N., Maier L.S. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 2006;116:3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wagner S., Hacker E., Maier L.S. Ca/calmodulin kinase II differentially modulates potassium currents. Circ. Arrhythm. Electrophysiol. 2009;2:285–294. doi: 10.1161/CIRCEP.108.842799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Picht E., DeSantiago J., Bers D.M. CaMKII inhibition targeted to the sarcoplasmic reticulum inhibits frequency-dependent acceleration of relaxation and Ca2+ current facilitation. J. Mol. Cell Cardiol. 2007;42:196–205. doi: 10.1016/j.yjmcc.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeSantiago J., Maier L.S., Bers D.M. Frequency-dependent acceleration of relaxation in the heart depends on CaMKII, but not phospholamban. J. Mol. Cell Cardiol. 2002;34:975–984. doi: 10.1006/jmcc.2002.2034. [DOI] [PubMed] [Google Scholar]
- 29.Valverde C.A., Mundiña-Weilenmann C., Mattiazzi A. Frequency-dependent acceleration of relaxation in mammalian heart: a property not relying on phospholamban and SERCA2a phosphorylation. J. Physiol. 2005;562:801–813. doi: 10.1113/jphysiol.2004.075432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chelu M.G., Wehrens X.H. Sarcoplasmic reticulum calcium leak and cardiac arrhythmias. Biochem. Soc. Trans. 2007;35:952–956. doi: 10.1042/BST0350952. [DOI] [PubMed] [Google Scholar]
- 31.Schlotthauer K., Bers D.M. Sarcoplasmic reticulum Ca2+ release causes myocyte depolarization. Underlying mechanism and threshold for triggered action potentials. Circ. Res. 2000;87:774–780. doi: 10.1161/01.res.87.9.774. [DOI] [PubMed] [Google Scholar]
- 32.Hund T.J., Rudy Y. Rate dependence and regulation of action potential and calcium transient in a canine cardiac ventricular cell model. Circulation. 2004;110:3168–3174. doi: 10.1161/01.CIR.0000147231.69595.D3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grandi E., Puglisi J.L., Bers D.M. Simulation of Ca-calmodulin-dependent protein kinase II on rabbit ventricular myocyte ion currents and action potentials. Biophys. J. 2007;93:3835–3847. doi: 10.1529/biophysj.107.114868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hund T.J., Decker K.F., Rudy Y. Role of activated CaMKII in abnormal calcium homeostasis and INa remodeling after myocardial infarction: insights from mathematical modeling. J. Mol. Cell Cardiol. 2008;45:420–428. doi: 10.1016/j.yjmcc.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hashambhoy Y.L., Winslow R.L., Greenstein J.L. CaMKII-induced shift in modal gating explains L-type Ca2+ current facilitation: a modeling study. Biophys. J. 2009;96:1770–1785. doi: 10.1016/j.bpj.2008.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu Y., Colbran R.J., Anderson M.E. Calmodulin kinase is a molecular switch for cardiac excitation-contraction coupling. Proc. Natl. Acad. Sci. USA. 2001;98:2877–2881. doi: 10.1073/pnas.051449198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang D., Zhu W.Z., Cheng H. Ca2+/calmodulin kinase II-dependent phosphorylation of ryanodine receptors suppresses Ca2+ sparks and Ca2+ waves in cardiac myocytes. Circ. Res. 2007;100:399–407. doi: 10.1161/01.RES.0000258022.13090.55. [DOI] [PubMed] [Google Scholar]
- 38.Koivumäki J.T., Korhonen T., Tavi P. Regulation of excitation-contraction coupling in mouse cardiac myocytes: integrative analysis with mathematical modeling. BMC Physiol. 2009;9:16. doi: 10.1186/1472-6793-9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Backs J., Backs T., Olson E.N. The δ-isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc. Natl. Acad. Sci. USA. 2009;106:2342–2347. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang R., Khoo M.S., Anderson M.E. Calmodulin kinase II inhibition protects against structural heart disease. Nat. Med. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 41.Gao L., Blair L.A., Marshall J. CaMKII-independent effects of KN93 and its inactive analog KN92: reversible inhibition of L-type calcium channels. Biochem. Biophys. Res. Commun. 2006;345:1606–1610. doi: 10.1016/j.bbrc.2006.05.066. [DOI] [PubMed] [Google Scholar]
- 42.Sag C.M., Wadsack D.P., Maier L.S. Calcium/calmodulin-dependent protein kinase II contributes to cardiac arrhythmogenesis in heart failure. Circ. Heart Fail. 2009;2:664–675. doi: 10.1161/CIRCHEARTFAILURE.109.865279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeng J., Rudy Y. Early afterdepolarizations in cardiac myocytes: mechanism and rate dependence. Biophys. J. 1995;68:949–964. doi: 10.1016/S0006-3495(95)80271-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanskanen A.J., Greenstein J.L., Winslow R.L. The role of stochastic and modal gating of cardiac L-type Ca2+ channels on early after-depolarizations. Biophys. J. 2005;88:85–95. doi: 10.1529/biophysj.104.051508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo T., Zhang T., Bers D.M. Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ. Res. 2006;99:398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- 46.Erickson J.R., Joiner M.L., Anderson M.E. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Christensen M.D., Dun W., Hund T.J. Oxidized calmodulin kinase II regulates conduction following myocardial infarction: a computational analysis. PLOS Comput. Biol. 2009;5:e1000583. doi: 10.1371/journal.pcbi.1000583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mangmool S., Shukla A.K., Rockman H.A. β-Arrestin-dependent activation of Ca2+/calmodulin kinase II after β1-adrenergic receptor stimulation. J. Cell Biol. 2010;189:573–587. doi: 10.1083/jcb.200911047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.