

Abstract
Mutations in rfx6 were recently associated with Mitchell-Riley syndrome, which involves neonatal diabetes, and other digestive system defects. To better define the function of Rfx6 in early endoderm development we cloned the Xenopus homologue. Expression of rfx6 begins early, showing broad expression throughout the anterior endoderm; at later stages rfx6 expression becomes restricted to the endocrine cells of the gut and pancreas. Morpholino knockdown of rfx6 caused a loss of pancreas marker expression, as well as other abnormalities. Co-injection of exogenous wild-type rfx6 rescued the morpholino phenotype in Xenopus tadpoles, whereas attempts to rescue the loss-of-function phenotype using mutant rfx6 based on Mitchell-Riley patients were unsuccessful. To better define the pleiotropic effects we performed microarray analyses of gene expression in knockdown foregut tissue. In addition to pancreatic defects, the microarray analyses revealed downregulation of lung, stomach and heart markers and an upregulation of kidney markers. We verified these results using RT-PCR and in situ hybridization. Based on the different rfx6 expression patterns and our functional analyses, we propose that rfx6 has both early and late functions. In early development Rfx6 plays a broad role, being essential for development of most anterior endodermal organs. At later stages however, Rfx6 function is restricted to endocrine cells.
Keywords: Rfx6, pancreas development, neonatal diabetes, Mitchell-Riley, organogenesis, Xenopus
Introduction
Neonatal diabetes is a rare presentation of diabetes mellitus (DM) that affects patients during the first six months of life. There are two predominant forms of neonatal diabetes, permanent (PNDM) and transient (TNDM). TNDM manifests early in life as insulin-dependent diabetes which goes into remission for several years; patients then often develop type 2 diabetes (Polak and Cave, 2007; Polak and Shield, 2004; von Muhlendahl and Herkenhoff, 1995). Approximately 68% of TNDM cases are caused by abnormalities at chromosome 6q24, with ZAC1 as the most likely candidate gene in this region (Arima et al., 2001; Gardner et al., 2000; Kamiya et al., 2000). Almost 25% of the remaining cases are caused by mutations in the genes KCNJ11 and ABCC8, which encode subunits of the β-cell potassium channels (Aguilar-Bryan and Bryan, 2008).
In contrast to TNDM, PNDM has no remission phase and patients remain insulin-dependent. The etiology of PNDM is only known in approximately half the cases (Hamilton-Shield, 2007). PNDM can typically be divided into three subgroups: disease caused by abnormal pancreas development, disease caused by reduced β-cell mass, and disease caused by β-cell dysfunction. Where PNDM is associated with abnormal pancreas development, mutations were identified in PTF1A (Sellick et al., 2004), PDX1 (Nicolino et al., 2010; Schwitzgebel et al., 2003; Stoffers et al., 1997), HNF1β (Yorifuji et al., 2004) and GLIS3 (Senee et al., 2006). Genes associated with decreased β-cell mass are EIF2AK3 (Delepine et al., 2000; Rubio-Cabezas et al., 2009b; Senee et al., 2004), insulin (Edghill et al., 2008; Polak et al., 2008; Stoy et al., 2007) and FOXP3 (Rubio-Cabezas et al., 2009a; Wildin et al., 2001). Genes associated with β-cell dysfunction in PNDM are glucokinase (Njolstad et al., 2003; Njolstad et al., 2001; Porter et al., 2005), KCNJ11 (Craig et al., 2009; Gloyn et al., 2004; Shimomura et al., 2010) and ABCC8 (Babenko et al., 2006; Ellard et al., 2007). However, the list of implicated genes is incomplete as the genetic basis of many PNDM cases has yet to be defined.
Mitchell-Riley is a recently described neonatal diabetes syndrome in which patients have severe neonatal diabetes and hypoplastic pancreas accompanied by duodenal or jejunal atresia and a small or absent gall bladder (Mitchell et al., 2004). Homozygosity mapping identified chromosomal regions linked to Mitchell-Riley syndrome, and sequencing candidate genes in these regions identified regulatory factor X 6 (rfx6) (Smith et al., 2010). Five distinct mutations in the coding sequence of Rfx6 were identified in six patients; four were homozygous and one was a compound heterozygote. Of the four homozygous mutations two were predicted to cause truncations, one at intron 2 affecting splicing, and another an out-of-frame deletion in exon 7. The other two homozygous mutations were mis-sense mutations, an R181Q in the DNA binding domain (DBD) and an S217P between the DBD and the dimerization domain. The heterozygous mutation, which consisted of two different mutations in rfx6 each on a different parental chromosome, consisted of a donor site-loss in intron 6 on one chromosome and a disruption of the intron 1 acceptor site on the other (Smith et al., 2010). Although it is likely that all of these mutations have a deleterious effect on pancreas development by way of producing a defective Rfx6 protein, there is no in vivo data to support this assumption.
The RFX family of transcription factors consists of 7 members (Aftab et al., 2008), all of which contain the winged-helix RFX DNA-binding domain; most also contain a dimerization domain enabling both hetero- and homodimeric interaction (Garvie et al., 2007; Horvath et al., 2004; Jabrane-Ferrat et al., 2002; Purvis et al., 2010; Wolfe et al., 2008). RFX family members are known to regulate various processes including spermatogenesis (Horvath et al., 2009; Horvath et al., 2004; Kistler et al., 2009; Wolfe et al., 2006; Wolfe et al., 2008), MHCII regulation (Garvie et al., 2007; Jabrane-Ferrat et al., 2002; Krawczyk et al., 2005; Nekrep et al., 2002; Niesen et al., 2009; Rousseau et al., 2004; Seguin-Estevez et al., 2009), and ciliogenesis (Ait-Lounis et al., 2007; Ashique et al., 2009; El Zein et al., 2009). The only RFX genes associated with pancreas development to date are rfx6 and rfx3, which have been shown to be required for appropriate β-cell formation and function (Ait-Lounis et al., 2007; Ait-Lounis et al., 2010; Smith et al., 2010; Soyer et al., 2010).
Recent studies have shown that rfx6 is required for pancreas development in mouse and zebrafish, though substantial interspecies differences have been identified (Smith et al., 2010; Soyer et al., 2010). In both species rfx6 is expressed in two waves, one early in development, before the expression of early pancreatic transcription factors such as pdx1 and ptf1a, and the second during endocrine cell development with expression continuing through to adulthood. Loss-of-function studies have demonstrated that rfx6 is required for development and maturation of endocrine cells in both mice and zebrafish, although with functional differences between the two species. In mice rfx6 is expressed broadly throughout the endoderm, whereas zebrafish rfx6 is exclusively expressed in the pancreas. Another substantial difference is that the zebrafish rfx6 knockdown led to only a small decrease in insulin expression and a dramatic decrease in expression of glucagon, somatostatin and ghrelin, whereas marker gene expression for all endocrine cell types, including insulin, was dramatically reduced in the mouse loss-of-function. Although all of these results demonstrate that rfx6 has an important role in endocrine cell development, the differences highlight the need for further studies of Rfx6.
To further define the role(s) of rfx6 in endoderm development we cloned the Xenopus laevis rfx6 homologue. We show that Xenopus rfx6 is initially expressed broadly in the anterior endoderm early in development (NF15), and that expression is later localized to the endocrine cells of the gut and pancreas. Morpholino-induced knockdown of rfx6 in Xenopus embryos induces a loss of pancreas marker gene expression. This morpholino phenotype can be rescued by co-injection of wild-type rfx6 mRNA. However, functional analyses of three mutated rfx6 mRNAs identified in Mitchell-Riley syndrome demonstrate that these mutants cannot rescue the morpholino phenotype. Microarray analyses of the loss-of-function (LOF) tissue at various developmental stages confirmed the loss of pancreatic marker gene expression, and revealed downregulation of genes expressed in other endoderm-derived organs in the anterior foregut –particularly stomach and lungs. Heart marker expression was also reduced, while there was an upregulation in kidney marker expression. These results demonstrate that Rfx6 is essential for pancreas development in Xenopus, and is implicated in normal development of other foregut organs; and that mutations identified in patients with Mitchell-Riley syndrome have a similar phenotype to null mutants, and thus are likely to cause the syndrome.
Materials and Methods
Cloning of Xenopus laevis rfx6
We initially identified the Xenopus tropicalis genome sequence for Rfx6 based on synteny mapping. Subsequently, we identified a partial Xenopus laevis clone (XL055a12) that contained the 3′end of the cDNA. The 3′ end of rfx6 was cloned using cDNA from wild-type NF35 embryos, and primers 1200for 5′ AGGCTAGTAAACAAAATGG 3′ and revUTR 5′ ACGTTTCCATAGGAGGTAGA 3′. The 5′ end of X. laevis Rfx6 was then cloned using 5′RACE with the following primers: RFXDC1-400rev 5′-CTCACTCTTCAACTTGCTAC-3′ and RFXDC1-200rev 5′-GAGCATAGAGTATGCATCGA-3′. The entire ORF of X. laevis Rfx6 was then cloned by PCR using the following primers RFXDC1-afor 5′ GGATGGTTGCATGGGCATT 3′ and RFXDC1-stoprev 5′ GGATGGTTGCATGGGCATT 3′ from NF41 whole gut cDNA using the LongRange PCR kit (Qiagen). The PCR product was cloned into pCS2+ and verified by sequencing. The mRNA sequence of X. laevis rfx6 has been deposited in GenBank with the accession number HQ665016.
To add a 5′ FLAG-tag to Rfx6 PCR primers FLAG-RFXDC1for 5′ AACCCGGTCGACTCCCGATTGAAAGG 3′ and T3 5′ ATTAACCCTCACTAAAGG 3′ were used to amplify rfx6 from the Rfx6-pCS2 vector. The PCR product was cloned into the CS107-FLAG vector. To create an inducible Rfx6 construct, rfx6 was amplified from Rfx6-pCS2 using the primers FLAG-RFXDC1for 5′ AACCCGGTCGACTCCCGATTGAAAGG 3′ and T3 5′ ATTAACCCTCACTAAAGG 3′. The PCR product was cloned upstream of the glucocorticoid receptor (GR) domain creating FLAG-Rfx6-GR and verified by sequencing.
Xenopus versions of the neonatal diabetes patient mutations were also cloned. The S217P mutant was created by introducing the mutation and an XmaI site. The gene was amplified in two fragments both using Rfx6-pCS2 as template. The 5′ fragment was amplified using primers SP6 5′ GATTTAGGTGACACTATAG 3′ and Rfx6StoPXmaIrev 5′ CAACTTGCTCCCGGGAAACCTTGTC 3′ (the reverse primer containing the restriction site and the mutation). The 3′ fragment was amplified using primers Rfx6StoPXmaIfwd 5′ GACAAGGTTTCCCGGGAGCAAGTTGA 3′ and CS2T7 5′ GTAATACGACTCACTATAG 3′. The two fragments were then ligated together into pCS2+ and the mutation verified by sequencing, and then subcloned with a 5′ FLAG-tag. To create an inducible S217P clone the GR domain was amplified from the Rfx6-GR vector using primers RQandSPmuts_GRfor 5′ CGTAAATCCTCAATGGCATC 3′ and RQandSPmuts_GRrev2 5′ GTATCTTATCAGGCCTGGATCTACG 3′. This PCR product was then cloned into the FLAG-S217P vector using EcoRV/StuI restriction enzymes giving the inducible mutant construct FLAG-Rfx6S217P-GR. The R181Q mutant was created by introducing the mutation and a KpnI site into primers. The gene was amplified in two fragments, both PCR reactions using FLAG-Rfx6 as a template. The 5′ fragment was amplified with primers SP6 5′ GATTTAGGTGACACTATAG 3′ and Rfx6R181QrevKpnI 5′ GAATGTCCTCGGGTACCAAGTTGTCTTGTCGTC 3′. The 3′ fragment was amplified with primers Rfx6R181QforKpnI 5′ CAAGAAGACTTGGTACCCGAGGACATTC 3′ and CS2T7 5′ GTAATACGACTCACTATAG 3′. The two fragments were cloned together into pCS2+. To create an inducible clone the GR domain was amplified from the Rfx6-GR vector using primers RQandSPmuts_GRfor 5′ CGTAAATCCTCAATGGCATC 3′ and RQandSPmuts_GRrev2 5′ GTATCTTATCAGGCCTGGATCTACG 3′. This PCR product was then cloned into the FLAG-R181Q vector using EcoRV/StuI restriction enzymes resulting in the inducible mutant construct FLAG-Rfx6R181Q-GR.
A mutant truncated after Exon 7 was created by amplifying the first 7 exons of Rfx6 from the Rfx6-pCS2 plasmid using primers SP6 5′ GATTTAGGTGACACTATAG 3′ primer and Rfx6ENDEX7 5′ TTTCTTTGGATATGCCCCCT 3′. The PCR product was ligated into pCS2+, and then subcloned with a 5′ FLAG-tag and verified by sequencing. To create an inducible clone the GR domain was amplified from Rfx6-GR using primers endEX7_GRfor 5′ CGAGTACCGTACGTACGAGAGATCT 3′ and endEX7_GRrev 5′ GCATTCTAGTTGTGGTTTGTCC 3′. This PCR product was cloned into the FLAG-EX7 vector using SnaBI, resulting in the inducible mutant construct FLAG-EX7-GR.
Probes and whole mount in situ hybridizations
The 3′ end of foxA2 was cloned using cDNA from wild-type NF35 embryos and primers FoxA2for 5′ GAATCCCATGAACACGTACATGA 3′ and FoxA2rev 5′ AGAGCCCAGGTGACAAGTCC 3′ designed based on BC155932. KcnJ1 was cloned using cDNA from wild-type NF35 embryos and primers KcnJ1for 5′ TGCAGCTACCTTCTTTCTGACA 3′ and KcnJ1rev 5′ GGGCACACCTATTCCTCAAA 3′ designed based on BC059301. The sox17α and sizzled plasmids were a gift from Aaron Zorn; all other genes were cloned previously, further details are available on request (Horb and Horb, 2010; Horb et al., 2009; Horb et al., 2003; Horb and Slack, 2002; Jarikji et al., 2009; Jarikji et al., 2007). PCR products were cloned into pCRII (Invitrogen) and confirmed by sequencing. Whole mount in situ hybridizations were performed as described using BM purple (Horb et al., 2003).
Antisense morpholino and mRNA injections
Antisense morpholino oligonucleotides were designed and manufactured by Gene Tools LLC. Morpholinos were designed against either the rfx6 translation start site (MO1) or the acceptor site of rfx6 intron 2 (MO2). An antisense mis-match morpholino (MM) was used as a control, containing 5 bp mis-match to the translation start site to disrupt binding. The sequences of the antisense morpholinos used are: MO1 5′ AATTGGCATTTCACCGGGTTCAGGC 3′; MO2 5′ AGAGAGCATTATACCTTTCCAAATG 3′; MM 5′ AATaGGgATTTgACCcGGTTCAcGC 3′ (mis-matched base pairs in lower case). Morpholinos were injected into the dorsal vegetal cells at the 8-cell stage. Morpholinos were manufactured with fluorescein-labeled oligonucleotides and targeting was verified by observing fluorescence after dissection of whole guts from injected embryos. For functional analysis we selected only samples for which the entire foregut was targeted. All mRNA for microinjection was created using the Ambion mMessage mMachine kit. To confirm targeting, experimental mRNAs were injected along with 400 pg gfp mRNA and targeting verified by observing appropriate fluorescence.
Tnt assay
The in vitro transcription and translation assay was performed using the TNT Quick Coupled Transcription/Translation System (Promega) following manufacturer’s instructions. Rfx6 mRNA cloned with a 3′ FLAG-tag was used with the start site morpholino (MO1) to confirm inhibition of translation, and the mis-match morpholino (MM) as a control. Western blots were performed using an anti-FLAG antibody.
Microarray analysis
MO1 and MM were injected into the dorsal vegetal blastomeres at the 8-cell stage. Anterior guts were collected at NF30, NF40 and NF44. Only samples that were targeted throughout the entire anterior gut were dissected. Sample sets (10 guts/set) were stored in RNAlater (Ambion); RNA extraction was performed using TRIzol (Invitrogen) and purified using the RNeasy Micro Kit (Qiagen). RNA analysis, cDNA preparation and hybridization were performed by Genome Québec (McGill University, Montréal).
Microarray results were analyzed using Affymetrix Expression Console and normalized using the Probe Logarithmic Intensity ERror estimation (PLIER) algorithm. Differential gene expression was analyzed using consecutive sampling with bin size of 25 (Guilbault et al., 2006; Novak et al., 2006a; Novak et al., 2006b; Novak et al., 2002). Representative standard deviations in each bin were calculated using non-linear regression to determine the boundaries of probability intervals. Candidate genes were selected as genes that lay beyond the probability interval of 0.9 in 6 or more comparisons. The microarray data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO series accession number GSE23642 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE23642).
Results
Rfx6 expression in Xenopus anterior endoderm becomes restricted as development proceeds
To identify Xenopus Rfx6 we used synteny mapping (human-Xenopus) to identify the genomic sequence for Xenopus tropicalis Rfx6. Using this sequence information we identified and cloned the full length Xenopus laevis ortholog of rfx6 (see materials and methods). The Xenopus Rfx6 protein shares 69% amino acid identity with human Rfx6, 68% with mouse and 63% with zebrafish. The Xenopus RFX domain is identical to the human DNA binding domain (DBD), and only one and two amino acids different to the mouse and zebrafish DBDs, respectively. Expression pattern analysis of rfx6 revealed that it was expressed in two distinct phases. In the first phase, rfx6 expression was found throughout the anterior endoderm in a broad domain from NF15 until NF34. At NF28 rfx6 expression begins to be localized to a dorsal and ventral region of the developing endoderm corresponding to the dorsal and ventral pancreatic buds (Fig. 1A–D). Expression then decreases at NF34 (Fig. 1I) and is no longer detectable by in situ at NF35 (data not shown). In the second phase, rfx6 was expressed in a punctate fashion throughout the gut and developing pancreas beginning at NF40, similar to that seen for other endocrine markers (Fig. 1E–H). Early in pancreas development (NF40), Xenopus rfx6 was expressed exclusively in the dorsal pancreas (Fig. 1E); by NF42 punctate expression of Rfx6 was also found in the ventral portion of the pancreas (Fig. 1F). This expression pattern is reminiscent of that seen with other endocrine progenitor markers such as insm1 suggesting that rfx6 is expressed in endocrine progenitors (Horb and Horb, 2010; Horb et al., 2009; Jarikji et al., 2009; Pearl et al., 2009).
Fig. 1. Rfx6 expression in the endoderm and isolated gut tissue.

(A) Sagittal bisection of an NF15 embryo showing an anteriorly localized stripe of rfx6 expression. (B) Sagittal bisection of an NF20 embryo showing rfx6 expression. (C) Transverse section through rfx6 expressing region of NF24 embryo showing broad expression in anterior endoderm. (D) NF28 embryo showing rfx6 expression restricted to a dorsal (arrow) and ventral (arrowhead) patch in the anterior foregut. (E) NF40 liver and pancreas showing rfx6 expression in the dorsal pancreas. (F) NF42 liver and pancreas showing rfx6 expression spreading to the ventral pancreas. (G) NF46 liver and pancreas showing rfx6 evenly expressed throughout the pancreas. (H) NF42 whole gut showing rfx6 expression in the dorsal pancreas, stomach and intestine. (A,B,D) anterior is left. (A-D) dorsal is up. (H) li, liver; p, pancreas; st, stomach. (I) RT-PCR of rfx6 in whole tadpoles at various stages throughout development.
Rfx6 is required for both anterior endoderm patterning and pancreas development
To determine if rfx6 is required for pancreas development we produced a knockdown phenotype using anti-sense morpholino oligonucleotides. One morpholino was designed to the translation start site (MO1) and a second designed to the acceptor site of intron 2 (MO2). As a control we used a 5 bp mis-match morpholino (MM) based on MO1. The ability of MO1 to block translation was tested with an in vitro transcription and translation assay (Fig. 2A), and RT-PCR was utilized to confirm that MO2 blocked intron splicing in vivo (Fig. 2B,C). The morpholinos were targeted to the anterior endoderm by injection into the two dorsal vegetal blastomeres at the 8-cell stage. Embryos injected with 25 ng of MO1 initially developed normally through gastrulation and neurulation with no change in expression of the general endoderm marker sox17α or the anterior endodermal genes hex, sizzled, foxA2 and hnf6 prior to NF20 (data not shown). However, beginning at NF25 we observed reduced expression of hnf6 and foxa2, but normal expression of hex, sizzled and sox17α (Fig. 2D–G, and data not shown). By NF35, expression of all anterior endodermal markers (hnf6, pdx1 and ptf1a) were reduced (Fig. 2H–K). To determine if differentiation of early beta cells was also affected by the knockdown of Rfx6, we examined whether initial insulin expression was reduced. By NF35, insulin expression is readily detected in the dorsal endoderm of MM morphants (Fig. 2N). However, in embryos injected with Rfx6 MO1 we did not detect any insulin expression (Fig. 2O). At later tadpole stages we also found reduced expression of all anterior endodermal markers. Within the pancreas, expression of the pancreatic transcription factors pdx1 and ptf1a was almost completely abolished, as was expression of the differentiation markers elastase and insulin (Fig. 3A–J). In addition, we did not detect expression of any endocrine markers (glucagon, somatostatin or neuroD) within the gastrointestinal tract (Fig. 3K–P). These results demonstrated that loss of rfx6 had no effect on early gastrula and neurula stages, but that rfx6 was specifically required beginning at early tailbud stages when regional specification of the anterior endoderm is solidified, and was also required for proper differentiation of most of the anterior endoderm.
Fig. 2. Rfx6 LOF leads to decreased expression of foregut and pancreas markers early in pancreas development.

(A) Western blot with anti-flag antibody of in vitro TnT assay of translation. This demonstrates that MO1 prevents translation of rfx6 mRNA, while the mismatch morpholino (MM) does not. Rfx6 mRNA, flag-tagged rfx6 mRNA plasmid; MM, mismatch morpholino. (B) Diagramatic representation of RT-PCR test of MO2 efficiency. (C) RT-PCR performed on wild-type (WT) and MO2-injected tadpoles. WT mRNA is processed leaving the reverse primer unable to bind resulting in no amplification in the Rfx6 panel. MO2 blocks mRNA processing at the intron 2 donor site (5′ end of the intron) as demonstrated by the presence of PCR product in MO2 lane. Ef1α is a housekeeping gene used as a loading control. (D,E) Hnf6 expression is reduced in the morphant foregut endoderm at NF25 (24/28 reduced). (F,G) FoxA2 expression is reduced in the morphant foregut endoderm at NF25 (17/28). (H,I) Hnf6 expression is reduced in the morphant foregut at NF35 (10/13 reduced). (J,K) Pdx1 expression is reduced in the morphant (17/19 reduced). (L,M) Ptf1a expression is reduced in both the dorsal (arrow) and ventral (arrowhead) pancreatic buds of the morphant (n=18). (N,O) Early insulin expression in the dorsal pancreatic bud (arrow) is lost in morphant tissue (56/62 reduced). Control tadpoles were injected with 25 ng mis-match morpholino.
Fig. 3. Rfx6 is required for pancreas differentiation.

Whole mount in situ hybridizations of NF42 whole guts from morphant (MO1) and mis-match (control) tadpoles. (A-D) Expression of pancreatic transcription factors pdx1 (n=18) and ptf1a (n=18) is almost completely abolished in morphant tissue. (E,F) Expression of acinar marker elastase is reduced (n=16). (G,H) Schematic illustrating organs in the whole gut. Pink, liver; red, gall bladder; blue, pancreas; yellow, stomach, duodenum and intestine. (I,J) β cell marker insulin is reduced (72/75 reduced). (K-N) Glucagon and somatostatin, both markers of endocrine cells in the pancreas and stomach are reduced (glucagon n=18, somatostatin 15/16 reduced). (O,P) NeuroD, developmental endocrine cell marker, is reduced (n=17).
Rfx6 knockdown phenotype is specific to loss of rfx6 expression
Since injection of unmodified Rfx6 resulted in an early gain-of-function phenotype, we confirmed that the knockdown phenotype was specifically related to the loss of Rfx6 with rescue experiments using a hormone-inducible Rfx6 (Rfx6-GR). We determined that 100 pg of rfx6-GR was sufficient to rescue the knockdown phenotype produced by 40 ng of MO2. Similar results were obtained with MO1, though the rescue was not as efficient (data not shown). Specifically, we found that Rfx6-GR could only rescue the morphant phenotype when dexamethasone was added at NF25; earlier induction resulted in a gain-of-function phenotype. To aid analysis, phenotypes were divided into three groups: normal, consisting of normal elastase expression such as that seen in the controls with normal gut morphology; moderate, with reduced elastase expression, abnormal gut morphology and a reduced pancreas size; and severe, where elastase expression was absent and foregut severely deformed so that organs could not readily be distinguished (see Fig. 4 for representative examples of each phenotype). When embryos were injected with MO2, elastase expression was reduced (n=47, 6 moderate, 41 severe), while in embryos co-injected with 100 pg of rfx6-GR and induced at NF25, elastase expression was partially restored (n=36, 20 normal, 15 moderate, 1 severe) (Fig. 4). In addition, we found insulin expression to be partially restored in the Rfx6-GR treated tadpoles (data not shown). These results demonstrate that the morpholino-injected phenotype is specifically due to loss of Rfx6. Furthermore, the fact that Rfx6-GR could only rescue the morphant phenotype when induced at NF25 agrees with our previous results in which we do not detect any reduction in gene expression in the anterior endoderm until NF25.
Fig. 4. Rfx6 mRNA rescues the morpholino knockdown phenotype.
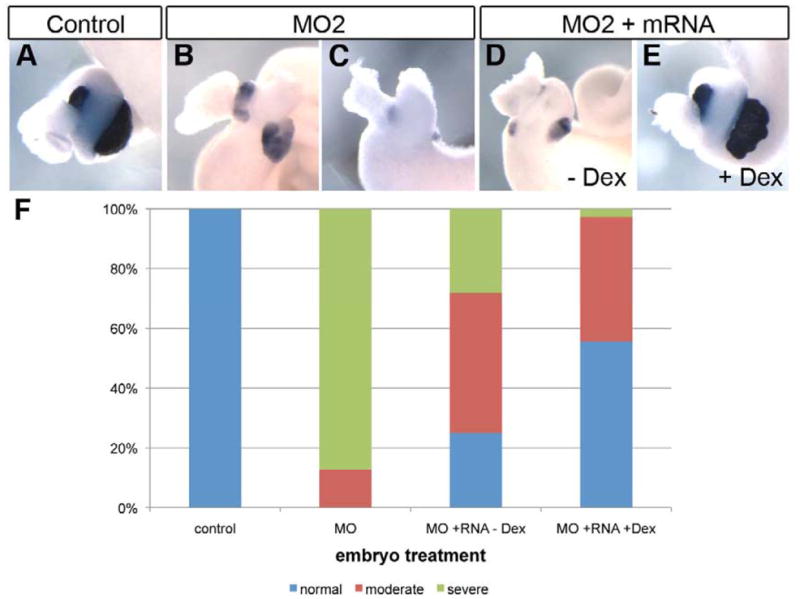
(A) Control whole gut stained for elastase expression (n=9). (B, C) Whole guts from embryos injected with 40 ng MO2, elastase expression is reduced (n=47, 6 moderate, 41 severe). (B) Moderate MO2 phenotype, (C) Severe MO2 phenotype. (D) Whole gut from embryo injected with 40 ng MO2 and 100 pg rfx6-GR mRNA. No dexamethosone was added. Elastase expression is reduced (n=32, 8 normal, 15 moderate, 9 severe). (E) Whole gut from embryo injected with 40 ng MO2 and 100 pg rfx6-GR mRNA. 2 μg/mL dexamethosone was added at NF25 to activate Rfx6-GR protein. Elastase expression is restored (n=36, 20 normal, 15 moderate, 1 severe). (F) Rescue results from all experimental whole guts. Severe phenotype refers to complete absence of elastase expression and gut malformation; moderate phenotype refers to reduced elastase expression and some gut malformation; normal refers to whole guts with normal levels of elastase expression and normal overall gut morphology.
Mutant rfx6 cannot rescue the loss-of-function phenotype thus rfx6 is a candidate gene for neonatal diabetes
Although mutations in rfx6 have been identified in patients with neonatal diabetes, their in vivo function was not tested (Smith et al., 2010). We used our morpholino-induced phenotype to determine if these mutations produced non-functional proteins, and therefore could contribute to or cause, neonatal diabetes. We tested the ability of three of the Rfx6 mutations in the context of the rescue experiment described above, using hormone-inducible versions for each construct. Control whole guts showed wild-type elastase expression (Fig. 5A, n=17); when 40 ng MO2 was injected elastase expression was reduced (Fig. 5B, n=26, 4 normal, 8 moderate, 14 severe). The first mutation we tested was the R181Q mutation, which lies in the DNA-binding domain (DBD), and based on the structure of DNA-bound human RFX1 DBD this residue makes direct contact with the DNA of the X-box (Gajiwala et al., 2000). The R181Q mutant introduces a charge reversal in a key DNA-binding residue. Co-injection of 100 pg R181Q mRNA did not rescue the knockdown phenotype (Fig. 5, n=16, 3 normal, 4 moderate, 9 severe). The second mutation tested, S217P, is located C-terminal to the DBD in an unknown functional domain; however, proline residues can introduce kinks into proteins and disrupt their structure. Co-injection of 100 pg of S217P was also unable to rescue the knockdown phenotype (Fig. 5E, n=16, 2 moderate, 14 severe). The third mutant construct we tested was the exon 7 truncation mutant, which still contains the DBD, but lacks the dimerization domain. RFX transcription factors are known to bind as homo- or heterodimers to alter gene expression and the expected inability of the exon 7 mutant to functionally dimerize appears to affect the function of Rfx6 as co-injection of 100 pg of the exon 7 mutant also did not rescue the morphant phenotype (Fig. 5E, n=16, 1 normal, 1 moderate, 14 severe). Taken together these results show that, unlike the wild-type Rfx6, these mutant constructs were unable to rescue the morphant phenotype, which suggests that these specific mutations identified in Mitchell-Riley patients do have an in vivo effect and contribute to disease progression.
Fig. 5. Mutant rfx6 mRNA cannot rescue the morpholino knockdown phenotype.
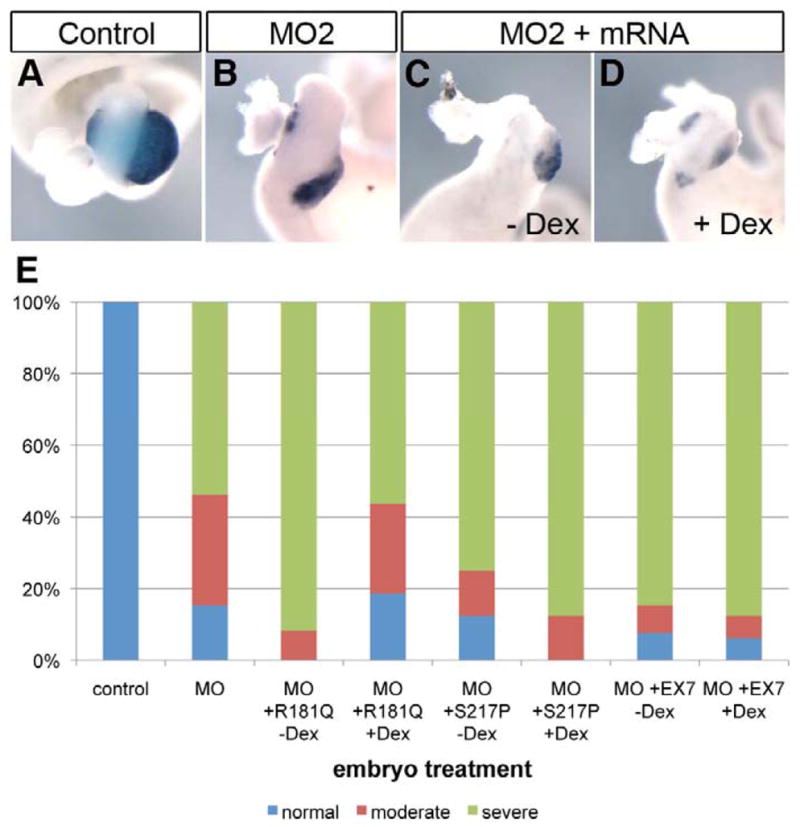
(A) Control whole gut stained for elastase expression (n=17). (B) Whole gut from embryo injected with 40 ng MO2, elastase expression is reduced (n=26, 4 normal, 8 moderate, 14 severe). (C) Whole gut from embryo injected with 40 ng MO2 and 100 pg rfx6R181Q-GR mRNA. No dexamethosone was added. Elastase expression is reduced (n=12, 1 moderate, 11 severe). (D) Whole gut from embryo injected with 40 ng MO2 and 100 pg rfx6R181Q-GR mRNA. 2 μg/mL dexamethosone was added at NF25 to activate Rfx6R181Q-GR protein. Elastase expression remains reduced (n=16, 3 normal, 4 moderate, 9 severe). (E) Rescue results for three rfx6 mutants: R181Q, S217P, and EX7 (Rfx6 is truncated after exon 7). Severe phenotype refers to complete absence of elastase expression and gut malformation; moderate phenotype refers to reduced elastase expression and some gut malformation; normal refers to whole guts with normal levels of elastase expression and normal overall gut morphology. S217P -dex n=16, 2 normal, 2 moderate, 14 severe. S217P +dex n=16, 2 moderate, 14 severe. EX7 –dex n=13, 1 normal, 1 moderate, 11 severe. EX7 +dex n=16, 1 normal, 1 moderate, 14 severe.
Loss of Rfx6 leads to loss of pancreas, stomach, lung and heart and an increase in kidney tissue
To gain further insights into the Rfx6 mechanism of action we performed a microarray on Rfx6 loss-of-function tissue at various stages throughout development (Fig. 6A). MO1 was used to create loss-of-function tadpoles, and MM was used to produce controls. We analyzed gene expression in the anterior foregut of control and morphant tadpoles at NF30, just after the pancreatic region was specified; at NF40, just after the pancreatic buds have fused; and at NF44, once the mature cell types had differentiated. The samples were hybridized to the Affymetrix Xenopus laevis GeneChip 2.0. Results were analyzed using Affymetrix Expression Console and normalized using the PLIER algorithm. Ratios of expression levels in MM samples compared to expression in MO1 samples were calculated and gene lists analyzed. In general, the microarray results concurred with our phenotypic characterization showing down-regulation of pancreas, stomach, intestine, heart and lung genes, whereas results for liver marker genes was inconclusive. Interestingly, the microarray data did show an increase in kidney marker genes (Table 1).
Fig. 6. LOF microarray experiment schematic and RT-PCR verification of selected genes.

(A) Schematic diagram of LOF microarray experiment. 8-cell stage embryos were injected with 25 ng of either Rfx6 start site morpholino (MO) or mis-match control (MM). Samples were collected at NF30, NF40 and NF44, RNA was extracted and hybridized to Affymetrix Xenopus laevis GeneChip 2.0. (B-D) RT-PCR verification of selected differentially expressed genes from the LOF microarray analysis. (B) NF30. (C) NF40. (D) NF44. +, microarray experiment showed upregulation, -, microarray experiment showed downregulation, NA, this probe was not on the microarray but was shown to be downregulated in in situ experiments, NC, the microarray experiment detected no change. Differential expression in RT-PCR findings are consistent with results seen in the microarray experiment. MM, mis-match control; MO, MO1; -RT, control reaction without reverse transcriptase.
Table 1. Genes differentially expressed in Rfx6 LOF microarray.
Genes have been sorted into categories based on the organ in which they are expressed. Fold change is relative to control samples, negative numbers represent down regulation in LOF tissue. NC, no change. NA, not applicable, the gene in question is not expressed that that time point. p < 0.05.
| unigene ID | fold change |
|||
|---|---|---|---|---|
| NF30 | NF40 | NF44 | ||
|
Pancreas | ||||
| amy2A | xl.21603 | NA | NA | −121.10 |
| Elas | xl.26506 | NA | −8.88 | −72.36 |
| DNase1-A | xl.932 | NC | NC | −27.66 |
| Cel | xl.24591 | NA | −10.85 | −23.57 |
| Sst | xl.13593 | NA | +2.30 | −15.00 |
| PDI | xl.80673 | NA | −13.94 | −11.46 |
| ctrb1 | xl.77315 | NA | −7.02 | −8.53 |
| Trypsin | xl.1858 | NA | NC | −4.94 |
| gcg-a | xl.193 | NA | NC | −4.51 |
| ins1-a | xl.817 | −2.21 | −5.84 | −4.03 |
| nm-a | xl.830 | NA | NC | −2.08 |
| Ptf1a | xl.29862 | NC | −3.62 | −3.77 |
| scg3-A | xl.15 | NA | NC | −2.10 |
| cela1 | xl.68767 | NA | −8.28 | NC |
| igf2-A | xl.23623 | −2.79 | −5.70 | NC |
| ctrl | xl.23397 | NA | −4.73 | NC |
| PP11-like | xl.82301 | NA | −4.69 | NC |
| hlxb9 | xl.55462 | NA | −3.17 | NC |
| cpa1 | xl.73839 | NA | −2.12 | NC |
| NeuroD | xl.26304 | −1.86 | NC | NC |
|
Stomach | ||||
| ATP4B | xl.26547 | NC | NC | −69.82 |
| ATP4A | xl.971 | NC | NC | −33.21 |
| tm4sf3 | xl.24486 | NC | NC | −10.26 |
| agr2 | xl.25847 | NC | +3.67 | −6.74 |
| Nkx6.2 | xl.55860 | NC | −6.92 | −3.98 |
| Sfrp2 | xl.15963 | NC | +2.16 | NC |
| PitX1 | xl.34927 | +1.75 | NC | NC |
|
Liver | ||||
| alb-b | xl.395 | NC | NC | −18.94 |
| adh8 | xl.53979 | NC | −5.52 | −2.24 |
| ca6 | xl.14854 | NC | −7.99 | NC |
| tm4sf4 | xl.53955 | NC | −5.21 | NC |
| ITIH3 | xl.5040 | NC | −4.48 | NC |
| fabp-a | xl.21513 | NC | −2.97 | NC |
| hnf4 | xl.1217 | +1.73 | NC | NC |
| adh6 | xl.48167 | NC | NC | +12.86 |
| arg1 | xl.1242 | NC | +2.16 | +16.96 |
|
Intestine | ||||
| villin1 | xl.81110, xl.24670 | NC | −4.21 | NC |
| crip1a | xl.48993 | NC | −3.01 | NC |
| xCad2 | xl.1358 | NC | NC | +3.08 |
| hand1 | xl.1098 | NC | NC | +3.47 |
| soat2 | xl.53150 | NC | NC | +6.64 |
| gamt | xl.11235 | NC | NC | +8.31 |
| darmin | xl.76581 | NC | NC | +8.59 |
|
Heart | ||||
| MLClv | xl.76359, xl.2576 | NC | −9.50 | −5.93 |
| MYH6 | xl.50419 | −5.73 | −3.95 | NA |
| tnni3 | xl.935 | NC | −2.93 | NA |
| tbx5 | xl.529 | −1.79 | −2.40 | NA |
| tnnc1 | xl.2228 | −3.97 | −2.28 | NA |
| tnnt2 | xl.23777 | −1.60 | −2.10 | NA |
| mlc1av-A | xl.11969 | −5.29 | −2.06 | NA |
| nkx2.5 | xl.22859 | −1.92 | −1.73 | NA |
| mlc2a | xl.76247 | −6.16 | NC | NA |
| MYH6 | xl.3161 | −6.14 | NC | NA |
| Mybpc3 | xl.2416 | −1.57 | NC | NA |
| nkx2.10-A | xl.623 | −1.57 | NC | NA |
| camk1 | xl.63784 | NC | +3.33 | +3.81 |
| MyHC | xl.80374 | NC | +1.79 | +3.88 |
|
Lung | ||||
| SFTPC | xl.55371 | NA | −15.60 | NA |
| cldn18 | xl.9841 | NA | −13.13 | NA |
| EST lung | xl.29467 | NA | −2.45 | NA |
| slc34a2 | xl.5596 | NA | −2.09 | NA |
| RSpo2 | xl.49496 | NA | −1.53 | NA |
|
Kidney | ||||
| tm4sf5 | xl.51559 | NC | −2.47 | NA |
| kcnj1 | xl.29882 | NC | +2.69 | NA |
| Gcm-1 | xl.16562 | NC | +3.13 | NA |
| UCP2 | xl.27125 | NC | +3.57 | NA |
| FoxC1 | xl.76079 | −1.75 | NC | NA |
| Ndrgl | xl.77767 | −1.62 | NC | NA |
| Nphsl | xl.14805 | −1.59 | NC | NA |
| Frzb2 | xl.619 | +1.71 | NC | NA |
| Hlbox1 | xl.1209 | +1.90 | NC | NA |
| tm6sf1 | xl.2482 | +2.17 | NC | NA |
| slc3a2 | xl.10583 | NC | NC | +2.34 |
| slc7a8 | xl.15844 | +2.11 | NC | +2.81 |
| slc43a2 | xl.5041 | NC | NC | +2.81 |
| PitX2A | xl.104 | NC | NC | +2.86 |
| Cln6 | xl.23306 | NC | NC | +4.93 |
| cldn4L1 | xl.21063 | NC | NC | +5.31 |
| xPox2 | xl.11968 | NC | +2.95 | +6.18 |
To confirm differential gene expression we performed RT-PCR analysis of selected genes identified as differentially expressed in the microarray (Fig. 6B). We were able to confirm the microarray data and found that genes differentially expressed in the microarray were also similarly differentially expressed in the morphant RT-PCRs (Fig. 6B–D). To further verify differential gene expression we performed in situ hybridization on control and morphant whole guts at NF42 using probes against selected differentially expressed genes confirmed by RT-PCR (Fig. 7). Stomach markers agr2 and frp5 were reduced, as was the lung marker surfactant C, and the heart marker nkx2.5 (Fig. 7A–H). In contrast, we found increased expression of kidney marker kcnj1, confirming the results observed in the microarray (Fig. 7I,J). Although expression of liver marker hex seemed unaffected by the loss of rfx6, the liver did appear smaller (Fig. 7K,L). Last, we also found that the gall bladder was unaffected, as the gall bladder component of hnf6 and sox17α expression was normal (Fig. 7M–P). Although expression of these markers was not changed in the microarray we included them in our in situ analysis because the gall bladder is often affected in Mitchell-Riley syndrome (Mitchell et al., 2004). In conclusion, the microarray data confirmed our phenotypic characterization and showed that rfx6 has a broader role in endoderm development than simply endocrine cells.
Fig. 7. In situ hybridization confirms differential expression of organ marker genes for stomach, lung, heart and kidney in LOF microarray.

(A,B) Isolated whole gut NF42 showing expression of stomach marker agr2 almost entirely abolished in morphant tissue (n=23). (C,D) Expression of stomach marker frp5 was reduced (n=18). (E-J) Whole tadpoles at NF42 with ventral skin removed for in situ hybridization. (E,F) Lung marker surfactant C (SurfC) is decreased in morphant tissue, closed arrows (10/14 reduced). (G,H) Heart marker nkx2.5 is decreased in morphant tissue, open arrow (8/14 reduced). (I,J) Kidney marker kcnJ1 is increased in morphant tissue, arrowhead (8/11 increased). (K,L) Expression of liver marker hex is unaffected by rfx6 knockdown (n=32). (M,N) Expression of gall bladder and duodenum marker hnf6 is reduced in duodenum but not gall bladder (17/18). (O,P) Gall bladder marker sox17α was not reduced (18/20 showed normal expression).
Discussion
Mitchell-Riley syndrome was recently described, as a neonatal diabetes syndrome that involves abnormalities of the anterior gut as well as diabetes (Mitchell et al., 2004). Patients with this syndrome are typically diagnosed within the first week of life and generally die within their first year of life (Smith et al., 2010). The early onset and severity of disease suggests that in patients with Mitchell-Riley syndrome the pancreas does not develop appropriately in utero. Homozygosity mapping of patients and unaffected family members indicated several chromosomal regions were potentially involved (Smith et al., 2010). The gene coding for the transcription factor Rfx6 was located in one of these regions.
The rfx6 expression pattern was first described in zebrafish and mice (Smith et al., 2010; Soyer et al., 2010). Rfx6 is initially expressed broadly in the anterior endoderm early in development and becomes more restricted as development progresses, eventually becoming restricted to the endodermal cells of the gut and pancreas (Smith et al., 2010; Soyer et al., 2010). There are some important differences in rfx6 expression between the species studied. Our studies show that Xenopus rfx6 is expressed in the pancreas, stomach and intestine but not in the liver (Fig. 1), whereas mouse studies have shown rfx6 is expressed throughout endoderm-derived tissues, including the liver (Smith et al., 2010). Zebrafish rfx6 expression is slightly different again, with expression limited solely to the pancreatic region (Soyer et al., 2010). These findings combined suggest that rfx6 has an important role in pancreas development, but that its function in other organs differs between animal species.
Loss of Rfx6 leads to a loss of endocrine cells in all studies to date, however the details differ between studies. In this study we showed that knockdown of Rfx6 leads to a loss of both endocrine and exocrine gene expression, including a drastic reduction in insulin expression. Similar experiments in zebrafish however, show only a slight reduction in insulin expression with a drastic decrease in expression of marker genes for other endocrine cell types (Soyer et al., 2010). In contrast, the mouse knockout showed that endocrine gene expression is lost, while exocrine cell types are unaffected (Smith et al., 2010). Our knockdown phenotype is more similar to the mouse knockout than the zebrafish knockdown, although our phenotype is more severe than both. It is possible that in mice and zebrafish other RFX proteins have overlapping functions, decreasing the severity of the knockout phenotype. Interestingly, the mouse knockout was created by ablating only the first five exons of rfx6, which removes the DNA-binding domain while the dimerization domain coding sequence remains intact. Consequently there is potential for a truncated C-terminal protein to be expressed, which could hypothetically interact with rfx6 binding partners, and explain the lack of a phenotype in murine exocrine cells. Thus this knockout might not accurately represent the null phenotype; a complete mouse knockout may give rise to results similar to what we report in Xenopus. The zebrafish Rfx6 knockdown study did not assess the effect on exocrine cell types, and thus exocrine gene expression may also have been lost in that system. In addition to the effects on pancreatic genes, our microarray analysis of the Xenopus LOF demonstrated that stomach, lung and heart were also lost in the Rfx6 knockdown. Other studies have not explored the effect of loss of Rfx6 on other organs. As Rfx6 is not expressed in tissues other than the pancreas in zebrafish it is unlikely that loss of Rfx6 would affect other tissues significantly. In the mouse, however, Rfx6 is expressed throughout the endoderm-derived organs yet the only mention of effect of LOF on non-pancreatic tissues is a distension of the gut in the knockout mouse (Smith et al., 2010). The zebrafish knockdown of Rfx6 was shown to prevent endocrine cells from maturing, leading to an increase in endocrine progenitor cells; though this did not appear to apply to beta cells, as insulin expression was not greatly decreased. Neither our study nor the mouse knockout however, showed an increase in progenitor cells. This suggests that the Xenopus knockdown phenotype is more similar to the mouse knockout than to the zebrafish knockdown.
Through rescuing the morpholino phenotype by co-injecting exogenous wild-type rfx6 we were able to verify that our rfx6 knockdown was specifically caused by a loss of rfx6. We used a dexamethosone-inducible mRNA construct and activated the protein at NF25, before pancreatic specification. We noted a mild rescue effect without addition of dexamethasone, suggesting that the system is somewhat leaky. We also examined whether the mutations found in rfx6 in Mitchell-Riley patients were functional in vivo by testing the ability of three of the four homozygous mutations to rescue the morpholino phenotype. We found that none of these mutant constructs (R181Q, S217P and the exon 7 truncation) were sufficient to attenuate Rfx6 function in vivo (Fig. 5). This strongly suggests that mutated rfx6 is responsible for at least some of the defects associated with Mitchell-Riley syndrome.
One of the benefits of using the Xenopus system to examine the functional relevance of specific transcription factors in endoderm organogenesis is the ability to control protein activity using hormone-inducible chimeric proteins. By employing this technique we were able to directly ascertain when Rfx6 function is required in early endoderm development. Specifically, we showed that Rfx6 could only rescue the knockdown phenotype when activated at NF25. This is exactly the stage when we first see reduced expression of hnf6 and foxA2. Our results differ from that seen in mice and zebrafish where Rfx6 was placed downstream of the transcription factor Neurogenin 3 (Ngn3), which is essential for development of endocrine cells (Gradwohl et al., 2000). Our results however, place Rfx6 upstream of Ngn3. We believe that our functional analysis of Rfx6 is specifically focused on the early function of Rfx6, while the mouse and zebrafish studies focused specifically on its function in endocrine cells. There is evidence supporting both placements of Rfx6. Positioning rfx6 downstream of ngn3 is supported by the fact that rfx6 was found in Ngn3+ cells in two screens, and that rfx6 is not expressed in the ngn3 knockout mouse (Smith et al., 2010; Soyer et al., 2010). The fact that pdx1 expression is decreased both in our loss-of-function study and the knockout mouse suggests that rfx6 lies upstream of pdx1, and hence also ngn3. Also supporting this is the fact that hnf6 expression is decreased in our loss-of-function tissue at NF25, and hnf6 has been shown to be upstream of pdx1 and ngn3 (Jacquemin et al., 2000; Jacquemin et al., 2003). These observations could also be explained by a dual role for rfx6: an early role in endoderm patterning and a later role specific to endocrine cell development and maturation. However, in order to better understand the role(s) of Rfx6, direct downstream targets need to be found. Additionally, a study separating the early and late functions of Rfx6 would help ascertain which functions are directly involved in pancreas development and precisely which missing functions cause the abnormalities seen in Mitchell-Riley syndrome, where rfx6 is mutated.
Acknowledgments
This work was supported by grants from the National Institutes of Health (DK077197), and the Canadian Diabetes Association (OG-3-09-2843-MH) to M.E.H. Esther Pearl is a postdoctoral fellow of Fonds de la recherche en santé du Québec (FRSQ). Special thanks go to Lori Horb for the Tnt assay, Dr Jaroslav P. Novak of GenexAnalysis (http://genexanalysis.net) for his mathematical analysis of microarray data, Dr Aaron Zorn for the sox17α and sizzled plasmids, and Frédéric Bourque for his care of the frogs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aftab S, Semenec L, Chu JS, Chen N. Identification and characterization of novel human tissue-specific RFX transcription factors. BMC Evol Biol. 2008;8:226. doi: 10.1186/1471-2148-8-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar-Bryan L, Bryan J. Neonatal diabetes mellitus. Endocr Rev. 2008;29:265–291. doi: 10.1210/er.2007-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ait-Lounis A, Baas D, Barras E, Benadiba C, Charollais A, Nlend Nlend R, Liegeois D, Meda P, Durand B, Reith W. Novel function of the ciliogenic transcription factor RFX3 in development of the endocrine pancreas. Diabetes. 2007;56:950–959. doi: 10.2337/db06-1187. [DOI] [PubMed] [Google Scholar]
- Ait-Lounis A, Bonal C, Seguin-Estevez Q, Schmid CD, Bucher P, Herrera PL, Durand B, Meda P, Reith W. The transcription factor Rfx3 regulates beta-cell differentiation, function, and glucokinase expression. Diabetes. 2010;59:1674–1685. doi: 10.2337/db09-0986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arima T, Drewell RA, Arney KL, Inoue J, Makita Y, Hata A, Oshimura M, Wake N, Surani MA. A conserved imprinting control region at the HYMAI/ZAC domain is implicated in transient neonatal diabetes mellitus. Hum Mol Genet. 2001;10:1475–1483. doi: 10.1093/hmg/10.14.1475. [DOI] [PubMed] [Google Scholar]
- Ashique AM, Choe Y, Karlen M, May SR, Phamluong K, Solloway MJ, Ericson J, Peterson AS. The Rfx4 transcription factor modulates Shh signaling by regional control of ciliogenesis. Sci Signal. 2009;2:ra70. doi: 10.1126/scisignal.2000602. [DOI] [PubMed] [Google Scholar]
- Babenko AP, Polak M, Cave H, Busiah K, Czernichow P, Scharfmann R, Bryan J, Aguilar-Bryan L, Vaxillaire M, Froguel P. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med. 2006;355:456–466. doi: 10.1056/NEJMoa055068. [DOI] [PubMed] [Google Scholar]
- Craig TJ, Shimomura K, Holl RW, Flanagan SE, Ellard S, Ashcroft FM. An in-frame deletion in Kir6.2 (KCNJ11) causing neonatal diabetes reveals a site of interaction between Kir6.2 and SUR1. J Clin Endocrinol Metab. 2009;94:2551–2557. doi: 10.1210/jc.2009-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet. 2000;25:406–409. doi: 10.1038/78085. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edghill EL, Flanagan SE, Patch AM, Boustred C, Parrish A, Shields B, Shepherd MH, Hussain K, Kapoor RR, Malecki M, MacDonald MJ, Stoy J, Steiner DF, Philipson LH, Bell GI, Hattersley AT, Ellard S. Insulin mutation screening in 1,044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes. 2008;57:1034–1042. doi: 10.2337/db07-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Zein L, Ait-Lounis A, Morle L, Thomas J, Chhin B, Spassky N, Reith W, Durand B. RFX3 governs growth and beating efficiency of motile cilia in mouse and controls the expression of genes involved in human ciliopathies. J Cell Sci. 2009;122:3180–3189. doi: 10.1242/jcs.048348. [DOI] [PubMed] [Google Scholar]
- Ellard S, Flanagan SE, Girard CA, Patch AM, Harries LW, Parrish A, Edghill EL, Mackay DJ, Proks P, Shimomura K, Haberland H, Carson DJ, Shield JP, Hattersley AT, Ashcroft FM. Permanent neonatal diabetes caused by dominant, recessive, or compound heterozygous SUR1 mutations with opposite functional effects. Am J Hum Genet. 2007;81:375–382. doi: 10.1086/519174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajiwala KS, Chen H, Cornille F, Roques BP, Reith W, Mach B, Burley SK. Structure of the winged-helix protein hRFX1 reveals a new mode of DNA binding. Nature. 2000;403:916–921. doi: 10.1038/35002634. [DOI] [PubMed] [Google Scholar]
- Gardner RJ, Mackay DJ, Mungall AJ, Polychronakos C, Siebert R, Shield JP, Temple IK, Robinson DO. An imprinted locus associated with transient neonatal diabetes mellitus. Hum Mol Genet. 2000;9:589–596. doi: 10.1093/hmg/9.4.589. [DOI] [PubMed] [Google Scholar]
- Garvie CW, Stagno JR, Reid S, Singh A, Harrington E, Boss JM. Characterization of the RFX complex and the RFX5(L66A) mutant: implications for the regulation of MHC class II gene expression. Biochemistry. 2007;46:1597–1611. doi: 10.1021/bi6023868. [DOI] [PubMed] [Google Scholar]
- Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JM, Molnes J, Edghill EL, Frayling TM, Temple IK, Mackay D, Shield JP, Sumnik Z, van Rhijn A, Wales JK, Clark P, Gorman S, Aisenberg J, Ellard S, Njolstad PR, Ashcroft FM, Hattersley AT. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med. 2004;350:1838–1849. doi: 10.1056/NEJMoa032922. [DOI] [PubMed] [Google Scholar]
- Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A. 2000;97:1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbault C, Novak JP, Martin P, Boghdady ML, Saeed Z, Guiot MC, Hudson TJ, Radzioch D. Distinct pattern of lung gene expression in the Cftr-KO mice developing spontaneous lung disease compared with their littermate controls. Physiol Genomics. 2006;25:179–193. doi: 10.1152/physiolgenomics.00206.2005. [DOI] [PubMed] [Google Scholar]
- Hamilton-Shield JP. Overview of neonatal diabetes. Endocr Dev. 2007;12:12–23. doi: 10.1159/000109601. [DOI] [PubMed] [Google Scholar]
- Horb LD, Horb ME. BrunoL1 regulates endoderm proliferation through translational enhancement of cyclin A2 mRNA. Dev Biol. 2010;345:156–169. doi: 10.1016/j.ydbio.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horb LD, Jarkji ZH, Horb ME. Xenopus insm1 is essential for gastrointestinal and pancreatic endocrine cell development. Dev Dyn. 2009;238:2505–2510. doi: 10.1002/dvdy.22071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horb ME, Shen CN, Tosh D, Slack JM. Experimental conversion of liver to pancreas. Curr Biol. 2003;13:105–115. doi: 10.1016/s0960-9822(02)01434-3. [DOI] [PubMed] [Google Scholar]
- Horb ME, Slack JM. Expression of amylase and other pancreatic genes in Xenopus. Mech Dev. 2002;113:153–157. doi: 10.1016/s0925-4773(02)00019-9. [DOI] [PubMed] [Google Scholar]
- Horvath GC, Kistler MK, Kistler WS. RFX2 is a candidate downstream amplifier of A-MYB regulation in mouse spermatogenesis. BMC Dev Biol. 2009;9:63. doi: 10.1186/1471-213X-9-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath GC, Kistler WS, Kistler MK. RFX2 is a potential transcriptional regulatory factor for histone H1t and other genes expressed during the meiotic phase of spermatogenesis. Biol Reprod. 2004;71:1551–1559. doi: 10.1095/biolreprod.104.032268. [DOI] [PubMed] [Google Scholar]
- Jabrane-Ferrat N, Nekrep N, Tosi G, Esserman LJ, Peterlin BM. Major histocompatibility complex class II transcriptional platform: assembly of nuclear factor Y and regulatory factor X (RFX) on DNA requires RFX5 dimers. Mol Cell Biol. 2002;22:5616–5625. doi: 10.1128/MCB.22.15.5616-5625.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemin P, Durviaux SM, Jensen J, Godfraind C, Gradwohl G, Guillemot F, Madsen OD, Carmeliet P, Dewerchin M, Collen D, Rousseau GG, Lemaigre FP. Transcription factor hepatocyte nuclear factor 6 regulates pancreatic endocrine cell differentiation and controls expression of the proendocrine gene ngn3. Mol Cell Biol. 2000;20:4445–4454. doi: 10.1128/mcb.20.12.4445-4454.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemin P, Lemaigre FP, Rousseau GG. The Onecut transcription factor HNF-6 (OC-1) is required for timely specification of the pancreas and acts upstream of Pdx-1 in the specification cascade. Dev Biol. 2003;258:105–116. doi: 10.1016/s0012-1606(03)00115-5. [DOI] [PubMed] [Google Scholar]
- Jarikji Z, Horb LD, Shariff F, Mandato CA, Cho KW, Horb ME. The tetraspanin Tm4sf3 is localized to the ventral pancreas and regulates fusion of the dorsal and ventral pancreatic buds. Development. 2009;136:1791–1800. doi: 10.1242/dev.032235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarikji ZH, Vanamala S, Beck CW, Wright CV, Leach SD, Horb ME. Differential ability of Ptf1a and Ptf1a-VP16 to convert stomach, duodenum and liver to pancreas. Dev Biol. 2007;304:786–799. doi: 10.1016/j.ydbio.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya M, Judson H, Okazaki Y, Kusakabe M, Muramatsu M, Takada S, Takagi N, Arima T, Wake N, Kamimura K, Satomura K, Hermann R, Bonthron DT, Hayashizaki Y. The cell cycle control gene ZAC/PLAGL1 is imprinted--a strong candidate gene for transient neonatal diabetes. Hum Mol Genet. 2000;9:453–460. doi: 10.1093/hmg/9.3.453. [DOI] [PubMed] [Google Scholar]
- Kistler WS, Horvath GC, Dasgupta A, Kistler MK. Differential expression of Rfx1-4 during mouse spermatogenesis. Gene Expr Patterns. 2009;9:515–519. doi: 10.1016/j.gep.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk M, Masternak K, Zufferey M, Barras E, Reith W. New functions of the major histocompatibility complex class II-specific transcription factor RFXANK revealed by a high-resolution mutagenesis study. Mol Cell Biol. 2005;25:8607–8618. doi: 10.1128/MCB.25.19.8607-8618.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell J, Punthakee Z, Lo B, Bernard C, Chong K, Newman C, Cartier L, Desilets V, Cutz E, Hansen IL, Riley P, Polychronakos C. Neonatal diabetes, with hypoplastic pancreas, intestinal atresia and gall bladder hypoplasia: search for the aetiology of a new autosomal recessive syndrome. Diabetologia. 2004;47:2160–2167. doi: 10.1007/s00125-004-1576-3. [DOI] [PubMed] [Google Scholar]
- Nekrep N, Jabrane-Ferrat N, Wolf HM, Eibl MM, Geyer M, Peterlin BM. Mutation in a winged-helix DNA-binding motif causes atypical bare lymphocyte syndrome. Nat Immunol. 2002;3:1075–1081. doi: 10.1038/ni840. [DOI] [PubMed] [Google Scholar]
- Nicolino M, Claiborn KC, Senee V, Boland A, Stoffers DA, Julier C. A novel hypomorphic PDX1 mutation responsible for permanent neonatal diabetes with subclinical exocrine deficiency. Diabetes. 2010;59:733–740. doi: 10.2337/db09-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesen MI, Osborne AR, Lagor WR, Zhang H, Kazemfar K, Ness GC, Blanck G. Technological advances in the study of HLA-DRA promoter regulation: extending the functions of CIITA, Oct-1, Rb, and RFX. Acta Biochim Biophys Sin (Shanghai) 2009;41:198–205. doi: 10.1093/abbs/gmp002. [DOI] [PubMed] [Google Scholar]
- Njolstad PR, Sagen JV, Bjorkhaug L, Odili S, Shehadeh N, Bakry D, Sarici SU, Alpay F, Molnes J, Molven A, Sovik O, Matschinsky FM. Permanent neonatal diabetes caused by glucokinase deficiency: inborn error of the glucose-insulin signaling pathway. Diabetes. 2003;52:2854–2860. doi: 10.2337/diabetes.52.11.2854. [DOI] [PubMed] [Google Scholar]
- Njolstad PR, Sovik O, Cuesta-Munoz A, Bjorkhaug L, Massa O, Barbetti F, Undlien DE, Shiota C, Magnuson MA, Molven A, Matschinsky FM, Bell GI. Neonatal diabetes mellitus due to complete glucokinase deficiency. N Engl J Med. 2001;344:1588–1592. doi: 10.1056/NEJM200105243442104. [DOI] [PubMed] [Google Scholar]
- Novak JP, Kim SY, Xu J, Modlich O, Volsky DJ, Honys D, Slonczewski JL, Bell DA, Blattner FR, Blumwald E, Boerma M, Cosio M, Gatalica Z, Hajduch M, Hidalgo J, McInnes RR, Miller MC, 3rd, Penkowa M, Rolph MS, Sottosanto J, St-Arnaud R, Szego MJ, Twell D, Wang C. Generalization of DNA microarray dispersion properties: microarray equivalent of t-distribution. Biol Direct. 2006a;1:27. doi: 10.1186/1745-6150-1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak JP, Miller MC, 3rd, Bell DA. Variation in fiberoptic bead-based oligonucleotide microarrays: dispersion characteristics among hybridization and biological replicate samples. Biol Direct. 2006b;1:18. doi: 10.1186/1745-6150-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak JP, Sladek R, Hudson TJ. Characterization of variability in large-scale gene expression data: implications for study design. Genomics. 2002;79:104–113. doi: 10.1006/geno.2001.6675. [DOI] [PubMed] [Google Scholar]
- Pearl EJ, Bilogan CK, Mukhi S, Brown DD, Horb ME. Xenopus pancreas development. Dev Dyn. 2009;238:1271–1286. doi: 10.1002/dvdy.21935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak M, Cave H. Neonatal diabetes mellitus: a disease linked to multiple mechanisms. Orphanet J Rare Dis. 2007;2:12. doi: 10.1186/1750-1172-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak M, Dechaume A, Cave H, Nimri R, Crosnier H, Sulmont V, de Kerdanet M, Scharfmann R, Lebenthal Y, Froguel P, Vaxillaire M. Heterozygous missense mutations in the insulin gene are linked to permanent diabetes appearing in the neonatal period or in early infancy: a report from the French ND (Neonatal Diabetes) Study Group. Diabetes. 2008;57:1115–1119. doi: 10.2337/db07-1358. [DOI] [PubMed] [Google Scholar]
- Polak M, Shield J. Neonatal and very-early-onset diabetes mellitus. Semin Neonatol. 2004;9:59–65. doi: 10.1016/S1084-2756(03)00064-2. [DOI] [PubMed] [Google Scholar]
- Porter JR, Shaw NJ, Barrett TG, Hattersley AT, Ellard S, Gloyn AL. Permanent neonatal diabetes in an Asian infant. J Pediatr. 2005;146:131–133. doi: 10.1016/j.jpeds.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Purvis TL, Hearn T, Spalluto C, Knorz VJ, Hanley KP, Sanchez-Elsner T, Hanley NA, Wilson DI. Transcriptional regulation of the Alstrom syndrome gene ALMS1 by members of the RFX family and Sp1. Gene. 2010;460:20–29. doi: 10.1016/j.gene.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau P, Masternak K, Krawczyk M, Reith W, Dausset J, Carosella ED, Moreau P. In vivo, RFX5 binds differently to the human leucocyte antigen-E, -F, and -G gene promoters and participates in HLA class I protein expression in a cell type-dependent manner. Immunology. 2004;111:53–65. doi: 10.1111/j.1365-2567.2003.01783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Minton JA, Caswell R, Shield JP, Deiss D, Sumnik Z, Cayssials A, Herr M, Loew A, Lewis V, Ellard S, Hattersley AT. Clinical heterogeneity in patients with FOXP3 mutations presenting with permanent neonatal diabetes. Diabetes Care. 2009a;32:111–116. doi: 10.2337/dc08-1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Patch AM, Minton JA, Flanagan SE, Edghill EL, Hussain K, Balafrej A, Deeb A, Buchanan CR, Jefferson IG, Mutair A, Hattersley AT, Ellard S. Wolcott-Rallison syndrome is the most common genetic cause of permanent neonatal diabetes in consanguineous families. J Clin Endocrinol Metab. 2009b;94:4162–4170. doi: 10.1210/jc.2009-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwitzgebel VM, Mamin A, Brun T, Ritz-Laser B, Zaiko M, Maret A, Jornayvaz FR, Theintz GE, Michielin O, Melloul D, Philippe J. Agenesis of human pancreas due to decreased half-life of insulin promoter factor 1. J Clin Endocrinol Metab. 2003;88:4398–4406. doi: 10.1210/jc.2003-030046. [DOI] [PubMed] [Google Scholar]
- Seguin-Estevez Q, De Palma R, Krawczyk M, Leimgruber E, Villard J, Picard C, Tagliamacco A, Abbate G, Gorski J, Nocera A, Reith W. The transcription factor RFX protects MHC class II genes against epigenetic silencing by DNA methylation. J Immunol. 2009;183:2545–2553. doi: 10.4049/jimmunol.0900376. [DOI] [PubMed] [Google Scholar]
- Sellick GS, Barker KT, Stolte-Dijkstra I, Fleischmann C, Coleman RJ, Garrett C, Gloyn AL, Edghill EL, Hattersley AT, Wellauer PK, Goodwin G, Houlston RS. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet. 2004;36:1301–1305. doi: 10.1038/ng1475. [DOI] [PubMed] [Google Scholar]
- Senee V, Chelala C, Duchatelet S, Feng D, Blanc H, Cossec JC, Charon C, Nicolino M, Boileau P, Cavener DR, Bougneres P, Taha D, Julier C. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet. 2006;38:682–687. doi: 10.1038/ng1802. [DOI] [PubMed] [Google Scholar]
- Senee V, Vattem KM, Delepine M, Rainbow LA, Haton C, Lecoq A, Shaw NJ, Robert JJ, Rooman R, Diatloff-Zito C, Michaud JL, Bin-Abbas B, Taha D, Zabel B, Franceschini P, Topaloglu AK, Lathrop GM, Barrett TG, Nicolino M, Wek RC, Julier C. Wolcott-Rallison Syndrome: clinical, genetic, and functional study of EIF2AK3 mutations and suggestion of genetic heterogeneity. Diabetes. 2004;53:1876–1883. doi: 10.2337/diabetes.53.7.1876. [DOI] [PubMed] [Google Scholar]
- Shimomura K, de Nanclares GP, Foutinou C, Caimari M, Castano L, Ashcroft FM. The first clinical case of a mutation at residue K185 of Kir6.2 (KCNJ11): a major ATP-binding residue. Diabet Med. 2010;27:225–229. doi: 10.1111/j.1464-5491.2009.02901.x. [DOI] [PubMed] [Google Scholar]
- Smith SB, Qu HQ, Taleb N, Kishimoto NY, Scheel DW, Lu Y, Patch AM, Grabs R, Wang J, Lynn FC, Miyatsuka T, Mitchell J, Seerke R, Desir J, Eijnden SV, Abramowicz M, Kacet N, Weill J, Renard ME, Gentile M, Hansen I, Dewar K, Hattersley AT, Wang R, Wilson ME, Johnson JD, Polychronakos C, German MS. Rfx6 directs islet formation and insulin production in mice and humans. Nature. 2010;463:775–780. doi: 10.1038/nature08748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soyer J, Flasse L, Raffelsberger W, Beucher A, Orvain C, Peers B, Ravassard P, Vermot J, Voz ML, Mellitzer G, Gradwohl G. Rfx6 is an Ngn3-dependent winged helix transcription factor required for pancreatic islet cell development. Development. 2010;137:203–212. doi: 10.1242/dev.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997;15:106–110. doi: 10.1038/ng0197-106. [DOI] [PubMed] [Google Scholar]
- Stoy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, Below JE, Hayes MG, Cox NJ, Lipkind GM, Lipton RB, Greeley SA, Patch AM, Ellard S, Steiner DF, Hattersley AT, Philipson LH, Bell GI. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci U S A. 2007;104:15040–15044. doi: 10.1073/pnas.0707291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Muhlendahl KE, Herkenhoff H. Long-term course of neonatal diabetes. The New England Journal of Medicine. 1995;333:704–708. doi: 10.1056/NEJM199509143331105. [DOI] [PubMed] [Google Scholar]
- Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, Bricarelli FD, Byrne G, McEuen M, Proll S, Appleby M, Brunkow ME. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- Wolfe SA, van Wert J, Grimes SR. Transcription factor RFX2 is abundant in rat testis and enriched in nuclei of primary spermatocytes where it appears to be required for transcription of the testis-specific histone H1t gene. J Cell Biochem. 2006;99:735–746. doi: 10.1002/jcb.20959. [DOI] [PubMed] [Google Scholar]
- Wolfe SA, Vanwert JM, Grimes SR. Transcription factor RFX4 binding to the testis-specific histone H1t promoter in spermatocytes may be important for regulation of H1t gene transcription during spermatogenesis. J Cell Biochem. 2008;105:61–69. doi: 10.1002/jcb.21793. [DOI] [PubMed] [Google Scholar]
- Yorifuji T, Kurokawa K, Mamada M, Imai T, Kawai M, Nishi Y, Shishido S, Hasegawa Y, Nakahata T. Neonatal diabetes mellitus and neonatal polycystic, dysplastic kidneys: Phenotypically discordant recurrence of a mutation in the hepatocyte nuclear factor-1beta gene due to germline mosaicism. J Clin Endocrinol Metab. 2004;89:2905–2908. doi: 10.1210/jc.2003-031828. [DOI] [PubMed] [Google Scholar]