

Abstract
Cutaneous squamous cell carcinomas (cSCCs) are the second most frequent cancers in fair-skinned populations; yet, because of their genetic heterogeneity, the key molecular events in cSCC tumorigenesis remain poorly defined. We have employed single nucleotide polymorphism microarray analysis to examine genome-wide allelic imbalance in 60 cSCCs using paired non-tumour samples. The most frequent recurrent aberrations were loss of heterozygosity (LOH) at 3p and 9p, observed in 39 (65%) and 45 (75%) tumours respectively. Microdeletions at 9p23 within the protein tyrosine phosphatase receptor type D (PTPRD) locus were identified in a total of 9 (15%) samples, supporting a tumour suppressor role for PTPRD in cSCC. In addition, microdeletions at 3p14.2 were detected in 3 (5%) cSCCs, implicating fragile histidine triad (FHIT) gene as a possible target for inactivation. Statistical analysis revealed that well-differentiated cSCCs demonstrated significantly fewer aberrations than moderately and poorly differentiated cSCCs; yet, despite a lower rate of allelic imbalance, some specific aberrations were observed equally frequently in both groups. No correlation was established between the frequency of chromosomal aberrations and immune or human papillomavirus status. Our data suggest that well differentiated tumours are a genetically distinct subpopulation of cSCC.
INTRODUCTION
Keratinocyte skin cancers, comprising basal cell carcinoma (BCC) and cutaneous squamous cell carcinoma (cSCC), represent an important cause of morbidity in fair-skinned populations and their incidence is increasing worldwide (de Vries et al., 2005). Ultraviolet radiation is the principal carcinogen implicated in keratinocyte skin cancer development (Armstrong et al., 2001). Substantial data indicate that dysregulation of the Sonic Hedgehog pathway is a key event in BCC tumorigenesis (reviewed in Boukamp, 2005). In contrast, cSCC display much more genetic heterogeneity, with several studies reporting recurrent aberrations on many chromosomes (Quinn et al., 1994; Ashton et al., 2003; Clausen et al., 2006; Purdie et al., 2007).
An etiological co-factor role in KSC has also been suggested for beta genus human papillomaviruses (HPVs). Beyond a demonstrated role in cSCC tumorigenesis in patients with the rare genetic disease epidermodysplasia verruciformis (Majewski and Jablonska, 1995) their oncogenic potential remains less well defined although the presence of beta HPV in normal skin has been shown to be a risk factor for keratinocyte skin cancers (Harwood et al., 2004). Organ transplant recipients (OTRs) receiving immunosuppression are at 100-fold increased risk of cSCC compared with immunocompetent (IC) individuals and their tumours may be more aggressive (Glover et al., 1997). It has been suggested that OTR cSCCs display fewer chromosomal aberrations than IC tumours (Rehman et al., 1997), possibly reflecting differing etiologies, but this observation remains to be confirmed.
SCC may be categorised as well, moderately and poorly differentiated depending on the degree of keratinization and cellular atypia. A greater percentage of poorly differentiated SCCs metastasize compared with well-differentiated counterparts (17% versus 0.6% respectively) (Breuninger et al, 1990). Previously, in a smaller series of tumours, we reported that well-differentiated cSCC display a lower rate of allelic imbalance than moderately or poorly differentiated tumours although certain specific aberrations occur equally frequently, suggesting that these tumours may constitute a genetically distinct subpopulation of cSCC (Purdie et al., 2007). However, this study was not sufficiently powered to enable stratification for two important potential confounding factors: HPV status and immune status. Here we have used microarray-based single nucleotide polymorphism (SNP) analysis to investigate whether a correlation exists between the genetic profile and immune status and/or HPV status of cSCC. In addition, we have used a larger series of samples to examine our previous hypothesis that well-differentiated tumours may be genetically distinct from other cSCCs.
RESULTS AND DISCUSSION
SNP microarray analysis was performed on a series of 60 primary cSCCs. Tumor DNA was analysed concomitantly with paired non-tumour DNA to permit the accurate identification of tumour-specific genetic events. Data included that from 16 cSCC previously analysed using 10K SNP microarrays (Purdie et al., 2007). Statistical analysis was restricted to gross chromosomal aberrations greater than approximately 600 kb since a comparison of data from two cSCC analysed on both 10K and 250K SNP microarrays revealed that aberrations of this size were reliably detected by both microarrays.
Histopathologic evaluation of the 60 cSCCs revealed that 29 were well-differentiated, 21 moderately differentiated and 10 poorly differentiated. Keratoacanthomas were specifically excluded from this study. Allelic imbalance was observed at 1 to 9 chromosomes (median of 5) in well-differentiated tumours compared with 5 to 21 chromosomes (median of 9) in moderately and poorly differentiated tumours (Table 1, complete data provided in Table S1). Well-differentiated cSCC had significantly fewer chromosomes displaying allelic imbalance than moderately or poorly differentiated cSCC (P< 0.001 in both cases) and a linear trend was shown to exist with respect to differentiation status (P<0.001). No significant difference existed between the numbers of chromosomes with aberrations in moderately and poorly differentiated cSCCs (P=0.645) nor was there any significant difference between these two groups when the comparison was restricted to the number of chromosomes showing LOH or gain events alone (P=1, P=0.092 respectively). Similar results were obtained when the 16 previously reported cSCCs were excluded from the analysis (Table S2). Despite a lower rate of allelic imbalance, certain specific aberrations were as frequent in well differentiated as in moderately or poorly differentiated tumours (Figure 1). For example, there was no significant difference between well-differentiated and other cSCC in the frequency of LOH at 3p (P=0.235) or gain at 3q (P=0.815) or 9q (P=0.839). Hierarchical clustering was used to investigate the genetic similarity between individual tumours. A high proportion of well differentiated cSCC clustered into a separate subset from the majority of moderately and poorly differentiated cSCCs (Figure 2). Taken together, these data support the hypothesis that well differentiated tumours demonstrate a characteristic genotype and constitute a discrete subpopulation of cSCCs.
TABLE 1.
Allelic Imbalance in 60 Squamous Cell Carcinomas
| ID1 | Allelic imbalance2 | Histological diagnosis | Immune status | HPV |
|---|---|---|---|---|
| 1 | 15 | Poorly differentiated3 | RT | + |
| 2 | 3 | Well differentiated4 | CT | − |
| 3 | 7 | Moderately differentiated | RT | + |
| 4 | 3 | Well differentiated | RT | + |
| 5 | 6 | Well differentiated | RT | − |
| 6 | 3 | Well differentiated | RT | + |
| 7 | 5 | Well differentiated | RT | + |
| 8 | 10 | Moderately differentiated | RT | − |
| 9 | 5 | Poorly differentiated | IC | + |
| 10 | 8 | Moderately differentiated | IC | + |
| 11 | 7 | Well differentiated | IC | + |
| 12 | 6 | Moderately differentiated | IC | + |
| 13 | 9 | Moderately differentiated | IC | + |
| 14 | 11 | Moderately differentiated | IC | + |
| 15 | 12 | Moderately differentiated | IC | − |
| 16 | 10 | Moderately differentiated | IC | + |
| 17 | 9 | Well differentiated | RT | + |
| 18 | 6 | Moderately differentiated | RT | + |
| 19 | 6 | Moderately differentiated | RT | + |
| 20 | 11 | Moderately differentiated | IC | − |
| 21 | 9 | Well differentiated | RT | + |
| 22 | 9 | Moderately differentiated | RT | + |
| 23 | 6 | Moderately differentiated | IC | + |
| 24 | 9 | Moderately differentiated | IC | − |
| 25 | 8 | Well differentiated | IC | + |
| 26 | 9 | Poorly differentiated | PUVA | − |
| 27 | 2 | Well differentiated | RT | + |
| 28 | 3 | Well differentiated | RT | − |
| 29 | 8 | Moderately differentiated | RT | − |
| 30 | 3 | Well differentiated | RT | + |
| 31 | 9 | Poorly differentiated | RT | + |
| 32 | 11 | Moderately differentiated | RT | − |
| 33 | 6 | Well differentiated | CT | + |
| 34 | 7 | Moderately differentiated | RT | − |
| 35 | 11 | Moderately differentiated | PUVA | − |
| 36 | 5 | Well differentiated | IC | + |
| 37 | 16 | Moderately differentiated | RT | + |
| 38 | 3 | Well differentiated | IC | − |
| 39 | 8 | Moderately differentiated | IC | + |
| 40 | 13 | Poorly differentiated | IC | + |
| 41 | 6 | Well differentiated | RT | − |
| 42 | 8 | Well differentiated | RT | + |
| 43 | 11 | Poorly differentiated | IC | + |
| 44 | 6 | Poorly differentiated | RT | + |
| 45 | 7 | Well differentiated | RT | + |
| 46 | 6 | Well differentiated | RT | − |
| 47 | 2 | Well differentiated | RT | + |
| 48 | 14 | Moderately differentiated | CLL | − |
| 49 | 2 | Well differentiated | RT | + |
| 50 | 7 | Well differentiated | CT | + |
| 51 | 6 | Well differentiated | RT | + |
| 52 | 17 | Poorly differentiated | RT | + |
| 53 | 5 | Well differentiated | RT | + |
| 54 | 1 | Well differentiated | RT | − |
| 55 | 2 | Well differentiated | RT | + |
| 56 | 4 | Well differentiated | RT | + |
| 57 | 21 | Poorly differentiated | RT | + |
| 58 | 4 | Well differentiated | RT | + |
| 59 | 5 | Poorly differentiated | IC | + |
| 60 | 5 | Well differentiated | RT | + |
Sample numbers 1-16 were published previously (Purdie et al., 2007)
Number of chromosomes with allelic imbalance
All tumours were invasive SCC
Well differentiated SCC did not include keratoacanthomas
RT, renal transplant recipient; CT, cardiac transplant recipient; IC, immunocompetent individual; PUVA, patients receiving psoralen and UVA photochemotherapy; CLL, chronic lymphocytic leukemia
Figure 1.

Ideogram summarising allelic imbalance in well differentiated cSCC (light coloured lines) and moderately and poorly differentiated cSCC (dark coloured lines). LOH events are indicated to the left of chromosomes with deletion shown in green and uniparental disomy in blue and gains are indicated to the right in red.
Figure 2.

Cluster dendrogram of cSCC. Distances between samples relates to agreement in aberration as a ratio of total number of aberrations. Well-differentiated cSCC are indicated in green, moderately differentiated in orange and poorly differentiated in red.
One early study using low resolution microsatellite analysis reported that the rate of LOH in OTR cSCCs was less than half of that observed in IC cSCCs and proposed that this reflected the influence of immune status on the molecular pathogenesis of tumours (Rehman et al., 1997). However, we observed no significant difference (P=0.396) between the number of chromosomes showing aberrations in OTR and IC tumours (Table 1, complete data provided in Table S1). As the earlier study focused on LOH alone, we also analysed LOH and gain events separately: no significant difference existed between the number of chromosomes showing LOH or gain in OTR and IC cSCC (P=0.450 and 0.639 respectively). Similar results were obtained when the 16 previously published cSCC were analysed separately from the other tumours (Table S2). A probable explanation for the discrepancy between these results and the previous finding lies in the fact that 60% (9/15) OTR cSCCs from the earlier study were well differentiated compared with only 35% (7/20) of IC cSCCs. Thus, we propose that the finding of an apparently lower frequency of allelic loss in the OTR tumours in this previous study was confounded by the genetic fingerprint associated with differentiation status rather than immune status per se.
The oncogenic potential of beta genus HPV, whose DNAs are prevalent in cutaneous SCC, remains unclear (reviewed in Nindl et al., 2007). Differences in chromosomal aberrations between HPV-positive and -negative cSCC could indicate diverse genetic mechanisms of tumour development, providing support for an etiological role for HPV. Statistical analysis established that no correlation existed between the number of chromosomes demonstrating aberrations and HPV status (P=0.847). Similar results were obtained when the 16 previously published cSCC were analysed separately from the other tumours (Table S2). Earlier data (Stockfleth et al., 2004) suggest that HPV DNA prevalence is lower in IC than OTR cSCC. A previous study from this laboratory using a degenerate nested PCR method detected beta HPV in 80% OTR cSCC, compared with only 30% IC cSCC (Harwood et al., 2000). Conversely, in the current study beta HPV DNAs were identified in similar proportions of IC (78%, 14/18) and OTR (74%, 29/39) tumours. One explanation for the discrepancy is our use here of a new and highly sensitive PCR-reverse hybridisation assay capable of identifying HPV DNA at levels below the detection threshold of earlier methods (de Koning et al., 2006), potentially resulting in a disproportionate increase in HPV detection among IC samples where viral copy number may be lower (Pfister, 2003; Stockfleth et al., 2004).
The most frequent aberration detected in the cSCC series was LOH at 9p, observed in 45 of 60 (75%) samples (Figure 1). Analysis of SNP call signal intensity data revealed that 20% (9/45) of 9p LOH events were copy number neutral and hence indicative of acquired uniparental disomy (UPD), extending our previous finding that UPD is a key mechanism of LOH in cSCC (Purdie et al., 2007). In the initial series of 16 cSCCs, we identified a frequent homozygous microdeletion at 9p23 within the PTPRD locus. These data implicated PTPRD as a candidate tumour suppressor gene in cSCC and raised the possibility that, in addition to the established CDKN2A tumour suppressor locus (Kubo et al., 1997), 9p LOH may be important for the inactivation of PTPRD. We have extended this finding by identifying PTPRD microdeletions in 6 of the 44 additional cSCCs analysed in the current study, making a total of 9 of 60 (15%) cSCCs (Table 2). Two of the 9 cSCC demonstrated metastatic potential and 4 of the remaining 7 cSCC with PTPRD microdeletions were diagnosed as poorly differentiated, although poorly differentiated tumours comprised a minority of our sample series (10/60 cSCCs); these data suggested that PTPRD inactivation may be associated with more aggressive tumours. We also observed homozygous microdeletions within the CDKN2A locus at 9p21.2 or 9p21.3 in 3 samples, confirming the previous finding that this tumour suppressor gene may be inactivated by both genetic and epigenetic mechanisms (Brown et al., 2004).
TABLE 2.
PTPRD microdeletions in 9 SCCs
| SCC ID | Start position1 | End position | Histological diagnosis |
|---|---|---|---|
| 3 | 9.08 2 10.42 |
9.22 10.51 |
Moderately differentiated3 |
| 9 | 9.16 | 9.76 | Poorly differentiated |
| 124 | 8.55 | 9.38 | Moderately differentiated |
| 21 | 8.98 | 9.87 | Well differentiated |
| 25 4 | 9.22 | 9.83 | Well differentiated |
| 26 | 9.13 | 9.99 | Poorly differentiated |
| 39 | 9.25 | 9.58 | Moderately differentiated |
| 40 | 9.47 | 9.86 | Poorly differentiated |
| 52 4 | 9.37 | 9.48 | Poorly differentiated |
Start and end positions of the microdeletion are indicated in Mb
Tumour demonstrated two regions of microdeletion
SCC T3 and T12 demonstrated metastatic potential as reported previously (Purdie et al, 2007)
Microdeletions in these tumours were hemizygous, all others were homozygous
LOH at 3p was another frequent aberration, observed in 39 (65%) tumours and involving the whole of 3p in 23 (59%) of these. Two cSCCs from patients 26 and 52 displayed a homozygous microdeletion at 3p14.2 on a background of LOH across 3p (Figure 3) while tumour T20 exhibited extensive LOH at the telomeric end of 3p (3pter-3p21.31) in conjunction with a homozygous 3p14.2 microdeletion in a heterozygous region of 3p. All microdeletions mapped to a region at 60.41-61.16 Mb, within the locus of FHIT gene. FHIT is a recognised tumour suppressor gene that is inactivated in several epithelial cancers including lung cancer (Sozzi et al., 1996) and breast cancer (Negrini et al, 1996), although its role in cSCC remains unclear. One previous study (Sikkink et al., 1997) reported that two of five cSCC tumours showed abnormal FHIT transcripts whereas another (Popp et al., 2002) found that three cSCC lines with 3p loss expressed normal FHIT transcripts, albeit at much reduced levels in two cases. However, the microsatellite marker analysis and comparative genomic hybridisation (CGH) used did not permit fine mapping of the genetic changes underlying these expression data. Our data from high resolution SNP microarray analysis provide the first reported example of a homozygous microdeletion within this locus in cSCC. Although it is possible that expression analysis may reveal abnormal FHIT transcripts in a larger subset of tumours, the low frequency of the 3p14.2 microdeletion within our samples, together with the observation that T20 displayed extensive LOH on 3p separate from the 3p14.2 microdeletion, suggests that FHIT is unlikely to be the only target on 3p. Indeed, early 3p deletion mapping studies in other cancers (Hibi et al., 1992; Maestro et al., 1993) have demonstrated the presence of three distinct deleted regions in several tumours, 3pter-3p24, 3p21 and 3p 14.2-3p12, implying that 3p loss may inactivate multiple tumour suppressor genes.
Figure 3.
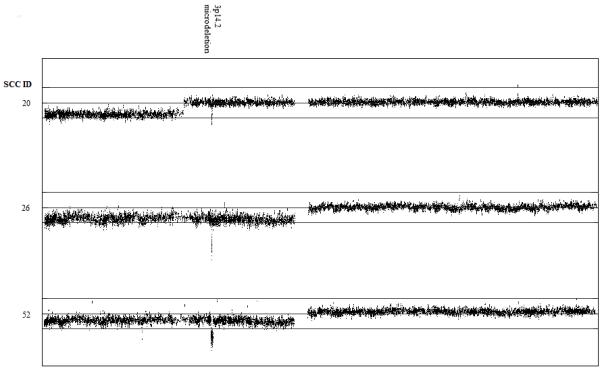
Display of copy number ratios on chromosome 3 reveals a 3p14.2 microdeletion within the FHIT locus. A running average of 2 consecutive tumour:non-tumour signal value ratios is plotted on a log2 scale according to chromosomal position. Upper line represents log2(2) and lower line represents log2(0.5). Microdeletions mapped to the following regions: SCC T20, 60.46-60.61 Mb; SCC T26, 60.41-60.58 Mb; SCC T52, 60.41-61.16 Mb.
Other recurrent events included LOH at 13q and 8p, each observed in 23 (38%) tumours and chromosomal gain on 3q (26 tumours; 43%), 8q (23 tumours; 38%) and 9q (21 tumours; 35%). Our data are consistent with the results of earlier research using polymorphic microsatellite markers or CGH to examine the genetics of cSCC (Quinn et al., 1994; Ashton et al., 2003), where allelic imbalance on chromosomes 3, 9 and 13 were among the predominant aberrations reported. In contrast, a recent study (Clausen et al., 2006) using CGH to examine cSCC including 11 tumours from our laboratory, found that 9p loss was a minor aberration, occurring in only 3.2% (1/31) SCC. Loss of 3p and 13q were also observed less frequently, in 19.4% (6/31) and 12.9% (4/31) tumours respectively. The most common chromosomal aberration was gain at 1p, identified in 41.9% (13/31) SCCs compared with only 9/60 (15%) samples in the present study. Overall more gains than losses of chromosomal material (ratio of 1.65:1) were reported using CGH (Clausen et al., 2006) whereas we observed that LOH (deletion and UPD) predominated over gain events by a ratio of 1.5:1 (Table S1). These discrepancies may in part reflect the limitations of CGH for identifying LOH and possible preferential detection of amplifications: deleted regions must be as large as 5-10 Mb to be detected whereas amplifications of 1 Mb can be identified (Forozan et al., 1997). Furthermore, CGH is unable to detect copy number-neutral LOH (UPD), which accounted for 20% of 9p LOH and 22% 13q LOH in the current study. Conversely, the lower frequency of 1p gain observed here combined with the overall predominance of LOH over amplifications detected implies that SNP microarray analysis may display greater sensitivity for identifying LOH than gain events. Methodological differences are likely to have been another contributory factor: in the two earlier investigations that reported a high frequency of LOH at 9p and other chromosomes (Quinn et al., 1994; Ashton et al., 2003) tumours were microdissected prior to DNA extraction as in our protocol, whereas the more recent CGH study (Clausen et al., 2006) performed analysis on a central section of the entire lesion. Depending upon the proportion of stromal tissue and hence non-tumour DNA present within samples, it seems possible that some LOH events may have been masked, whereas gains of chromosomal material might still be detected if the degree of amplification were sufficiently large.
In summary, we have used genome-wide SNP microarray analysis to confirm our previous hypothesis that well differentiated tumours are a genetically distinct subpopulation of cutaneous SCC. Contrary to earlier research (Rehman et al., 1997), we found no significant difference in the frequency of allelic imbalance in OTR and IC cSCCs nor were we able to establish any correlation between the frequency of chromosomal aberrations and HPV status. Extensive LOH at 3p and 9p were observed in a majority of samples, consistent with previous data. Loss of 9p may entail the inactivation of PTPRD, recently implicated as a candidate tumour suppressor gene in cSCC (Purdie et al., 2007). Here, we demonstrate PTPRD microdeletions in 9 of 60 (15%) cSCCs and identify FHIT on 3p14.2 as another possible target for inactivation. Our findings demonstrate the effectiveness of genome-wide SNP microarray analysis as a means of unravelling the genetic complexity of cSCC.
MATERIALS AND METHODS
A total of 60 primary cSCCs were analysed, including 16 primary cSCCs described in an earlier study (Purdie et al., 2007). Of these tumours, 42 were from immunocompromised individuals (39 were receiving immunosuppressive therapy (3 cardiac and 36 renal transplant recipients), 2 were receiving psoralen and UVA photochemotherapy for psoriasis and one had chronic lymphocytic leukaemia). Ethical approval for this investigation was obtained from the East London and City Health Authority local ethics committee and the study was conducted according to the Declaration of Helsinki Principles. All patients participating in the study provided written, informed consent.
All histopathologic diagnoses were confirmed by a single experienced dermatopathologist (author RC). Samples were enriched for cSCC keratinocytes, DNA extracted from cSCC and non-tumour control blood samples and subjected to 250K Nsp SNP array analysis as previously described (Purdie et al., 2007). The presence of beta HPV was investigated in cSCC DNA samples using the RHA kit skin (beta) HPV (Diassay B.V., Rijswijk, Netherlands) according to the manufacturer’s instructions.
Statistical analyses were carried out using Poisson regression. We used a generalised linear model with a Poisson distribution imposed on the number of chromosomes per patient with at least one event, and a log link function. Sequential analysis of deviance was carried out on the model which was fitted with terms representing differentiation effect, immune effect and HPV effect respectively, using a Chi-squared test on the deviances. To test for a linear trend with respect to differentiation status, P-values were calculated against the null hypothesis that the linear trend of count against differentiation status had zero slope. This analysis was all carried out using the “glm” function within the statistical environment R (R Development Core Team, 2007). For individual comparisons amongst the differentiation subgroups, we used Tukey contrasts between the pairwise comparisons, using the multcomp (Hothorn et al, 2007) package within R to adjust for the multiple comparison situation. Hierarchical clustering was carried out in R using hclust with an asymmetrical binary metric between samples, where the asymmetric binary distance between pairs of samples was calculated as the number of aberrations observed in just one sample expressed as a proportion of the number of aberrations in one or both of the samples. Inter-cluster similarities were calculated using Ward’s agglomeration method (Ward, 1963).
Supplementary Material
ACKNOWLEDGEMENTS
Supported by: Cancer Research UK, British Skin Foundation Small Grant Award reference S/BS3, Research Advisory Board of St Bartholomews and The Royal London Charitable Foundation reference RAC 404.
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest
REFERENCES
- Armstrong BK, Kricker A. The epidemiology of UV induced skin cancer. J Photochem Photobiol B. 2001;63:8–18. doi: 10.1016/s1011-1344(01)00198-1. [DOI] [PubMed] [Google Scholar]
- Ashton KJ, Weinstein SR, Maguire DJ, Griffiths LR. Chromosomal aberrations in squamous cell carcinoma and solar keratoses revealed by comparative genomic hybridization. Arch Dermatol. 2003;139:876–82. doi: 10.1001/archderm.139.7.876. [DOI] [PubMed] [Google Scholar]
- Brown VL, Harwood CA, Crook T, Cronin JG, Kelsell DP, Proby CM. p16INK4a and p14ARF tumor suppressor genes are commonly inactivated in cutaneous squamous cell carcinoma. J Invest Dermatol. 2004;122:1284–92. doi: 10.1111/j.0022-202X.2004.22501.x. [DOI] [PubMed] [Google Scholar]
- Breuninger H, Black B, Rassner G. Microstaging of squamous cell carcinomas. Am J Clin Path. 1990;92:624–7. doi: 10.1093/ajcp/94.5.624. [DOI] [PubMed] [Google Scholar]
- Boukamp P. Non-melanoma skin cancer: what drives tumour development and progression? Carcinogenesis. 2005;26:1657–67. doi: 10.1093/carcin/bgi123. [DOI] [PubMed] [Google Scholar]
- Clausen OP, Aass HC, Beigi M, Purdie KJ, Proby CM, Brown VL, et al. Are keratoacanthomas variants of squamous cell carcinomas? A comparison of chromosomal aberrations by comparative genomic hybridization. J Invest Dermatol. 2006;126:2308–15. doi: 10.1038/sj.jid.5700375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning M, Quint W, Struijk L, Kleter B, Wanningen P, van Doorn L-J, et al. Evaluation of a novel highly sensitive broad-spectrum PCR-reverse hybridization assay for detection and identification of beta-papillomavirus DNA. J Clin Microbiol. 2006;44:1792–800. doi: 10.1128/JCM.44.5.1792-1800.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries E, van de Poll-Franse LV, Louwman WJ, de Gruijl FR, Coebergh JW. Predictions of skin cancer incidence in the Netherlands up to 2015. Br J Dermatol. 2005;152:481–8. doi: 10.1111/j.1365-2133.2005.06386.x. [DOI] [PubMed] [Google Scholar]
- Forozan F, Karhu R, Kononen J, Kallioniemi A, Kallioniemi OP. Genome screening by comparative genomic hybridization. Trends Genet. 1997;13:405–9. doi: 10.1016/s0168-9525(97)01244-4. [DOI] [PubMed] [Google Scholar]
- Glover MT, Deeks JJ, Raftery MJ, Cunningham J, Leigh IM. Immunosuppression and risk of non-melanoma skin cancer in renal transplant recipients. Lancet. 1997;349:398. doi: 10.1016/S0140-6736(97)80015-3. [DOI] [PubMed] [Google Scholar]
- Harwood CA, Surentheran T, McGregor JM, Spink PJ, Leigh IM, Breuer J, et al. Human papillomavirus infection and non-melanoma skin cancer in immunosuppressed and immunocompetent individuals. J Med Virol. 2000;61:289–97. doi: 10.1002/1096-9071(200007)61:3<289::aid-jmv2>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Harwood CA, Surentheran T, Sasieni P, Proby CM, Bordea C, Leigh IM, et al. Increased risk of skin cancer associated with the presence of epidermodysplasia verruciformis human papillomavirus types in normal skin. Br J Dermatol. 2004;150:949–57. doi: 10.1111/j.1365-2133.2004.05847.x. [DOI] [PubMed] [Google Scholar]
- Hibi K, Takahashi T, Yamakawa K, Ueda R, Sekido Y, Ariyoshi Y, et al. Three distinct regions involved in 3p deletion in human lung cancer. Oncogene. 1992;7:445–9. [PubMed] [Google Scholar]
- Hothorn T, Bretz F, Westfall P, Heiberger RM. Multcomp: simultaneous inference for general linear hypotheses. R package version 0.992-8. 2007 [Google Scholar]
- Kubo Y, Urano Y, Matsumoto K, Ahsan K, Arase S. Mutations of the INK4a locus in squamous cell carcinomas of the human skin. Biochem Biophys Res Commun. 1997;232:38–41. doi: 10.1006/bbrc.1997.6217. [DOI] [PubMed] [Google Scholar]
- Maestro R, Gasparotto D, Vukosavljevic T, Barzan L, Sulfaro S, Boiocchi M. Three discrete regions of deletion at 3p in head and neck cancers. Cancer Res. 1993;53:5775–9. [PubMed] [Google Scholar]
- Majewski S, Jablonska S. Epidermodysplasia verruciformis as a model of human papillomavirus-induced genetic cancer of the skin. Arch Dermatol. 1995;131:1312–8. [PubMed] [Google Scholar]
- Negrini M, Monaco C, Vorechovsky I, Ohta M, Druck T, Baffa R, et al. The FHIT gene at 3p14.2 is abnormal in breast carcinomas. Cancer Res. 1996;56:3173–9. [PubMed] [Google Scholar]
- Nindl I, Gottschling M, Stockfleth E. Human papillomaviruses and non-melanoma skin cancer: basic virology and clinical manifestations. Dis Markers. 2007;23:247–59. doi: 10.1155/2007/942650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister H. Human papillomavirus and cancer. J Natl Cancer Inst Monogr. 2003;31:52–6. doi: 10.1093/oxfordjournals.jncimonographs.a003483. [DOI] [PubMed] [Google Scholar]
- Popp S, Waltering S, Herbst C, Moll I, Boukamp P. UV-B-type mutations and chromosomal imbalances indicate common pathways for the development of Merkel and skin squamous cell carcinomas. Int J Cancer. 2002;99:352–60. doi: 10.1002/ijc.10321. [DOI] [PubMed] [Google Scholar]
- Purdie KJ, Lambert SR, Teh MT, Chaplin T, Molloy G, Raghavan M, et al. Allelic imbalances and microdeletions affecting the PTPRD gene in cutaneous squamous cell carcinomas detected using single nucleotide polymorphism microarray analysis. Genes Chromosomes Cancer. 2007;46:661–9. doi: 10.1002/gcc.20447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn AG, Sikkink S, Rees JL. Basal cell carcinomas and squamous cell carcinomas of human skin show distinct patterns of chromosome loss. Cancer Res. 1994;54:4756–9. [PubMed] [Google Scholar]
- R Development Core Team . R: a language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2007. ISBN 3-900051-07-0, URL http://www.R-project.org. [Google Scholar]
- Rehman I, Quinn AG, Takata M, Taylor AE, Rees JL. Low frequency of allelic loss in skin tumours from immunosuppressed individuals. Br J Cancer. 1997;76:757–9. doi: 10.1038/bjc.1997.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikkink SK, Rehman I, Rees JL. Deletion mapping of chromosome 3p and 13q and preliminary analysis of the FHIT gene in human nonmelanoma skin cancer. J Invest Dermatol. 1997;109:801–5. doi: 10.1111/1523-1747.ep12340991. [DOI] [PubMed] [Google Scholar]
- Sozzi G, Veronese ML, Negrini M, Baffa R, Cotticelli MG, Inoue H, et al. The FHIT gene at 3p14.2 is abnormal in lung cancer. Cell. 1996;85:17–26. doi: 10.1016/s0092-8674(00)81078-8. [DOI] [PubMed] [Google Scholar]
- Stockfleth E, Nindl I, Sterry W, Ulrich C, Schmook T, Meyer T. Human papillomaviruses in transplant-associated skin cancers. Dermatol Surg. 2004;30:604–9. doi: 10.1111/j.1524-4725.2004.00144.x. [DOI] [PubMed] [Google Scholar]
- Ward JH. Hierarchical grouping to optimise an objective function. J Am Stat Assoc. 1963;58:236–44. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.