

Abstract
Estrogens act upon nuclear estrogen receptors (ER) to ameliorate cell-mediated autoimmune disease. As most immunomodulatory effects of estrogens in EAE have been attributed to the function of ER-α, we previously demonstrated that ER-β ligand treatment reduced disease severity without affecting peripheral cytokine production or levels of CNS inflammation, suggesting a direct neuroprotective effect; however, the effect of ER-β treatment on the function of immune cells within the target organ remained unknown. Here, we used adoptive transfer studies to show that ER-β ligand treatment was protective in the effector, but not the induction phase of EAE, as shown by decreased clinical disease severity with the preservation of axons and myelin in spinal cords. The analysis of the immune cell infiltrates in the CNS revealed that while ER-β ligand treatment did not reduce overall levels of CNS inflammation, there was a decrease in the DC percentage, and these CNS DC had decreased TNF-α production. Finally, experiments using DC deficient in ER-β revealed that the expression of ER-β on DC was essential for protective effects of ER-β ligand treatment in EAE. Our results demonstrate for the first time an effect of ER-β ligand treatment in vivo on DC in the target organ of a prototypic cell-mediated autoimmune disease.
Keywords: Cell-trafficking, Dendritic cells, EAE/MS, Rodents
Introduction
Pregnancy confers protection in a variety of cell-mediated autoimmune diseases in humans and in their respective animal models, including psoriasis, myasthenia gravis, Grave’s disease, rheumatoid arthritis, and multiple sclerosis (MS) [1–4]. Late pregnancy in humans has been associated with a decrease in Th1 immune responses. In MS, the reduction in Th1 immunity during late pregnancy is paralleled by a reduction in relapses [5]. Estrogen treatment in the MS mouse model, experimental autoimmune encephalomyelitis, has been shown to reduce clinical disease by inhibiting a variety of disease-promoting mechanisms, including reductions in proinflammatory cytokines, chemokines, and migration factors, as well as increases in CD4+CD25+Foxp3+ T regulatory cells [6–10]. Estrogens signal primarily through two nuclear receptor subtypes, estrogen receptor (ER)-α and -β, whereas more rapid membrane effects have also been described [11, 12]. Although both ER are expressed in all immune cell types, most of the protective effects of estrogen treatment in EAE have been shown to be mediated through ER-α without evidence for involvement of ER-β signaling [13–15]. Recently, our lab has shown that ER-β ligand treatment during EAE reduced clinical disease relatively late and preserved axon densities despite a lack of an effect on decreasing CNS inflammation and altering peripheral cytokine production. This suggested a neuroprotective effect that was independent of influences on the peripheral immune system [16]. However, an effect of ER-β ligand treatment on the composition and the function of immune cells in the target organ during EAE remained unknown.
There is a great deal of evidence that APC localized to the CNS at sites of immune cell infiltration play a pivotal role in the outcome of neuroinflammation. The induction of EAE requires priming of antigen-specific CD4+ T cells (TC) in secondary lymphoid tissues, and re-activation of these CD4+ TC at the target organ by professional APC. DC can drive Th-cell differentiation and are potent APC that can influence innate and adaptive immune responses. DC in the healthy CNS normally reside in the meninges and around CNS blood vessels. Recent studies have shown that during adaptive immunity, mature myeloid DC preferentially accumulate at the perivascular inflammatory foci of the spinal cords during peak EAE disease severity, inducing the production of effector TC in the CNS [17–19]. In a model where DC were the only cells expressing MHCII molecules, DC alone were sufficient to initiate EAE [20]. Furthermore, during relapse-remitting EAE, CNS myeloid DC were shown to initiate epitope spreading, leading to expansion of encephalitogenic CD4+ TC that can induce clinical relapses [17]. Therefore, DC in the target organ are central to the immunopathogenesis of EAE and other Th1-mediated autoimmune diseases.
DC express ER-α and ER-β in many stages of development, and it is thought that ER signaling is involved in the development and function of these cells. In vivo and in vitro studies have revealed that estrogen-dependent DC development and maturation is in part mediated through ER-α [21, 22]. In autoimmunity, one study has shown that estrogen acting through ER-α can inhibit the development of EAE by reducing the number of DC in the secondary lymphoid organ during the priming phase [23]. In contrast, little is known about the role of ER-β during immune cell development, and even less is known about the role of ER-β on DC in the target organ during autoimmune disease. In the present study, we examined the effect of ER-β ligand treatment on immune cells in the CNS during chronic EAE. Our data demonstrate for the first time a role for ER-β in vivo on DC in the target organ of a prototypic cell-mediated autoimmune disease and thereby present a novel therapeutic target for future treatment of such diseases.
Results
ER-β ligand treatment during the effector phase of EAE reduces disease severity
To determine whether ER-β ligand treatment affects the induction or effector phase of EAE, adoptive transfer studies were performed in which donor (Fig. 1A) or recipient (Fig. 1C) mice were treated with ER-β ligand or vehicle. As shown in Fig. 1B, ER-β ligand treatment in the induction phase of EAE (in donor mice) did not alter the ability of autoantigen-stimulated lymph node cells (LNC) to induce EAE upon adoptive transfer to naïve, untreated recipient mice. Specifically, adoptive transfer of immune cells from ER-β ligand-treated donor mice resulted in EAE disease severity in recipients that was comparable to disease induced by immune cells from vehicle-treated donors. As a positive control for detecting a treatment effect, ER-α ligand treatment in the induction phase of EAE decreased encephalitogenicity, as shown by decreased disease in naïve recipients (Fig. 1B).
Figure 1.
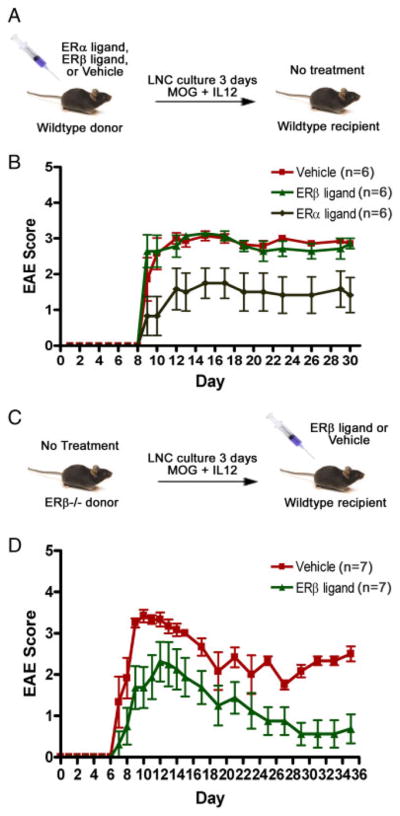
ER-β ligand treatment effects in the induction versus the effector phase of adoptive EAE. (A) Induction phase treatment. WT C57BL/6 mice were adoptively transferred with LNC from ER-β ligand-or vehicle-treated WT donor mice immunized with myelin oligodendrocyte glycoprotein (MOG) 35–55 peptide. (B) EAE score of mice treated as described in (A). ER-α ligand treatment was used as the positive control (p = 0.05, repeated one-way ANOVA). (C) Effector phase treatment. Thy1-YFP tg mice treated with ER-β ligand were adoptively transferred with LNC from untreated ER-β−/− mice immunized with autoantigen. (D) EAE score of mice treated as described in (C) (p = 0.04, days 25–35, repeated one-way ANOVA). Each group consisted of 6–7 mice in each experiment. Data are representative of three independent experiments.
To determine whether ER-β ligand may instead function in the effector phase of EAE, ER-β−/− mice were immunized and their autoantigen-stimulated LNC adoptively transferred into ER-β-ligand-treated recipient mice (Fig. 1C). The use of ER-β−/− mice in the induction phase eliminated any possible effects of ER-β ligand treatment on donor cells in recipient mice. In contrast to no effect of ER-β ligand treatment during the induction phase, ER-β ligand treatment during the effector phase (in recipient mice) decreased EAE disease severity as compared with vehicle-treated EAE mice (Fig. 1D). These results demonstrated that ER-β ligand treatment in the effector phase, but not the induction phase, reduced the severity of clinical EAE. These data show that ER-β ligand treatment does not act through priming of antigen-specific effector TC during induction of the immune response, but rather reduces the ability of such effectors to invoke disease in the target organ.
ER-β ligand treatment preserved axonal densities and myelin staining in the spinal cord
Next, neuropathology was assessed in mice treated with ER-β ligand during the effector phase of adoptive EAE. Neurons and axons in spinal cord sections of ER-β ligand and vehicle-treated animals that received ER-β−/− donor LNC were visualized by neurofilament-200 (NF200) staining (Fig. 2A, top). In addition, these recipient mice carried a transgene for yellow fluorescent protein (YFP) under the control of the neuronal-specific Thy1 promoter; thus, YFP expression was used to confirm NF200 immunofluorescent staining. NF200 immunoreactivity completely overlapped with YFP expression (not shown). Quantification of NF200 staining revealed significantly reduced axonal densities in vehicle-treated mice with adoptive EAE compared with that of healthy controls, whereas ER-β ligand-treated EAE mice demonstrated preservation of axonal densities to levels comparable to that of healthy controls (Fig. 2B, left).
Figure 2.
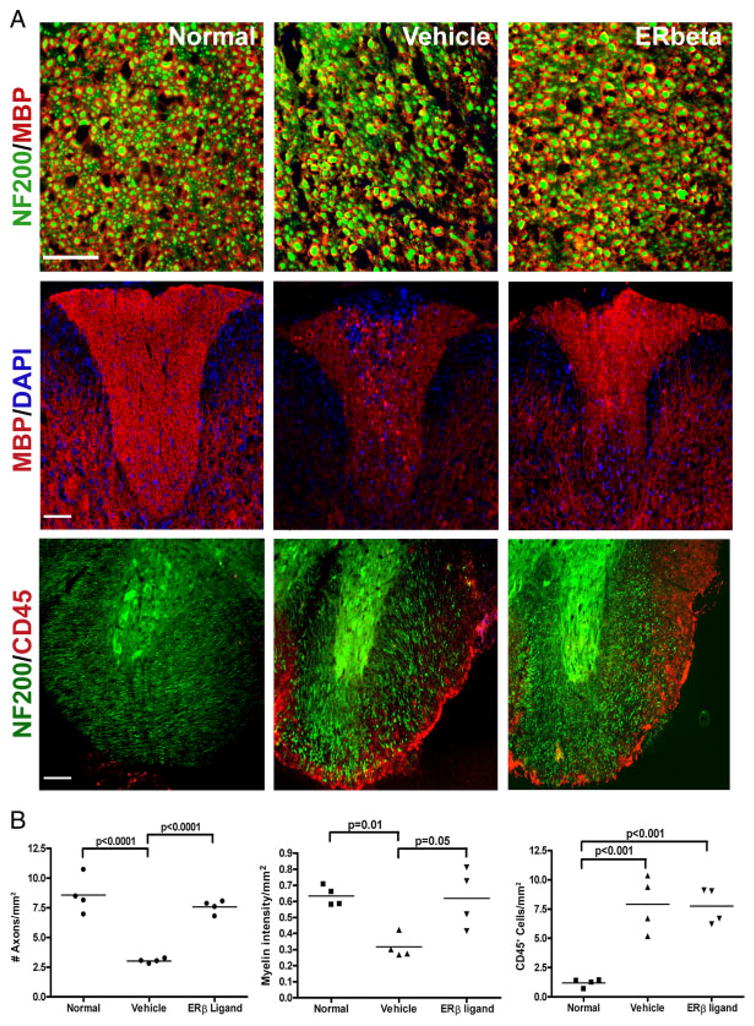
ER-β ligand treatment during the effector phase of EAE: effects on axonal densities. (A, top row) Representative 40 × captures of spinal cord sections at the thoracic lateral funiculus of control (left), vehicle-treated (middle), and ER-β ligand-treated (right) EAE mice sacrificed at day 35 post-adoptive transfer. Myelin and axons were stained with MBP (red) and NF200 (green), respectively. Scale bar, 50 μm. (A, middle row) Representative 10 × captures of the dorsal column of spinal cord sections stained with MBP (red) and DAPI (blue) for cell nuclei. Scale bar, 100 μm. (A, bottom row) Representative 10 × confocal images of spinal cord lateral funiculus cross-sections at the thoracic level were stained with NF200 (green) and pan-leukocyte marker CD45 (red). Scale bar, 100 μm. (B) Quantification of axonal densities (left), myelin staining intensity (middle), and CD45 immunoreactivity (right) as shown in (A). Four mice in each treatment group were examined with three sections per mouse for a total of 12 sections analyzed for each treatment group. p-values were determined by one-way ANOVA. Data are representative of three independent experiments.
Since myelin is integral to proper saltatory conduction along axons, myelin staining intensity was also examined in these spinal cords. Consistent with a decrease in axonal density, vehicle-treated EAE mice also exhibited decreased myelin basic protein (MBP) staining intensity when compared with healthy controls. In contrast, ER-β ligand treatment significantly preserved MBP staining intensity as compared with vehicle treatment (Fig. 2A and B, middle). These results showed that ER-β ligand treatment in the effector phase of adoptive EAE preserved myelin and axons. Despite this neuroprotection, ER-β ligand treatment did not prevent the accumulation of inflammatory infiltrates in the CNS of mice in the effector phase of adoptive EAE (Fig. 2A, bottom). Both ER-β ligand and vehicle-treated EAE mice had levels of CNS inflammation that were significantly increased compared with healthy controls (Fig. 2B, right). Together, these data demonstrated that ER-β ligand treatment during the effector phase of EAE resulted in neuroprotection, despite the accumulation of CNS inflammation.
ER-β ligand treatment altered immune cell composition in the CNS of mice with EAE
Although ER-β ligand treatment of EAE mice did not result in a decrease in the level of CNS inflammation, it remained possible that the cellular composition of the inflammation was affected by the treatment. Thus, CNS infiltrates were characterized for cellular composition in experiments where ER-β ligand was administered only during the effector phase of adoptive EAE, to recipient mice. In these experiments, mice were treated during the effector phase with either ER-β ligand or vehicle, and at disease onset immune cells from the CNS were isolated and assessed by flow cytometry. Confirming immunohistochemistry data in Fig. 2, there were no appreciable differences in the expression of CD45 in the CNS between ER-β ligand and vehicle-treated groups when assessed by flow cytometry (Fig. 3B). Further, although ER-β ligand treatment in the effector phase of EAE had no effect on the levels of CD4+ or CD8+ TC in the CNS of EAE mice (Fig. 3C), there was a significant decrease in the percentage of CD11b/CD11c+ DC (Fig. 3D and E). Notably, ER-β ligand treatment did not alter the percentage of CD4+CD25hi-Foxp3+ T regulatory cells that could potentially suppress encephalitogenic TC in the CNS (not shown). Naïve mice did not show detectable levels of TC or DC in the CNS. Further analysis of CD11b/CD11c+ DC in the CNS of EAE mice revealed that ER-β ligand treatment appeared to decrease MHCII expression when compared with vehicle-treated mice, but there were no differences in the level of expression of the costimulatory molecules CD80 and CD86 on DC between treatment groups (Supporting Information Fig. 1). Altogether, these results showed that the cellular composition of CNS inflammation in EAE was affected by ER-β ligand treatment during the effector phase. Specifically, ER-β ligand treatment decreased the percentage of CD11b/CD11c+ DC in the CNS.
Figure 3.
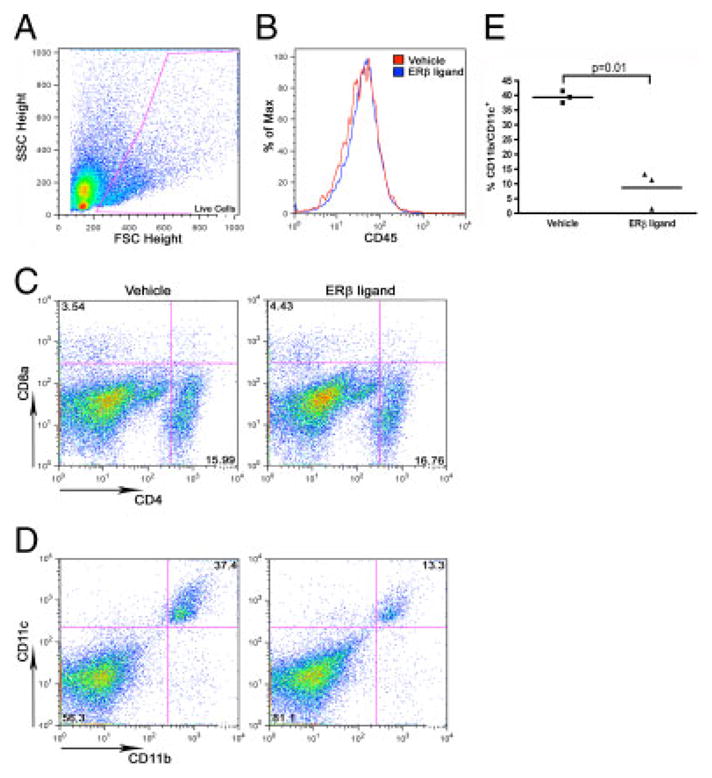
ER-β ligand treatment effects on DC in the CNS of EAE mice. Thy1-YFP transgenic mice treated with vehicle or ER-β ligand were adoptively transferred with autoantigen-stimulated ER-β−/− LNC (3 × 106 cells/mouse). CNS immune cells were pooled from 7–10 animals at disease onset (10 days post-adoptive transfer) and analyzed by flow cytometry. Cells were stained with CD45, and CD4 and CD8α, or CD11b and CD11c and gated on live cell populations. (A) Forward scatter (FSC) versus side scatter (SSC) dot plot with gate on live cells that were further analyzed in (B)–(D). (B) Histogram of CD45+ infiltrating cells in the CNS of ER-β (blue) and vehicle (red) treated EAE mice. (C) Dot plots of CD4+ or CD8+ T-cell populations in the CNS of ER-β ligand (right) versus vehicle-treated (left) EAE mice. (D) Dot plots of CD11b/CD11c+ DC in the CNS of ER-β ligand (right) versus vehicle-treated (left) EAE mice. (E) Statistical analysis of the percentage of CD11b/CD11c+ DC in the CNS of ER-β ligand (right) versus vehicle-treated (left) EAE mice (p = 0.01, paired t-test). In all experiments, three samples were examined for each treatment group. Dot plots and histograms are representative of results of three independent experiments.
ER-β ligand treatment decreased the production of TNF-α by DC in the CNS of EAE mice
We next asked whether ER-β ligand treatment might affect cytokine production by DC in the target organ. We focused on TNF-α because TNF-α is known to mediate demyelination and axonal transection in EAE [24, 25], and we had observed protection of myelin and axons with ER-β ligand treatment (Fig. 2). DC were sorted ex vivo from the CNS of ER-β ligand and vehicle-treated mice at disease onset and TNF-α mRNA levels were quantified by RT-PCR. TNF-α mRNA levels were reduced by 40% in CD11b/CD11c+ DC derived from ER-β ligand-treated EAE mice as compared with vehicle-treated (Fig. 4A). Together, these data showed that in addition to reducing the number of DC in the target organ (Fig. 3), ER-β ligand treatment also reduced their ability to make TNF-α.
Figure 4.
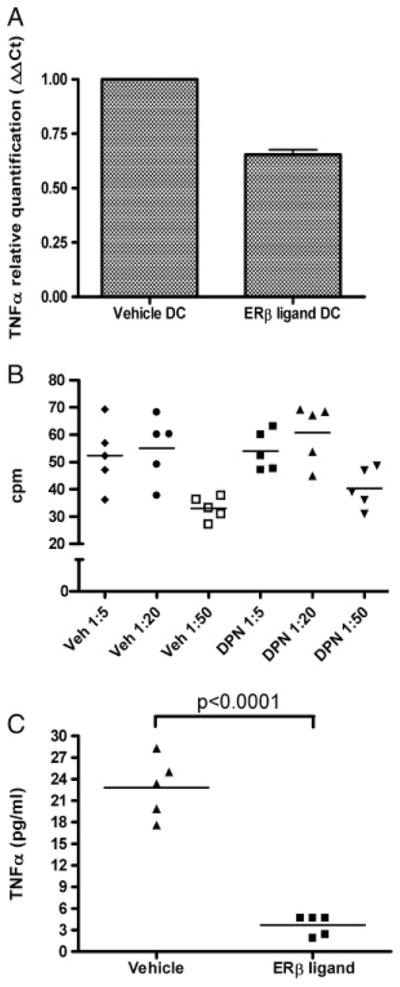
ER-β ligand treatment effects on the production of TNF-α by CNS dendritic cells. (A) Relative quantification of TNF-α mRNA levels in CD11b/CD11c+ DC (left) isolated from the CNS of EAE mice treated with vehicle or ER-β ligand at disease onset (10 days post-adoptive transfer). GAPDH served as an internal control. The results are presented with the vehicle-treated group normalized to a level of one, and ER-β ligand-treated group as a fraction thereof. Data show mean+SD pooled from three independent experiments. (B) Proliferation of DC/TC co-culture. DC were sorted from the CNS of ER-β ligand or vehicle-treated EAE mice, whereas TC were sorted from lymph nodes of a separate group of untreated immunized mice ten days post-immunization. CNS DC and LN TC were re-stimulated with autoantigen at ratios of 1:5, 1:20, and 1:50, with 1 × 105 T-cells/well. (C) TNF-α levels from DC/TC co-culture. TNF-α levels were measured from supernatants of cultures of DC/TC at the 1:5 ratio (p<0.0001, Student’s t-test). CNS DC were pooled from two mice for one sample for a total of five samples from ten mice in each treatment group. LN TC were collected and pooled from ten untreated mice.
ER-β ligand treatment of DC decreased the in vitro production of TNF-α during MOG-specific stimulation
To further determine whether ER-β ligand treatment in vivo induced functional changes in CNS DC, we performed DC/TC co-cultures. DC were derived from the CNS of ER-β ligand or vehicle-treated EAE mice, whereas autoantigen-primed TC were obtained from LN of untreated mice immunized with autoantigen. Consistent with the previous studies using co-cultures [26], autoantigen stimulation of co-cultures resulted in proliferation at DC/TC ratios of 1:5 and 1:20, but not at 1:50. Notably, there was no difference in this proliferation when comparing DC derived from ER-β ligand versus vehicle-treated mice (Fig. 4B). However, when TNF-α levels were examined in supernatants, decreased levels of TNF-α were found in cultures that contained DC derived from the CNS of ER-β ligand-treated, as compared with vehicle-treated mice (Fig. 4C). In this experiment, it is possible that the source of TNF-α may be DC and TC. As TNF-α can mediate demyelination and axonal transection in EAE [27, 28], effects on TNF-α production when DC were treated with ER-β ligand were consistent with reduced demyelination and axonal loss in ER-β ligand-treated EAE mice (Fig. 2).
ER-β expression in DC is essential for ER-β ligand-mediated protection in EAE
Finally, to determine the functional significance of ER-β expression on DC during ER-β ligand treatment in EAE, we performed another set of adoptive transfer studies using donor cells that lacked ER-β expression in DC. Immunized ER-β−/− and WT donors LNC were sorted for CD11b/CD11c+ DC and CD11b/CD11c− (non-DC) fractions. The DC fractions were from ER-β−/− or WT mice, whereas non-DC fractions were all from WT mice. Cells from various ER-β−/− and WT donors were mixed with the ratios of DC (3%) and non-DC (97%) based on the immune cell composition of non-manipulated immunized donor LNC, then stimulated with autoantigen before adoptive transfer into ER-β ligand- or vehicle-treated recipient mice (Fig. 5A). As shown in Fig. 5B, ER-β ligand-treated mice adoptively transferred with WT DC (green) had reduced EAE disease severity compared with ER-β ligand-treated mice that were adoptively transferred ER-β−/− DC (orange). These results demonstrated that ER-β ligand treatment during the effector phase of EAE acts at least in part on ER-β-expressing DC.
Figure 5.
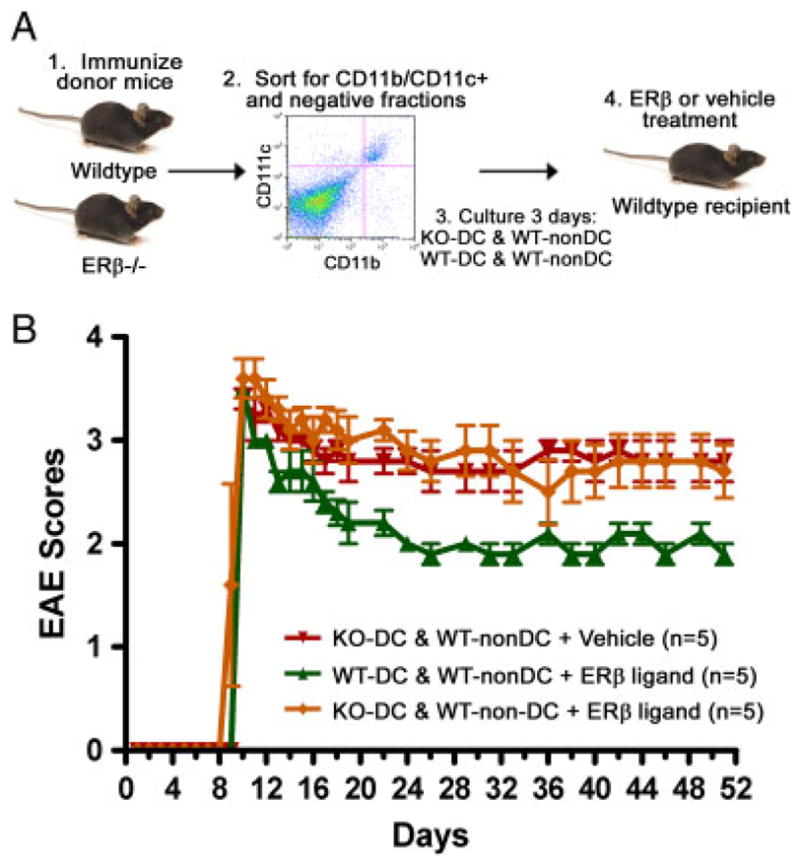
The role of ER-β expression on DC in the protective effect of ER-β ligand treatment in EAE. (A) LNC from immunized WT and ER-β−/− donor mice were sorted for CD11b/CD11c+ DC and CD11b/CD11c− (non-DC) populations, then various DC and non-DC were mixed as indicated, restimulated with autoantigen for 3 days, and adoptively transferred to ER-β ligand or vehicle-treated recipient mice. (B) EAE score of mice treated as described in (A) (p<0.0001, repeated one-way ANOVA).
Discussion
Previously, our lab showed that ER-β ligand treatment was neuroprotective in active EAE without altering cytokine production of autoantigen-specific immune cells in the periphery and without reducing the level of CNS inflammation. Specifically, ER-β ligand treatment preserved axon densities and myelin staining late in disease despite persistent inflammation in the CNS [16]. However, it remained unknown whether qualitative differences might exist in the inflammatory infiltrates of ER-β ligand-treated EAE mice. Therefore, in the present study, we examined immune cells in the CNS of EAE mice treated with ER-β ligand. We found that ER-β ligand treatment conferred clinical protection in the effector phase of adoptive EAE and reduced the percentage of DC in the target organ. DC isolated from the CNS of ER-β ligand-treated EAE mice exhibited decreased TNF-α production. Finally, we showed that ER-β ligand treatment in EAE conferred disease protection through ER-β expressed on DC. This is the first study elucidating an in vivo immunomodulatory role for ER-β during autoimmune demyelinating disease.
DC are emerging as critical mediators of inflammation in a variety of organ-specific autoimmune diseases such as rheumatoid arthritis, psoriasis, and EAE due to their efficient antigen-presenting ability [20, 26, 28–31]. CNS DC are critical to EAE pathogenesis, as DC infiltrates in the CNS during EAE preferentially localize with effector TC at sites of inflammation and they alone can activate infiltrating naïve TC to differentiate and perpetuate inflammation [20, 28]. Our finding of quantitative and qualitative effects of ER-β ligand treatment on CNS DC, which occurred in a setting of improved clinical and neuropathologic disease corroborates other studies showing that CNS DC play a critical role in EAE disease severity [32–34]. Further, ER-β ligand treatment can now be considered as a novel treatment strategy targeting DC in the CNS.
DC are excellent targets for organ-specific autoimmune diseases for several reasons. Within the innate arm of immunity, DC represent a potent population of APC that are not only capable of inducing innate immune responses in the periphery but also are essential for the perpetuation of adaptive immune responses in the target organ during autoimmunity. Thus, an advantage of targeting DC is the prevention of the recruitment of adaptive immune responses. Treatments targeting DC represent a more selective strategy and an advantage over treatments that involve pan immunosuppression or the depletion of T or B cells since these latter methods are associated with adverse effects such as increased susceptibility to infection. Thus far, there has only been one molecule, CEP-701, an flt-3 ligand inhibitor, developed to selectively target DC [35]. Although CEP-701 has been shown to reduce EAE disease severity, the side effects of this novel compound in humans remain to be determined. Recently, another group has found that the injection of neural stem/precursor cells could hamper the maturation of DC and thus the development of EAE, but the therapeutic use of neural stem/precursor cells remains unknown [32]. ER-β ligand treatment presents a relatively safe candidate for DC modulation because estrogen treatments have been widely used in humans for decades, and the adverse effects of estrogen treatment on the breast and uterus lining are mediated through ER-α, not ER-β.
Distinct protective mechanisms have been shown for ER-α and ER-β during autoimmune demyelinating disease, and it is possible that antagonistic effects of ER-α and ER-β may also exist. Antagonistic effects of ER-α and ER-β intracellularly have been reported whereby ER-β can lead to transdominant negative regulation of ER-α [36]. In the uterus, a tissue replete with both receptors, ER-β ligand treatment is known to antagonize the ER-α-mediated increase in uterine weight [37]. In the immune system, estrogens are known to modulate many immune cell types, including the development of bone marrow-derived DC into fully functional APC [21]. In vitro studies have identified ER-α as a critical mediator of these developmental events during immunity, whereas the role of ER-β was not previously found [38]. Our in vivo data implicate the role of ER-β in the attenuation of DC function in the target organ during the effector phase of auto-immune demyelinating disease. ER-α signaling is critical to hematopoietic cell differentiation into DC, and while this is conducive to generating an effective immune response, an overproduction of immunogenic DC may lead to autoimmunity. We speculate that ER-β may serve as a negative regulator of ER-α-mediated DC development and maturation during health, and that autoimmunity may ensue when ER-β-mediated regulation fails.
The role of cytokines in the neuropathologic outcome of neuroinflammatory diseases has long been recognized. An important functional consequence of ER-β ligand treatment on DC may be the ability to reduce TNF-α production by DC in the target organ. TNF-α, an inflammatory cytokine, is produced broadly by the immune system and has been associated with increased neuroinflammation in several acute and chronic neurological disorders, including traumatic brain injury, ischemia, Parkinson’s disease, Alzheimer’s disease, amyotrophic lateral sclerosis, and MS [39–41]. In MS patients, CSF and serum levels of TNF-α are elevated compared with healthy subjects, and a rise in TNF-α in PBMCs has also been shown to precede clinical relapses [25, 42]. TNF-α signaling through the neurotrophin receptor p55 in neurons and glia can mediate glutamate toxicity or lead to the activation of apoptotic signaling cascades (NF-κB, JNK, or p38 pathway) [42, 43]. Notably, estradiol’s protective effect in EAE has been attributed in part to its ability to inhibit the production of proinflammatory cytokines such as TNF-α from peripheral immune cells, and this has been shown to be mediated through ER-α [43, 44]. Our results demonstrating an ER-β ligand-mediated reduction TNF-α in DC in the CNS in vivo, and in DC:TC cultures in vitro, which correlated with sparing of myelin and axons, together demonstrate a previously unknown immunomodulatory capacity for ER-β treatment. Notably, because ER-β is broadly expressed in the CNS on neurons, astrocytes, and oligodendrocytes, our findings do not preclude additional neuroprotective mechanisms as well. Nevertheless, our findings clearly support the notion that ER-β ligand treatment should now be considered a potential strategy to attenuate DC function in the target organ of autoimmune demyelinating diseases.
Materials and methods
Animals
Female ER-β homozygous knockout mice were purchased from Taconic Farms (Germantown, NY, USA), and female WT C57BL/6 and B6.Cg-Tg (Thy1-YFP) 16Jrs/J mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Animals were maintained under standard conditions in a 12-h dark/light cycle with access to food and water ad libitum. All procedures were done in accordance with the guidelines of the National Institutes of Health and the Chancellor’s Animal Research Committee of the University of California, Los Angeles Office for the Protection of Research Subjects.
Adoptive EAE and hormone manipulations
Animals were subcutaneously injected with myelin oligodendrocyte glycoprotein (MOG), amino acids 35–55 (200 μg/animal, American Peptides) emulsified in complete Freund’s adjuvant and supplemented with Mycobacterium tuberculosis H37Ra (200 μg/animal, Difco Laboratories) over four draining inguinal and axillary LN sites in a volume of 0.1 mL/mouse. Animals were either treated with vehicle consisting of 10% molecular-grade ethanol (EM Sciences) and 90% Miglylol 812N liquid oil (Sasol North America) or the ER-β ligand, Diarylproprionitrile (Tocris Biosciences) diluted with vehicle at a dose of 8 mg/kg/day for seven days before immunization or adoptive transfer of in vitro stimulated lymphocytes. This dose of ER-β ligand had previously been shown to induce a known biological response in vivo on a positive control tissue, specifically the ability to block ER-α-mediated increases in uterine weight [16, 37]. For adoptive transfer, LNC were cultured in 24-well plates at a concentration of 4 × 106 cells/mL of complete RPMI medium containing 5% heat-inactivated FBS, 1 mM sodium pyruvate, L-glutamine, 2ME, NEAA, Pen-strep, and 25 mM HEPES buffer. For adoptive transfer of ER-β−/− or WT DC and non-DC mixture, LNC obtained from ER-β−/− or WT mice were first separated by flow cytometry cell sorting (see Cell Sorting and RT-PCR). Subsequently, WT non-DC were cultured with 3% ER-β−/− or WT DC. Cells were stimulated with 25 μg/mL MOG, amino acids 35–55, and 20 ng/mL recombinant mouse IL-12 (BD Biosciences and Biolegend) for 72 h at 37° C, 5% CO2. On the third day of culture, LNC were washed with 1 × PBS and each animal received 3 × 106 cells in 0.3 mL ice-cold injection-grade 1 × PBS by i.p. injection. Animals were monitored daily for EAE signs based on a standard EAE 0–5 scale scoring system: 0—healthy, 1—complete loss of tail tonicity, 2—loss of righting reflex, 3—partial paralysis, 4—complete paralysis of one or both hind limbs, and 5—moribund.
Mononuclear cell isolation
To isolate mononuclear cells from the brain and spinal cord, animals were deeply anesthetized with isoflurane and perfused transcardially with ice-cold 1 × PBS for 20–30 min. Brains were dissected and spinal cords were flushed with 1 × PBS into complete RPMI medium (Lonza). CNS tissues from each group (n = 7) were pooled to achieve a sufficient amount of immune cells for in vitro cell culture or flow cytometric analysis. CNS tissues were digested with Liberase Blendzyme I (Roche Applied Science), DNaseI (Invitrogen), and 1 mM MgCl2 (Sigma) in HBSS for 30 min at 37°C, then passed through a wire mesh screen, followed by 100, 70, and 40 μm nylon cell strainers to obtain single cell suspensions. Cells were washed in complete RPMI medium and suspended in 50% Percoll (GE Healthcare Biosciences) medium in HBSS. Mononuclear cells were collected at the 63:50% interface of a 63:50:30% Percoll step gradient following 30 min centrifugation at 1800 rpm at 4°C. Inguinal and axillary LN and spleens were passed through a wire mesh, followed by 70 and 40 μm nylon cell strainers. To remove erythrocytes, splenocytes were suspended in complete RPMI medium, overlaid at 1:1 ratio onto Lymphoprep (Accurate Chemical) medium and mononuclear cells were collected at the Lymphoprep/RPMI interface following 30 min centrifugation at 1200 rpm in 4°C.
DC and TC co-culture
CD11c+ DC were isolated from the CNS of 20 mice ten days post-immunization with 200 μg MOG, amino acids 35–55, in complete Freund’s adjuvant. These mice had been treated in vivo with either ER-β ligand or vehicle beginning 7 days prior to immunization. Another group of ten untreated mice were also immunized with MOG 35–55 and LNC sorted for CD3+ TC. Subsequently, cells were co-cultured in 96-well plates for 96 h at 37° C, 5% CO2 in the presence of 25 μg/mL MOG, amino acids 35–55, at ratios of 1:5, 1:20, and 1:50 DC/TC, with each well containing 1 × 105 TC. At 72 h, 1 μCi of 3H-thymidine was added to the wells. At 96 h, supernatants were collected and the cells were harvested for a proliferation assay using a Betaplate counter (Wallac, Model 1205).
Cell sorting and RT-PCR
All cell sorting for in vitro cell culture and RT-PCR was performed in the UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research Flow Cytometry Core Facility that is supported by National Institutes of Health awards CA-16042 and AI-28697, and by the JCCC, the UCLA AIDS Institute, the David Geffen School of Medicine at UCLA, and the UCLA Chancellor’s Office. Cells were surface labeled for CD11b-FITC and CD11c-APC double-positive DC or CD3-APC (Biolegend) positive TC using the FACSAria II cytometer and FACSDiva software, version 6.1. RT-PCR for mouse TNF-α mRNA levels in CNS CD11b/CD11c+ DC was performed by SABiosciences (Frederick, MD, USA) using the Delta–Delta count method and mouse GAPDH as the control.
Flow cytometry
Mouse mononuclear cells or splenocytes were collected on a 96 v-shaped plate (Titertek) for flow cytometric analysis. Single cell suspensions in FACS buffer (2% FBS in PBS) were incubated with anti-CD16/32 at 1:100 dilution for 20 min at 4°C to block Fc receptors, centrifuged, and resuspended in FACS buffer with the following Ab added at 1:100 dilution for 30 min at 4°C: anti-CD11b, anti-CD11c, anti-CD8, anti-CD4, anti-CD25, anti-CD80, anti-CD86, anti-MHCII, and Rat-IgG1, -IgG2a, and -IgG2b isotype controls (Biolegend). Cells were subsequently washed twice in FACS buffer and then acquired on FACSCalibur (BD Biosciences) and analyzed by FlowJo software (Treestar). Quadrants were determined using cells labeled with appropriate isotype control Ab. All flow cytometry figures represent best of three experiments.
Histological preparation
Mice were deeply anesthetized in isoflurane and perfused transcardially with ice-cold 1 × PBS for 20–30 min, followed by 10% formalin for 10–15 min. Spinal cords were dissected and submerged in 10% formalin overnight at 4°C, followed by 30% sucrose for 24 h. Spinal cords were cut in thirds and embedded in a 75% gelatin/15% sucrose solution. Forty-micrometer thick free-floating spinal cord cross-sections were obtained with a microtome cryostat (Model HM505E) at −20°C. Tissues were collected serially and stored in 1 × PBS with 1% sodium azide in 4°C until immunohistochemistry.
Immunohistochemistry
Prior to histological staining, 40-μm thick free-floating sections were thoroughly washed with 1 × PBS to remove residual sodium azide. In the case of anti-MBP labeling, tissue sections undergo an additional 2-h incubation with 5% glacial acetic acid in 100-proof ethanol at room temperature, followed by 30 min incubation in 3% hydrogen peroxide in PBS. All tissue sections were permeabilized with 0.3% Triton X-100 in 1 × PBS and 2% normal goat serum for 30 min at room temperature and blocked with 10% normal goat serum in 1 × PBS for 2 h or overnight at 4° C. The following primary Ab were used: anti-MBP at 1:1000 dilutions, anti-CD45 at 1:500 dilutions (Chemicon), and anti-NF200 at 1:750 dilutions (Sigma). Tissues labeled with anti-MBP continue with a secondary Ab labeling step consisting of 1 h incubation with biotinylated IgG Ab at 1:1000 dilutions (Vector Labs), followed by 1.5-h incubation with strepavidin Ab conjugated to Alexa 647 fluorochrome (Chemicon). All other tissues follow with secondary Ab conjugated to TRITC or Cy5 (Vector Labs and Chemicon) for 1.5 h. To assess the number of cells, a nuclear stain DAPI (2 ng/mL; Molecular Probes) was added 10 min prior to final washes after secondary Ab incubation. Sections were mounted on slides, allowed to semi-dry, and cover slipped in fluoromount G (Fisher Scientific). IgG-control experiments were performed for all primary Ab, and only non-immunoreactive tissues under these conditions were analyzed.
Microscopy
Stained sections were examined and photographed using a confocal microscope (Leica TCS-SP, Mannheim, Germany) or a fluorescence microscope (BX51WI; Olympus, Tokyo, Japan) equipped with Plan Fluor objectives connected to a camera (DP70; Olympus). Digital images were collected and analyzed using Leica confocal and DP70 camera software. Images were assembled using Adobe Photoshop (Adobe Systems, San Jose, CA, USA).
Quantification
To quantify immunohistochemical staining results, three spinal cord cross-sections at the T1–T5 level from each mouse (n = 3) were captured under microscope at 10 × or 40 × magnification using the DP70 Image software and a DP70 camera (both from Olympus). All images in each experimental set were captured under the same light intensity and exposure limits. Image analysis was performed using ImageJ Software v1.30, downloaded from the NIH website (http://rsb.info.nih.gov/ij). Axonal densities were calculated by counting the number of NF200+ cells in a 40 × image over the area of the captured tissue section. Inflammatory infiltrates were quantified by measuring the intensity of CD45 staining in captured 10 × images.
Statistical analysis
EAE severity significance was determined by repeated measures one-way ANOVA. Immunohistochemical and flow cytometry data were analyzed by bootstrap one-way ANOVA and paired t-test, respectively. For these analyses, the mean or median was used as the comparator, and the F-stat equation was modified such that absolute values replaced the squaring of values. For bootstrap one-way ANOVA, post hoc analysis was performed on F-stat values and significance was determined at the 95% confidence interval.
Supplementary Material
Acknowledgments
The authors acknowledge Tina Chung, BS for technical laboratory assistance and Stefan Gold, PhD for helpful suggestions and discussions. The support for this work was provided by National Institutes of Health grant K24NS062117 and National Multiple Sclerosis Society grants RG 3593, 4033, and CA1028 to R.R.V., as well as by the Skirball Foundation, the Hilton Foundation, and the Sherak Family Foundation.
Abbreviations
- ER
estrogen receptor
- LNC
lymph node cells
- MBP
myelin basic protein
- MOG
myelin oligodendrocyte glycoprotein
- NF200
neurofilament-200
- TC
T cells
Footnotes
Conflict of Interest: The authors declare no financial or commercial conflict of interest.
Supporting Information available online
References
- 1.Murase JE, Chan KK, Garite TJ, Cooper DM, Weinstein GD. Hormonal effect on psoriasis in pregnancy and post partum. Arch Dermatol. 2005;141:601–606. doi: 10.1001/archderm.141.5.601. [DOI] [PubMed] [Google Scholar]
- 2.Djelmis J, Sostarko M, Mayer D, Ivanisevic M. Myasthenia gravis in pregnancy: report on 69 cases. Eur J Obstet Gynecol Reprod Biol. 2002;104:21–25. doi: 10.1016/s0301-2115(02)00051-9. [DOI] [PubMed] [Google Scholar]
- 3.LeBeau SO, Mandel SJ. Thyroid disorders during pregnancy. Endocrinol Metab Clin North Am. 2006;35:117–136. vii. doi: 10.1016/j.ecl.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 4.Russell AS, Johnston C, Chew C, Maksymowych WP. Evidence for reduced Th1 function in normal pregnancy: a hypothesis for the remission of rheumatoid arthritis. J Rheumatol. 1997;24:1045–1050. [PubMed] [Google Scholar]
- 5.Confavreux C, Hutchinson M, Hours MM, Cortinovis-Tourniaire P, Moreau T, Grp PMS. Rate of pregnancy-related relapse in multiple sclerosis. N Engl J Med. 1998;339:285–291. doi: 10.1056/NEJM199807303390501. [DOI] [PubMed] [Google Scholar]
- 6.Ito A, Bebo BF, Matejuk A, Zamora A, Silverman M, Fyfe-Johnson A, Offner H. Estrogen treatment down-regulates TNF-alpha production and reduces the severity of experimental autoimmune encephalomyelitis in cytokine knockout mice. J Immunol. 2001;167:542–552. doi: 10.4049/jimmunol.167.1.542. [DOI] [PubMed] [Google Scholar]
- 7.Jansson L, Olsson T, Holmdahl R. Estrogen induces a potent suppression of experimental autoimmune encephalomyelitis and collagen-induced arthritis in mice. J Neuroimmunol. 1994;53:203–207. doi: 10.1016/0165-5728(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 8.Bebo BF, Fyfe-Johnson A, Adlard K, Beam AG, Vandenbark AA, Offner H. Low-dose estrogen therapy ameliorates experimental autoimmune encephalomyelitis in two different inbred mouse strains. J Immunol. 2001;166:2080–2089. doi: 10.4049/jimmunol.166.3.2080. [DOI] [PubMed] [Google Scholar]
- 9.Gold S, Sasidar MV, Morales LBJ, Du S, Tiwari-Woodruff S, Sicotte NL, Voskuhl R. Estriol treatment decreases matrix metalloproteinase (MMP)-9 in autoimmune demyelinating disease through estrogen receptor (ER)-alpha. Lab Invest. 2009;89:1076–1183. doi: 10.1038/labinvest.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim S, Voskuhl RR. Decreased IL-12 production underlies the decreased ability of male lymph node cells to induce experimental autoimmune encephalomyelitis. J Immunol. 1999;162:5561–5568. [PubMed] [Google Scholar]
- 11.Yates MA, Li Y, Chlebeck PJ, Offner H. GPR30, but not estrogen receptor-alpha, is crucial in the treatment of experimental autoimmune encephalomyelitis by oral ethinyl estradiol. BMC Immunol. 2010;11:20–25. doi: 10.1186/1471-2172-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang C, Dehghani B, Li Y, Kaler LJ, Proctor T, Vandenbark AA, Offner H. Membrane estrogen receptor regulates experimental autoimmune encephalomyelitis through up-regulation of programmed death 1. J Immunol. 2009;182:3294–3303. doi: 10.4049/jimmunol.0803205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morales LBJ, Loo KK, Liu HB, Peterson C, Tiwari-Woodruff S, Voskuhl RR. Treatment with an estrogen receptor alpha ligand is neuroprotective in experimental autoimmune encephalomyelitis. J Neurosci. 2006;26:6823–6833. doi: 10.1523/JNEUROSCI.0453-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voskuhl R, Loo KK, Palaszynski K, Ashouri J, Lubahn D, Liu HB. Estrogen receptor-alpha mediates estrogen’s immune protection in EAE. Mult Scler. 2004;171:6936–6940. doi: 10.4049/jimmunol.171.12.6936. [DOI] [PubMed] [Google Scholar]
- 15.Liu HB, Loo KK, Palaszynski K, Ashouri J, Lubahn DB, Voskuhl RR. Estrogen receptor alpha mediates estrogen’s immune protection in autoimmune disease. J Immunol. 2003;171:6936–6940. doi: 10.4049/jimmunol.171.12.6936. [DOI] [PubMed] [Google Scholar]
- 16.Tiwari-Woodruff S, Morales LB, Lee R, Voskuhl RR. Differential neuroprotective and antiinflammatory effects of estrogen receptor (ER)alpha and ERbeta ligand treatment. Proc Natl Acad Sci USA. 2007;104:14813–14818. doi: 10.1073/pnas.0703783104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4(+) T-H-17 cells in relapsing EAE. Nat Immunol. 2007;8:172–180. doi: 10.1038/ni1430. [DOI] [PubMed] [Google Scholar]
- 18.Serafini B, Columba-Cabezas S, Di Rosa F, Aloisi F. Intracerebral recruitment and maturation of dendritic cells in the onset and progression of experimental autoimmune encephalomyelitis. Am J Pathol. 2000;157:1991–2002. doi: 10.1016/S0002-9440(10)64838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deshpande P, King IL, Segal BM. Cutting edge: CNS CD11c+ cells from mice with encephalomyelitis polarize Th17 cells and support CD25+CD4+T cell-mediated immunosuppression, suggesting dual roles in the disease process. J Immunol. 2007;178:6695–6699. doi: 10.4049/jimmunol.178.11.6695. [DOI] [PubMed] [Google Scholar]
- 20.Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, Becher B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005;11:328–334. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- 21.Paharkova-Vatchkova V, Maldonado R, Kovats S. Estrogen preferentially promotes the differentiation of CD11c(+) CD11b(intermediate) dendritic cells from bone marrow precursors. J Immunol. 2004;172:1426–1436. doi: 10.4049/jimmunol.172.3.1426. [DOI] [PubMed] [Google Scholar]
- 22.Zhang QH, Hu YZ, Cao J, Zhong YQ, Zhao YF, Mei QB. Estrogen influences the differentiation, maturation and function of dendritic cells in rats with experimental autoimmune encephalomyelitis. Acta Pharmacol Sin. 2004;25:508–513. [PubMed] [Google Scholar]
- 23.Liu HY, Buenafe AC, Matejuk A, Ito A, Zamora A, Dwyer J, Vandenbark AA, Offner H. Estrogen inhibition of EAE involves effects on dendritic cell function. J Neurosci Res. 2002;70:238–248. doi: 10.1002/jnr.10409. [DOI] [PubMed] [Google Scholar]
- 24.Achiron A, Gurevich M, Magalashvili D, Kishner I, Dolev M, Mandel M. Understanding autoimmune mechanisms in multiple sclerosis using gene expression microarrays: treatment effect and cytokine-related pathways. Clin Dev Immunol. 2004;11:299–305. doi: 10.1080/17402520400001603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brosnan CF, Selmaj K, Raine CS. Hypothesis – a role for tumor necrosis factor in immune-mediated demyelination and its relevance to multiple-sclerosis. J Neuroimmunol. 1988;18:87–94. doi: 10.1016/0165-5728(88)90137-3. [DOI] [PubMed] [Google Scholar]
- 26.McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11:335–339. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- 27.Dittel BN, Visintin I, Merchant RM, Janeway CA. Presentation of the self antigen myelin basic protein by dendritic cells leads to experimental autoimmune encephalomyelitis. J Immunol. 1999;163:32–39. [PubMed] [Google Scholar]
- 28.Zozulya AL, Ortler S, Lee J, Weidenfeller C, Sandor M, Wiendl H, Fabry Z. Intracerebral dendritic cells critically modulate encephalitogenic versus regulatory immune responses in the CNS. J Neurosci. 2009;29:140–152. doi: 10.1523/JNEUROSCI.2199-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zaba LC, Fuentes-Duculan J, Eungdamrong NJ, Abello MV, Novitskaya I, Pierson KC, Gonzalez J, et al. Psoriasis is characterized by accumulation of immunostimulatory and Th1/Th17 cell-polarizing myeloid dendritic cells. J Invest Dermatol. 2009;129:79–88. doi: 10.1038/jid.2008.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murugaiyan G, Mittal A, Weiner HL. Increased osteopontin expression in dendritic cells amplifies IL-17 production by CD4(+) T cells in experimental autoimmune encephalomyelitis and in multiple sclerosis. J Immunol. 2008;181:7480–7488. doi: 10.4049/jimmunol.181.11.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santiago-Schwarz F, Anand P, Liu S, Carsons SE. Dendritic cells (DCs) in rheumatoid arthritis (RA): progenitor cells and soluble factors contained in RA synovial fluid yield a subset of myeloid DCs that preferentially activate Th1 inflammatory-type responses. J Immunol. 2001;167:1758–1768. doi: 10.4049/jimmunol.167.3.1758. [DOI] [PubMed] [Google Scholar]
- 32.Pluchino S, Zanotti L, Brambilla E, Rovere-Querini P, Capobianco A, Alfaro-Cervello C, Salani G, et al. Immune regulatory neural stem/precursor cells protect from central nervous system autoimmunity by restraining dendritic cell function. PLoS One. 2009;4:e5959. doi: 10.1371/journal.pone.0005959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skarica M, Wang T, McCadden E, Kardian D, Calabresi PA, Small D, Whartenby KA. Signal transduction inhibition of APCs diminishes th17 and Th1 responses in experimental autoimmune encephalomyelitis. J Immunol. 2009;182:4192–4199. doi: 10.4049/jimmunol.0803631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furtado GC, Pina B, Tacke F, Gaupp S, van Rooijen N, Moran TM, Randolph GJ, et al. A novel model of demyelinating encephalomyelitis induced by monocytes and dendritic cells. J Immunol. 2006;177:6871–6879. doi: 10.4049/jimmunol.177.10.6871. [DOI] [PubMed] [Google Scholar]
- 35.Whartenby KA, Calabresi PA, McCadden E, Nguyen B, Kardian D, Wang TH, Mosse C, et al. Inhibition of FLT3 signaling targets DCs to ameliorate autoimmune disease. Proc Natl Acad Sci USA. 2005;102:16741–16746. doi: 10.1073/pnas.0506088102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hall JM, McDonnell DP. The estrogen receptor beta-isoform (ER beta) of the human estrogen receptor modulates ER alpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology. 1999;140:5566–5578. doi: 10.1210/endo.140.12.7179. [DOI] [PubMed] [Google Scholar]
- 37.Frasor J, Barnett DH, Danes JM, Hess R, Parlow AF, Katzenellenbogen BS. Response-specific and ligand dose-dependent modulation of estrogen receptor (ER) alpha activity by ERbeta in the uterus. Endocrinology. 2003;144:3159–3166. doi: 10.1210/en.2002-0143. [DOI] [PubMed] [Google Scholar]
- 38.Carreras E, Turner S, Paharkova-Vatchkova V, Mao A, Dascher C, Kovats S. Estradiol acts directly on bone marrow myeloid progenitors to differentially regulate GM-CSF or Flt3 ligand-mediated dendritic cell differentiation. J Immunol. 2008;180:727–738. doi: 10.4049/jimmunol.180.2.727. [DOI] [PubMed] [Google Scholar]
- 39.McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation. 2008;5:45. doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raine CS, Bonetti B, Cannella B. Multiple sclerosis: expression of molecules of the tumor necrosis factor ligand and receptor families in relationship to the demyelinated plaque. Rev Neurol. 1998;154:577–585. [PubMed] [Google Scholar]
- 41.Karni A, Abraham M, Monsonego A, Cai GF, Freeman GJ, Hafler D, Khoury SJ, Weiner HL. Innate immunity in multiple sclerosis: myeloid dendritic cells in secondary progressive multiple sclerosis are activated and drive a proinflammatory immune response. J Immunol. 2006;177:4196–4202. doi: 10.4049/jimmunol.177.6.4196. [DOI] [PubMed] [Google Scholar]
- 42.Ware CF. Network communications: lymphotoxins, LIGHT, and TNF. Annu Rev Immunol. 2005;23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719. [DOI] [PubMed] [Google Scholar]
- 43.Eissner G, Kolch W, Scheurich P. Ligands working as receptors: reverse signaling by members of the TNF superfamily enhance the plasticity of the immune system. Cytokine Growth Factor Rev. 2004;15:353–366. doi: 10.1016/j.cytogfr.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 44.Gold SM, Voskuhl RR. Estrogen treatment in multiple sclerosis. J Neurol Sci. 2009;286:99–103. doi: 10.1016/j.jns.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.