

Abstract
Membranous nephropathy (MN) is one of the commonest glomerular diseases, typically presenting in older males with nephrotic syndrome. The development and characterization of animal models of MN, in particular, the passive Heymann nephritis model (PHN), has greatly advanced our understanding of this disease. In this review we discuss the different animal models of human MN that are available, with an emphasis on the PHN model, including technical issues, the typical disease course and its application to human disease.
Keywords: membranous nephropathy, podocyte, glomerulus, proteinuria, Heymann nephritis, animal models
Introduction
Membranous nephropathy is a leading cause of nephrotic syndrome in older adults and is defined by the presence of subepithelial immune deposits (between the glomerular basement membrane (GBM) and the podocyte) (Figure 1). The subsequent expansion of the GBM results in a membrane-like (membranous) thickening of the glomerular capillary wall. It is mostly idiopathic, although about 20% of cases are associated with clinical conditions such as cancer, infection, autoimmune disease or drugs. Although animal models of some secondary causes of MN have been described, in particular, lupus membranous nephritis, in this review we will focus only on models of idiopathic MN. To date there are no transgenic or knockout models of MN, and all the examples in this review are inducible models.
Figure 1. Human Membranous Nephropathy Pathology.
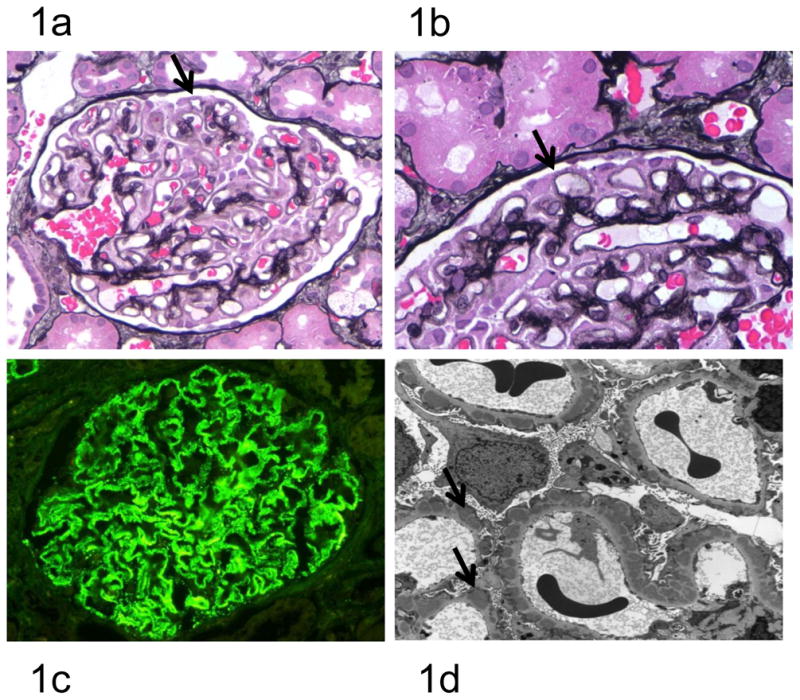
At early stages, the glomeruli and interstitium look essentially normal, but as the disease progresses, the thickening of capillary loops becomes evident due to the accumulation of sub-epithelial immune complexes and the deposition of new basement membrane material by the podocyte (Figure 1a, ×400). Staining with silver methenamine may reveal characteristic spikes (arrowed) representing basement membrane material projecting between the immune deposits (Figure 1b, ×1000). Immunofluorescence reveals finely granular deposits of IgG (predominantly IgG4) in a subepithelial distribution on the outer surface of the GBM (Figure 1c). Electron microscopy reveals the characteristic subepithelial immune deposits (arrowed) (Figure 1d).
Pathogenesis of Idiopathic Membranous Nephropathy
Membranous nephropathy is characterized by the development of immune complexes in the subepithelial space on the outer surface of the glomerular basement membrane (GBM). These immune deposits consist of immunoglobulin (IgG, predominantly IgG4), complement components (C3, C5b-9), and presumably antigen. In idiopathic MN, autologous antibodies cross the glomerular endothelial cell and GBM and react to an endogenous antigen on the podocyte foot process in a mechanism similar to the Heymann nephritis model.[1] The first antigen to be clearly described in cases of human MN was neutral endopeptidase, initially identified as the target antigen in several cases of neonatal MN.[2] More recently, the M-type phospholipase A2 receptor (PLA2R) has been identified as the antigen in most cases of idiopathic MN.[3] Immune complexes may also form in situ due to the reaction of antibody to an exogenous antigen which has become trapped in the subepithelial space (planted antigen). This mechanism may explain the association of membranous nephropathy with some infections and certain drugs[4].
In MN, C3 and C5b-9 can be detected in subepithelial immune deposits.[5] Activation of complement by the mannose binding lectin pathway is suggested by the common finding of C4b deposition and the identification of MBL in the glomeruli of idiopathic MN.[6,7] Following C5b-9 insertion into the lipid bilayer of the podocyte cell membrane, instead of cell lysis, a series of signaling events result in cell activation and changes in podocyte structure and function. These include the generation of matrix components leading to thickening of the GBM; the production of inflammatory mediators; a reduction in podocyte number; alterations in the slit diaphragm; and foot process effacement (reviewed in[8]).
Rat models of Membranous Nephropathy
Passive Heymann Nephritis
Since its establishment, the Heymann model has been used to study mechanisms of cellular injury as well as the podocyte’s responses to injury.
Active versus Passive Heymann Nephritis
An active immune model utilizes the production of autologous antibodies to renal antigen(s) by the animal’s own immune system. Heymann et al. first described the injection of kidney extracts with Freund’s adjuvant to induce nephrotic syndrome in 1959 (Active Heymann Nephritis).[1] Freund’s adjuvant is used as a non-specific immune system stimulant.
A passive model of immunization utilizes antiserum to an antigen(s) generated in another animal (often another species including rabbits, goats, or sheep), which are then injected into a rat or mouse to elicit immune complex formation. The initial 5-7 day period of a passive model is referred to as the heterologous phase as it relies on an immune response to the antibody injected from a foreign or heterologous source. Subsequently, the rat or mouse mounts its own immune response to the deposited heterologous antibody and thereby enters the autologous phase of disease. In early studies, an insoluble sub-fraction from rat proximal tubule brush borders termed fraction 1A (Fx1A) was isolated and utilized to produce antibodies for a passive heterologous model termed Passive Heymann Nephritis (PHN).[9-11] This resulted in the development of subepithelial deposits of IgG, C3 and C5b-9 associated with heavy proteinuria. The immune deposits form in situ due to the binding of circulating IgG to antigenic epitopes expressed on the foot processes of the glomerular epithelial cell.
Background of the rat model of PHN
In Heymann nephritis the antibodies are targeted against the Heymann nephritis antigenic complex (HNAC) consisting of two proteins, megalin and receptor associated protein (RAP).[12] Megalin is a ~600 kDa molecular weight glycoprotein belonging to the LDL receptor family, which acts as a multi-functional receptor facilitating endocytosis. It is expressed in both the brush border of proximal tubular cells and in the clathrin-coated pits on the sole of podocyte foot processes. Megalin forms a heterodimeric complex with RAP, and several epitopes on this complex seem to be involved in the formation of the immune deposits. [13] Notably, in animal models, active immunization with megalin alone or passive immunization with anti-megalin antibody result in the accumulation of immune deposits in the subepithelial space, but no C3 or C5b-9 deposition occurs and no proteinuria occurs.[14] We now recognize that other antibodies in the anti-Fx1A fraction inhibit complement regulatory proteins on the podocyte leading to local activation of complement via the alternate or mannose binding lectin pathways.
Technical aspects of the PHN model (reviewed in [15,16])
Because PHN has been so widely studied over the years, a variety of methods for inducing disease have been utilized. Various strains such as Sprague Dawley, Wistar, Munich Wistar, Lewis and Piebold Viral Glaxo (PVG) have been utilized. Generally male rats are used, but female rats are also used, including pregnant females.[17] Male (200g-300g) and female (100-200g) rats typically receive a single dose of heterologous Fx1A antibody produced in rabbits or sheep. Uninephrectomy,[18] as well as multiple injections followed by sensitization with rabbit IgG[19] have been utilized to produce accelerated models of PHN. Antibody is administered either intravenously through the tail vein or intraperitoneally. Doses range from 2-7ml/Kg when serum is administered and 20-240mg/Kg when isolated IgG is administered, the dose determined empirically for each batch of antibody. Controls consist of saline or PBS, pre-immune serum or pre-immune IgG from sheep or rabbits prior to Fx1A immunizations, as well as serum or IgG from non-immunized rabbits or sheep. However, an important control is to co-administer cobra venom factor (CVF), which depletes C3 and C5 fragments thereby decreasing terminal membrane attack complex formation.
Development of the anti-Fx1A antibody
In order to establish the rat model of PHN, one must first produce the Fx1A antigen. Rat cortices are pressed through a 150um mesh and re-suspended in saline. Glomerular and tubular fragments are removed by low speed centrifugation and the supernatant containing the renal tubular epithelial fraction (Fx1) is sedimented by centrifugation. The pelleted fraction (Fx1A), containing organelles and cell membranes is washed in water and used for the production of heterologous antibody.[10] The Fx1A antigen is combined with Freund’s and injected into either rabbits or sheep. Additionally, gp330, one of the pathogenic antigens in Fx1A, has also been used to produce heterologous antibodies, which can induce PHN.[20,21] The heterologous antibody against Fx1A should be injected into rats either as serum, plasma, or isolated IgG. A dose response/time course should be performed to establish the optimal dose and onset of proteinuria. Finally, it is important to measure proteinuria in all rats, as in our experience, only 80% of injected animals actually get proteinuric, and thus the non-proteinuric animals should be excluded. Moreover, all animals need to be tested for antibody deposition in the glomerulus to insure that the there was appropriate binding of the Fx1A antibody.
Expected outcomes of the rat model of PHN
PHN is a progressive model of glomerular injury. Following injection of anti-Fx1A antibody, circulating gp330 antibodies bind to megalin expressed in the clathrin-coated pits along the sides and bases of podocyte foot processes. The antibody is capped and shed into the subepithelial space, where it deposits in the lamina rara externa of the glomerular basement membrane (GBM). Immune deposits continue to accumulate in this fashion until they obscure the slit diaphragm. [12] Sub-lethal complement activation occurs on effected podocytes causing damage and intracellular signaling activation. [22,23] It has been well documented that deposition of the terminal components of complement (C5b-9) occurs not only in PHN[24] but also in MN in man.[25] Moreover, C5b-9 is required in some models of PHN [26]. However, it should be noted that in some experimental models of PHN utilizing the C6 deficient PVG strain of rats, disease formation and progression occurs in the absence of C5b-9.[27] Four to seven days after injection, the onset of proteinuria occurs, but this varies depending on initiating antibody and typically can range between 100mg/day and 500mg/day. This level of proteinuria persists throughout the autologous phase of the model. While the only histological changes are GBM thickening at the light microscopy level, foot process effacement can be observed by electron microscopy (EM). Recently it has been demonstrated that changes in nephrin at the slit diaphragm can be detected prior to the onset of proteinuria.[28,29] Eventually glomerular and tubulointerstitial sclerosis occurs as a result of ongoing proteinuria and podocyte injury. [8]
Key lessons from the PHN model
The PHN model elegantly demonstrates the paradigm of an in situ immune complex disease due to antibodies targeted to a podocyte antigen (megalin/receptor associated protein) analogous to human MN. The central role of complement, and the podocyte response to injury have been extensively studied in this model. Therapeutic strategies have also been investigated in the model including blockade of the RAAS, anti-oxidant therapy, protease inhibitors, growth factor inhibitors, among others. In the PHN model, the administration of scavengers of ROS (deferioxamine, DMTU), inhibitors of lipid peroxidation or inhibitors of xanthine oxidase have all been shown to markedly decrease urine protein excretion.
Advantages of the PHN model include its nearly identical pathology to human MN; it is relatively easy to produce the heterologous antibody and induce disease in rats; and the time course for the onset and progression of disease is relatively short (days-weeks). However, there are some clear limitations. The most obvious is that although megalin is expressed in human tubular brush border, it is not expressed on human podocytes and therefore cannot be the pathogenic human antigen. Tubular immune deposits are also found in this model unlike human MN. Also, the dominant immunoglobulin deposited in human MN is IgG4, which does not fix complement, unlike the antibodies in the PHN model, and there is evidence that the central role of C5b-9 may have been over-emphasized.[27]
Other Rat Models of Membranous Nephropathy
Active Heymann Nephritis Model [1]
The Active Heymann Nephritis model was the first to be developed, and more closely resembles human membranous because it is actually induced by the rats own immune system rather than passively planting antibody. However, it is now rarely used as it takes substantially longer to induce disease, is much more variable, and the immunizations have to be repeatedly administered into the foot pads, which can be uncomfortable for the rats.
Anti-dipeptidyl peptidase IV (DPPIV) rat model[30,31]
Dipeptidyl peptidase IV (DPPIV) is another major antigen of Fx1A fraction of tubular brush border which is expressed by podocytes, but is also found circulating in serum. Injection of rabbit anti-DPPIV antibody into 6 week old Lewis rats results in the development of proteinuria on day 1, peaking day 2 followed by a gradual decline. Notably, unlike Heymann nephritis, the proteinuria and IgG deposition occur in the absence of glomerular C3 deposition and the immune deposits are transient, disappearing from the glomeruli within 5 days.
Mouse models of Membranous Nephropathy
Although we and others have made attempts, there are few published reports of a robust mouse model of MN.
Cationic BSA mouse model[32]
In this recent study, three different strains of mice (ICR, BALB/c and C57BL6) were pre-immunized with cationic bovine serum albumin (cBSA) every other day for a week. Two weeks later, mice were re-immunized with cBSA in Freund’s adjuvant. Both ICN and BALB/c mice developed disease, however BALB/c mice required a higher dose. In this study mice developed features of MN including severe proteinuria, diffuse thickening of the GBM, subepithelial deposits, and GBM spikes.
Anti-renal tubular antigen model[33]
In this study, a single injection of rabbit antiserum against homologous, pronase-digested, renal tubular antigens was given to C57.BlIO mice. While subepithelial deposits of rabbit IgG were described, only transient albuminuria occurred, and there was no spike formation, podocyte vacuolization, nor foot process fusion. The authors speculated that because the subclass of rabbit anti-Fx1A was IgG1, little complement fixation occurred and the model lacked many of the hallmarks of MN.[33] In unpublished data, the authors of this review have tried a similar approach utilizing both rabbit and sheep antibodies to mouse pronase-digested, renal tubular antigens in C57BL6 mice. Virtually identical results were obtained to this study.
Aminopeptidase A (APA) model[34]
In this model a transient complement independent albuminuria associated with binding of antibody to podocytes has been described secondary to monoclonal antibodies to APA.[34] More recent data suggests anti-APA antibodies may induce proteinuria secondary to interference with the CD2AP/nephrin/podocyte slit diaphragm complex. [35]
Non-rodent Models of Membranous Nephropathy
Rats and mice are the preferred animals to model human disease in view of their availability, costs, short gestation period, and the greater ability to manipulate these animals. Other animal models of MN have been developed including a rabbit model using injected cationic bovine serum albumin (cBSA)[36], but this is now rarely used. More recently, the transfer of anti-NEP antibodies has produced a disease similar to MN in rabbits.[2] A pig model of MN was also developed fortuitously during studies of xenograft tolerance.[37] In this model, heterologous (goat) antibodies to angiotensin converting enzyme (ACE), which is expressed by porcine podocytes (but not, rat, mouse or rabbit), led to the development of subepithelial immune deposits and proteinuria.
Future Directions
The identification of phospholipase A2 receptor as the target antigen in most patients with idiopathic MN has re-energized the field and opened up exciting new avenues for investigation. We can expect the development of new animal models based on this antigen. Unfortunately, neither rodents nor rabbits express PLA2R on the podocyte surface, limiting anti-PLA2R transfer models, and transgenic models are now in development. Other potential podocyte antigens in human MN including aldose reductase, and anti-manganese superoxide dismutase (SOD2),[38] may also lend themselves to the development of instructive models.
It may be that some of these models will eventually succeed the PHN model, but PHN is still going strong after 50 years of investigation, and likely will continue to educate us for many years to come.
Figure 2. Generation of PHN model.

Step 1 Antigen Isolation: Fraction 1a (Fx1a), which contains gp330, is isolated from rat proximal tubule brush border. [10] Step 2 Antibody Production: A sheep is immunized subcutaneously with Fx1a emulsified in complete Freund’s adjuvant for the first immunization and incomplete Freund’s for at least 3 subsequent immunizations. Sheep antibodies to Fx1a are produced by the sheep and the antibody is obtained by plasmapheresis to generate large volumes of plasma containing the antibody. Whole plasma or isolated IgG are then used to inject into rats. Step 3 Antibody Testing: Antibody tolerance, route of administration (IV or IP) and optimal dose are determined through a series of dose response tests. Once the optimal parameters to induce the model are established, a study can be performed.
Figure 3. Periodic Acid Schiff staining of rat PHN kidneys.

The GBM lining the outer aspect of the capillary loops is smooth and thin in rats administered control sheep serum at 1 month (A, 400× and 1000× magnification) and 6 months (C, 400× and 1000× magnification). The GBM lining the outer aspect of the capillary loops is thicker (B, 400× and 1000× magnification, arrow heads) and some podocytes have abnormal shape (B, thin arrow) in rats administered sheep-anti Fx1a (PHN) at 1 month. Following six months of PHN and ongoing proteinuria, some glomeruli have become sclerotic (D, 400× and 1000× magnification).
Table 1.
Animal Models of Membranous Nephropathy
| Animal | Antigen | Glomerular Immune Deposits | Tubular Immune Deposits | Comments | Ref |
|---|---|---|---|---|---|
| Rat | Megalin (PHN) | +++P | ++P | Pro: Currently best model, widely used, nearly identical pathology, easy to produce, short time frame. Con: PHN antigen (megalin) not found in human MN |
[12] |
| Rat | Megalin (AHN) | +++P | ++P | Longer to produce, more variable, more discomfort for rats | [1] |
| Rat | DPPIV | +T | +T | Transient model, few immune deposits | [31,39] |
| Mouse | Cationic BSA | ++P | - | Best mouse model, limited experience. | [32] |
| Mouse | Renal tubular antigen | ++P | ++P | Subepithelial deposits, but minimal proteinuria | [33,40] |
| Mouse | Aminopeptidase A | ++P | +++P | Proteinuria marked, but complement independent | [34,35] |
| Rabbit | NEP | +T | - | Rarely used | [2] |
| Rabbit | DPPIV | ++P | - | Rarely used | [41,42] |
| Rabbit | ACE | +T | +++P | Rarely used | [41,43,44] |
| Pig | ACE | ++P | +++P | Rarely used | [37] |
CD26: dipeptidyl peptidase IV, NEP: neutral endopeptidase, P: persistent, large diffuse; T: small focal transient. (Adapted from Table 1 Maruyama S et al, J Am Soc Nephrol 1999[37]).
Acknowledgments
This work was supported by National Institutes of Health grants to S.J.S. (DK60525, DK56799, DK 51096), and by the American Diabetes Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Heymann W, et al. Production of nephrotic syndrome in rats by Freund’s adjuvants and rat kidney suspensions. Proc Soc Exp Biol Med. 1959;100 (4):660–664. doi: 10.3181/00379727-100-24736. [DOI] [PubMed] [Google Scholar]
- 2.Debiec H, et al. Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N Engl J Med. 2002;346 (26):2053–2060. doi: 10.1056/NEJMoa012895. [DOI] [PubMed] [Google Scholar]
- 3.Beck LH, Jr, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361 (1):11–21. doi: 10.1056/NEJMoa0810457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jefferson JA, Couser WG. Therapy of membranous nephropathy associated with malignancy and secondary causes. Semin Nephrol. 2003;23 (4):400–405. doi: 10.1016/s0270-9295(03)00055-x. [DOI] [PubMed] [Google Scholar]
- 5.Brenchley PE, et al. Urinary C3dg and C5b-9 indicate active immune disease in human membranous nephropathy. Kidney Int. 1992;41 (4):933–937. doi: 10.1038/ki.1992.143. [DOI] [PubMed] [Google Scholar]
- 6.Lhotta K, et al. Glomerular deposition of mannose-binding lectin in human glomerulonephritis. Nephrol Dial Transplant. 1999;14 (4):881–886. doi: 10.1093/ndt/14.4.881. [DOI] [PubMed] [Google Scholar]
- 7.Moseley HL, Whaley K. Control of complement activation in membranous and membranoproliferative glomerulonephritis. Kidney Int. 1980;17 (4):535–544. doi: 10.1038/ki.1980.62. [DOI] [PubMed] [Google Scholar]
- 8.Shankland SJ. The podocyte’s response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 2006;69 (12):2131–2147. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- 9.Edgington TS, et al. Autologous immune complex nephritis induced with renal tubular antigen. I. Identification and isolation of the pathogenetic antigen. J Exp Med. 1968;127 (3):555–572. doi: 10.1084/jem.127.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edgington TS, et al. Characterization and isolation of specific renal tubular epithelial antigens. J Immunol. 1967;99 (6):1199–1210. [PubMed] [Google Scholar]
- 11.Feenstra K, et al. Experimental glomerulonephritis in the rat induced by antibodies directed against tubular antigens. I. The natural history: a histologic and immunohistologic study at the light microscopic and the ultrastructural level. Lab Invest. 1975;32 (2):235–242. [PubMed] [Google Scholar]
- 12.Farquhar MG, et al. The Heymann nephritis antigenic complex: megalin (gp330) and RAP. J Am Soc Nephrol. 1995;6 (1):35–47. doi: 10.1681/ASN.V6135. [DOI] [PubMed] [Google Scholar]
- 13.Kerjaschki D, et al. Identification of a pathogenic epitope involved in initiation of Heymann nephritis. Proc Natl Acad Sci U S A. 1992;89 (23):11179–11183. doi: 10.1073/pnas.89.23.11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raychowdhury R, et al. Induction of Heymann nephritis with a gp330/megalin fusion protein. Am J Pathol. 1996;148 (5):1613–1623. [PMC free article] [PubMed] [Google Scholar]
- 15.Pippin JW, et al. Inducible rodent models of acquired podocyte diseases. Am J Physiol Renal Physiol. 2009;296 (2):F213–229. doi: 10.1152/ajprenal.90421.2008. [DOI] [PubMed] [Google Scholar]
- 16.Kerjaschki D, Neale TJ. Molecular mechanisms of glomerular injury in rat experimental membranous nephropathy (Heymann nephritis) J Am Soc Nephrol. 1996;7 (12):2518–2526. doi: 10.1681/ASN.V7122518. [DOI] [PubMed] [Google Scholar]
- 17.Faas MM, et al. Pregnancy aggravates proteinuria in subclinical glomerulonephritis in the rat. J Lab Clin Med. 1999;134 (3):267–274. doi: 10.1016/s0022-2143(99)90207-x. [DOI] [PubMed] [Google Scholar]
- 18.Benigni A, et al. Angiotensin-converting enzyme inhibition prevents glomerular-tubule disconnection and atrophy in passive Heymann nephritis, an effect not observed with a calcium antagonist. Am J Pathol. 2001;159 (5):1743–1750. doi: 10.1016/s0002-9440(10)63021-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagao T, et al. Effect of DP-1904, a thromboxane A2 synthase inhibitor, on passive Heymann nephritis in rats. Eur J Pharmacol. 1996;316 (1):73–80. doi: 10.1016/s0014-2999(96)00662-0. [DOI] [PubMed] [Google Scholar]
- 20.Makker SP, Tramontano A. Differential capacity of anti-RAP and anti-megalin antibodies to produce progressive passive Heymann nephritis - implications for the pathogenesis of idiopathic human membranous glomerulonephritis. J Pathol. 2006;210 (3):282–287. doi: 10.1002/path.2058. [DOI] [PubMed] [Google Scholar]
- 21.Zoja C, et al. Indomethacin reduces proteinuria in passive Heymann nephritis in rats. Kidney Int. 1987;31 (6):1335–1343. doi: 10.1038/ki.1987.147. [DOI] [PubMed] [Google Scholar]
- 22.Cybulsky AV, et al. Experimental membranous nephropathy redux. Am J Physiol Renal Physiol. 2005;289 (4):F660–671. doi: 10.1152/ajprenal.00437.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Susani M, et al. Antibodies to glycolipids activate complement and promote proteinuria in passive Heymann nephritis. Am J Pathol. 1994;144 (4):807–819. [PMC free article] [PubMed] [Google Scholar]
- 24.Cybulsky AV, et al. Complement-induced glomerular epithelial cell injury. Role of the membrane attack complex in rat membranous nephropathy. J Clin Invest. 1986;77 (4):1096–1107. doi: 10.1172/JCI112408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cosyns JP, et al. Immunohistochemical analysis of C3 cleavage fragments, factor H, and the C5b-9 terminal complex of complement in de novo membranous glomerulonephritis occurring in patients with renal transplant. Clin Nephrol. 1986;26 (4):203–208. [PubMed] [Google Scholar]
- 26.Baker PJ, et al. Depletion of C6 prevents development of proteinuria in experimental membranous nephropathy in rats. Am J Pathol. 1989;135 (1):185–194. [PMC free article] [PubMed] [Google Scholar]
- 27.Spicer ST, et al. Induction of passive Heymann nephritis in complement component 6-deficient PVG rats. J Immunol. 2007;179 (1):172–178. doi: 10.4049/jimmunol.179.1.172. [DOI] [PubMed] [Google Scholar]
- 28.Nakatsue T, et al. Nephrin and podocin dissociate at the onset of proteinuria in experimental membranous nephropathy. Kidney Int. 2005;67 (6):2239–2253. doi: 10.1111/j.1523-1755.2005.00328.x. [DOI] [PubMed] [Google Scholar]
- 29.Saran AM, et al. Complement mediates nephrin redistribution and actin dissociation in experimental membranous nephropathy. Kidney Int. 2003;64 (6):2072–2078. doi: 10.1046/j.1523-1755.2003.00305.x. [DOI] [PubMed] [Google Scholar]
- 30.Natori Y, Shindo N. Proteinuria induced by anti-dipeptidyl peptidase IV (gp108); role of circulating and glomerular antigen. Clin Exp Immunol. 1994;95 (2):327–332. doi: 10.1111/j.1365-2249.1994.tb06532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ronco P, et al. A monoclonal antibody to brush border and passive Heymann nephritis. Clin Exp Immunol. 1984;55 (2):319–332. [PMC free article] [PubMed] [Google Scholar]
- 32.Chen JS, et al. Mouse model of membranous nephropathy induced by cationic bovine serum albumin: antigen dose-response relations and strain differences. Nephrol Dial Transplant. 2004;19 (11):2721–2728. doi: 10.1093/ndt/gfh419. [DOI] [PubMed] [Google Scholar]
- 33.Assmann KJ, et al. Membranous glomerulonephritis in the mouse. Kidney Int. 1983;24 (3):303–312. doi: 10.1038/ki.1983.159. [DOI] [PubMed] [Google Scholar]
- 34.Assmann KJ, et al. A nephritogenic rat monoclonal antibody to mouse aminopeptidase A. Induction of massive albuminuria after a single intravenous injection. J Exp Med. 1992;175 (3):623–635. doi: 10.1084/jem.175.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dijkman HB, et al. Podocyte changes after induction of acute albuminuria in mice by anti-aminopeptidase A mAb. Nephron Exp Nephrol. 2003;94 (3):e85–93. doi: 10.1159/000072026. [DOI] [PubMed] [Google Scholar]
- 36.Border WA, et al. Induction of membranous nephropathy in rabbits by administration of an exogenous cationic antigen. J Clin Invest. 1982;69 (2):451–461. doi: 10.1172/JCI110469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maruyama S, et al. Membranous glomerulonephritis induced in the pig by antibody to angiotensin-converting enzyme: considerations on its relevance to the pathogenesis of human idiopathic membranous glomerulonephritis. J Am Soc Nephrol. 1999;10 (10):2102–2108. doi: 10.1681/ASN.V10102102. [DOI] [PubMed] [Google Scholar]
- 38.Prunotto M, et al. Autoimmunity in membranous nephropathy targets aldose reductase and SOD2. J Am Soc Nephrol. 21(3):507–519. doi: 10.1681/ASN.2008121259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miettinen A, et al. Monoclonal antibodies against membrane proteins of the rat glomerulus. Immunochemical specificity and immunofluorescence distribution of the antigens. Am J Pathol. 1990;137 (4):929–944. [PMC free article] [PubMed] [Google Scholar]
- 40.van Leer EH, et al. Redistribution of glomerular dipeptidyl peptidase type IV in experimental lupus nephritis. Demonstration of decreased enzyme activity at the ultrastructural level. Lab Invest. 1993;68 (5):550–556. [PubMed] [Google Scholar]
- 41.Tauc M, et al. Characterization of monoclonal antibodies specific for rabbit renal brush-border hydrolases: application to immunohistological localization. J Histochem Cytochem. 1988;36 (5):523–532. doi: 10.1177/36.5.2895788. [DOI] [PubMed] [Google Scholar]
- 42.Verroust PJ. Kinetics of immune deposits in membranous nephropathy. Kidney Int. 1989;35 (6):1418–1428. doi: 10.1038/ki.1989.143. [DOI] [PubMed] [Google Scholar]
- 43.Fukatsu A, et al. Local formation of immune deposits in rabbit renal proximal tubules. Kidney Int. 1988;34 (5):611–619. doi: 10.1038/ki.1988.225. [DOI] [PubMed] [Google Scholar]
- 44.Matsuo S, et al. Glomerulonephritis induced in the rabbit by antiendothelial antibodies. J Clin Invest. 1987;79 (6):1798–1811. doi: 10.1172/JCI113021. [DOI] [PMC free article] [PubMed] [Google Scholar]