

Abstract
Germline deletion of the myostatin gene results in hyperplasia and hypertrophy of the tension-generating (extrafusal) fibres in skeletal muscle. As this gene is expressed predominantly in myogenic tissues it offers an excellent model with which to investigate the quantitative relationship between muscle and axonal development. Here we show that skeletal muscle hyperplasia in myostatin null mouse is accompanied by an increase in nerve fibres in major nerves of both the fore- and hindlimbs. We show that axons within these nerves undergo hypertrophy. Furthermore, we provide evidence that the age-related neural atrophic process is delayed in the absence of myostatin. Finally, we show that skeletal muscle hyperplasia in the myostatin null mouse is accompanied by an increase in the number of muscle spindles (also called stretch receptors or proprioceptors). However, our work demonstrates that the mechanisms regulating intrafusal fibre hyperplasia and hypertrophy differ from those that control the aetiology of extrafusal fibres.
Keywords: axon, muscle, myostatin, nerve fibre, proprioceptor, skeletal, spindle
Introduction
The relationship between nerves and muscles has fascinated biologists for almost 500 years, ever since the first detailed description by Fallopius of the muscular innervation by the cranial nerves (reviewed by Steinberg, 2002). Key work carried out at the beginning of the last century by Shorey, amongst others, demonstrated that nerves and muscles are not only linked in terms of being part of a functional unit (to initiate contraction) but also influence each other's development (Shorey, 1909). Studies performed by Hamburger (1934) developed these ideas further by demonstrating that the survival of motor neurons is regulated by target tissues in the embryonic limb. A key breakthrough in this field came from the study of Hollyday & Hamburger (1976) who transplanted an additional limb field to the flank of a developing chick and discovered that this operation resulted in an increase of the motor neuron population. The most significant finding from this landmark study was that the increase in cell number was not due to an expansion of the progenitor pool but rather to a decrease in the degree of cell death, which would normally eliminate motor neurons that failed to form stable synaptic connections. The identity of the tissues that promoted motor neuron survival was established through tissue ablation studies in which the somites, the source of skeletal muscle, were shown to have a major influence on motor neuron death (Phelan & Hollyday, 1991).
Today it is generally believed that the vertebrate body generates considerably more motor neurons than are needed, and their survival is dependent on the limited production of trophic factors from muscle and Schwann cells (Sendtner et al., 2000; Strelau et al., 2009). The exact number of cells that are eliminated is thought to be in the region of 50–60% compared to those that survive (Lance-Jones, 1982). It is believed that trophic factors, expressed at elevated levels at the neuromuscular junction, are delivered to the cell body through the deployment of retrograde transport mechanisms along the axon (Bartlett et al., 1998; Pu et al., 1999; Reynolds et al., 2000). These suggestions have been assimilated into the trophic factor access hypothesis (reviewed by Banks & Chamberlain, 2005; Oppenheim et al., 1999). Numerous trophic factors have been shown to promote motor neuron survival, including insulin-like growth factor (IGF)-1 and ciliary neurotrophic factor (CNTF), often most efficacious when used in combination. Furthermore, there is evidence that certain motor neuron sub-types may depend on specific combinations of trophic factors to survive (Forger et al., 2003).
Many experiments have been performed in which the number of muscle fibres was reduced and the outcome on motor neuron number assessed. It was shown that the number of surviving motor neurons decreased when the number of muscle fibres was reduced (Habgood et al., 1984; Tanaka & Landmesser, 1986; Grieshammer et al., 1998). In contrast, very few experiments have determined axon numbers in nerves following manipulations that increased the number of muscle fibres. Such studies would be valuable as they would not only validate the trophic factor access hypothesis but, importantly, would determine whether the extra surviving cells can be maintained over extended periods.
We here exploit the phenotype of the myostatin null mouse specifically to determine the effect of a genetic manipulation that results in increased muscle fibre number on nerve fibre development in two major limb nerves. The radial and ischiatic nerves were chosen as representative elements in the anterior and posterior, respectively, of the mouse. The radial is the largest nerve in the brachial plexus. It conducts action potentials that mediate the contraction of extensor muscles of the upper arm and forearm, as well as relaying cutaneous sensation from the dorsal autopod. The ischiatic nerve is the broadest nerve in the body and is a continuation of the sacral plexus. It innervates thigh and shank muscles as well as relaying cutaneous sensation (Gray et al., 1995). Myostatin (GDF-8) is a member of the transforming growth factor-beta (TGF-β) super-family of proteins that negatively regulate skeletal muscle mass (McPherron et al., 1997). Genetic deletion of myostatin in mice results in muscle enlargement, which is brought about by enlargement (hypertrophy) and increase in the number (hyperplasia) of tension-generating fibres (McPherron et al., 1997; Elashry et al., 2009). The mutant is particularly valuable for this study as the gene is expressed predominantly in muscle, not in connective tissue or in tissues of the central nervous system.
Muscle fibres of skeletal muscle perform two major functions. The vast majority generate tension through contraction. The fibres responsible for generating tension are called extrafusal fibres. They are categorised in terms of their myosin heavy chain (MHC) content and, in mice, are classed as oxidative (type I and type IIa), intermediate (type IIx) or glycolytic (type IIb). However, another population of contractile protein-rich fibres are also present in skeletal muscle. These muscle spindles (also called stretch receptors or proprioceptors) are essential for the optimal working of the tissues as they prevent overstretching by acting as mechano-sensors (Maier, 1997). Muscle spindles are composed of a connective tissue capsule that surrounds a varying number of discrete muscle fibres called intrafusal fibres. In contrast, extrafusal fibres contain only one muscle fibre, surrounded by a connective tissue layer. Two forms of muscle spindles have been identified based on the organisation of the nuclei; nuclear bag fibres, where the nuclei are grouped at the equatorial region, and nuclear chain fibres, where the nuclei are centrally located and arranged in a chain-like fashion. Typically, a type Ia sensory fibre projects from the non-contracting portion of the muscle spindle to the CNS, relaying information about the degree of contraction in extrafusal fibres.
Here we report that the deletion of myostatin, which causes muscle fibre hyperplasia, results in an increase in the number of axons present in both the radial and ischiatic nerves. Previous work has shown that the diameter of a motor neuron is a reflection of the size of the motor unit. Fast (glycolytic) muscle fibres are part of large motor units in which the axon tends to have a large diameter. In contrast, slow (oxidative) fibres are part of small motor units and are innervated by small-diameter neurons (Carmeli & Reznick, 1994). We report that the large glycolytic muscle fibre type profile of myostatin null animals is reflected in an increase in the average axon diameter in both the radial and ischiatic nerves. Lastly, we show that the increase in extrafusal fibre number is accompanied by an increase in the number of muscle spindles.
Material and methods
Animals
C57/BL6 and transgenic mstn−/− male mice (minimum of n=3) were bred in the Biological Resource Unit at the University of Reading. Mice were housed with food and water ad libitum until they reached the required age.
Muscle tissue collection
All the mice were killed through schedule one procedures. Two muscles, the flexor digitorum superficialis (FDS) and the flexor digitorum brevis (FDB), from the forelimbs and hindlimbs, respectively, were carefully dissected, snap frozen with liquid nitrogen pre-cooled with isopentane and kept at −80 °C until processing. These muscles were chosen as they have a high sensory function and are rich in muscle spindles.
Nerve tissue collection
The radial nerve and ischiatic nerve from the forelimb and hindlimbs, respectively, were dissected before their branching points. From the anatomical point of view, the radial nerve is the largest of the brachial plexus nerves. It passes behind the axillary artery to the caudal surface of the humerus to traverse from the medial to the lateral surface of the upper forelimb. The ischiatic nerve runs under the biceps femoris and semitendinosus muscles. Before the knee joint, the nerve divides into two main branches, the tibial and the peroneal nerves (Gray's Anatomy, 1995). Both the radial and ischiatic nerves were identified, isolated and fixed in situ by the addition of a few drops of 2.5% glutaraldehyde in 0.1 m phosphate buffer (pH. 7.3) prior to resection. Sections 2 mm in length were fixed in 2.5% glutaraldehyde in 0.1 m phosphate buffer (pH. 7.3) for 3 days, ethanol-dehydrated, washed in resin (LR medium resin kit; TAAB, UK) and then equilibrated in resin overnight at 4 °C. The samples were placed in moulds before the addition of hardener (acrylic resin accelerator; TAAB). The resin blocks were sectioned at 1 μm thickness using an ultra-microtome with a disposable glass knife.
Histological analysis
Resin sections were left to dry on a hot surface (60 °C) overnight. Subsequently, sections were covered with 1% toluidine blue with 1% sodium borate for 1 min, washed in distilled water, dried and mounted using DPX, as previously performed (Illanes et al., 1990).
Immunohistochemistry
Frozen muscle samples were rapidly embedded in tissue tech freezing medium (Jung) using dry ice-cooled ethanol. Whole muscles were cryosectioned and placed on polylysine-coated slides from the tendon of origin to the tendon of insertion to get a single 10-μm section for every 50 μm of muscle length. The sections were air-dried for at least 2 h before staining. The sections were blocked in wash buffer [5% foetal calf serum (v/v) in phosphate-buffered saline containing 0.05% Triton X-100]. Basal lamina and myosin heavy chain isoforms (MHC) type I-, IIa- and IIb-expressing fibres were identified using rabbit anti-laminin (1 : 200) polyclonal antibody (Sigma) and A4.840 IgM (1 : 1), A4.74 IgG (1 : 4) and BFF3 IgM (1 : 1) supernatant monoclonal primary antibodies, respectively (Developmental Studies Hybridoma Bank). Primary antibodies were visualised using Alexa Fluor 488 goat anti-mouse IgG (Molecular Probes A11029, 1 : 200) and Alexa Fluor 633 goat anti-mouse IgM (Molecular Probes A21046, 1 : 200) secondary antibodies. All antibodies were diluted in wash buffer 30 min prior to use.
Transmission electron microscopy
The nerve sections were stained with lead acetate. Transmission electron microscopy was carried out using a Philips CM 20 with an accelerating voltage of 80 kV.
Digital image analysis
Images of toluidine blue-stained sections were captured using a light microscope under an oil immersion lens (× 100) with an Axiocam digital camera. Quantification of nerve fibres was performed manually as previously described (Illanes et al., 1990). Measurement of the nerve fibre cross-sectional area (CSA) was carried out via manual drawing around each fibre using Zeiss axiovision software version 4.7. First, the total CSA of the nerve fibre was determined by measuring the area inside the outer limit of the myelin sheath. Secondly, the axonal CSA was measured. Finally, the myelin sheath CSA was calculated by subtracting the nerve fibre value from the axon area (Fig. 2K). Images of stained FDS and FDB muscle transverse sections were captured using a Zeiss Axioscop2 fluorescent microscope. Images of whole cross-sections were reconstructed using adobe photoshop CS. Quantification of proprioceptors and the number of intrafusal fibres was performed by following the same muscle spindle on serial sections through the entire length of the muscle. Intrafusal fibre cross-sectional area measurements were performed manually using Zeiss axiovision software version 4.7. All FDS and FDB fibres negative for MHC IIa and IIb on double-stained sections were considered to be of a type IIX phenotype by subtraction of the number of type I-stained fibres from serial muscle sections.
Fig. 2.
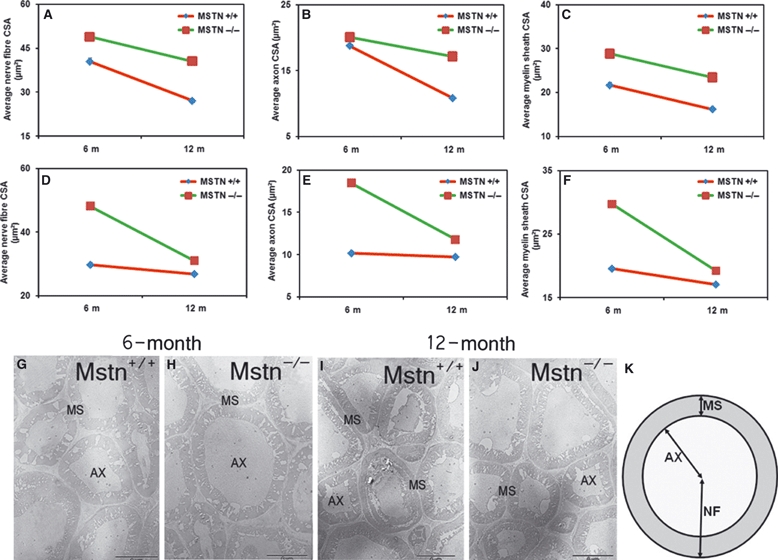
Myostatin deletion increases nerve fibre CSA. Average nerve fibre CSA (axons and myelin sheath) of the radial (A) and ischiatic (D) nerves of 6- and 12-month-old mstn+/+ and mstn−/− mice. (A) mstn−/− mice show a significant increase in nerve fibre CSA compared to mstn+/+ counterparts (P < 0.001). Significant age-related changes in CSA were observed in both genotypes analysed (P < 0.001). Average axon CSA of the radial (B) and ischiatic (E) nerves of 6- and 12-month-old mstn+/+ and mstn−/− mice. Two-way anova for both nerves showed significant changes by genotype, age and interaction of the two parameters. Average myelin sheath CSA of the radial (C) and ischiatic (F) nerves of 6- and 12-month-old mstn+/+ and mstn−/− mice. Two-way anova for both nerves showed significant changes by genotype, age and interaction of the two parameters. (G–J) TEM images showing sections of radial nerve from 6- and 12-month-old mstn+/+ and mstn−/− mice. Images from 6-month-old mstn−/− mice show an increase in the CSA of axons compared to age-matched mstn+/+ animals. Fibres from 12-month-old mstn−/− radial nerves show a reduction in the CSA compared to those from 6-month-old animals. AX, axon; MS, myelin sheath. (K) Diagrammatic illustration showing the technique used for nerve fibre (NF), axon (AX) and myelin sheath (MS) cross-sectional area measurements. Scale bar: 5 μm.
Statistical analysis
Quantification of the total axon number and cross-sectional area of both genotype (wild type vs. myostatin null) and age (6 vs. 12 months old) was analysed using two-way analysis of variance (anova). Assessment of the number of proprioceptors, the number of intrafusal fibres and the CSA of intrafusal fibres was carried out using a two-tailed Student t-test, where P ≤ 0.05 was considered to be significant.
Results
Myostatin deletion increases the number of the axons in fore- and hindlimb nerves
It is well established that myostatin (GDF-8) inhibition increases the muscle mass through hyperplasia and/or hypertrophy (McPherron et al., 1997; Elashry et al., 2009). Development of muscle has been shown to influence motor neurons but this paradigm has not been examined in mstn−/− animals. To this end we chose two major nerves that innervate the muscles of the fore- (radial) and hindlimb (ischiatic). Our analysis showed that myostatin deletion resulted in 21.4 ± 2.8% increases in the number of axons in the radial nerve and 14.4 ± 0.8% increases in the ischiatic nerve at 6 months of age. A similar increase was observed at 12 months of age (Fig. 1A–C).
Fig. 1.
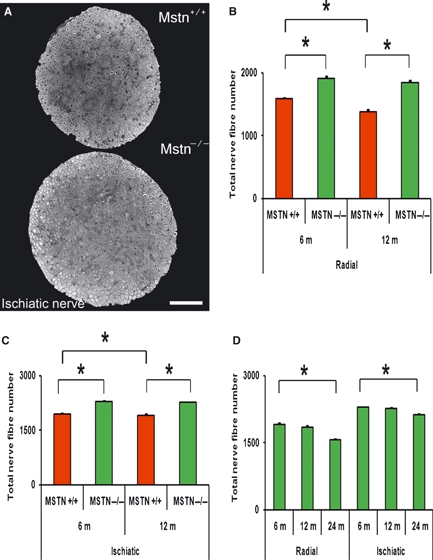
Myostatin deletion increases the number of nerve fibres. (A) Representative images showing transverse sections through the ischiatic nerve of 6-month-old mstn+/+ and mstn−/− mice. Myostatin deletion-induced hyperplasia results in an increased cross-sectional area. Scale bar: 20 μm. (B,C) Quantification of the average nerve fibre number in the radial (B) and ischiatic (C) nerves of 6- and 12-month-old mstn+/+ (red) and mstn−/− (green) mice (n = 3 mice per group). Higher values were observed in mstn−/− nerves at both ages compared to age-matched mstn+/+ counterparts. Two-way anova shows a significant genotype-dependent increase in nerve fibre number (P < 0.001). mstn+/+ animals 12 months old showed decreases in the number of nerve fibres for both radial (B) and ischiatic (C) nerves when compared to 6-month-old animals of equivalent genotype (n = 3 mice per group), but not mstn−/− animals. Two-way anova shows significant age-dependent decreases in nerve fibre number (P = 0.001 for both nerves). (D) Average nerve fibre number in the radial and ischiatic nerves of 6-, 12- and 24-month-old mstn−/− mice. One-way anova was performed to measure the effect of age on nerve fibre number of the mstn−/− mice (6- vs. 12- vs. 24-month-old mice). Significant reductions in the number of axons from 6 to 24 m were observed (n = 3 animals at each time point; *P < 0.001). Error bars are mean ± SEM.
Myostatin deletion delays the age-related reduction of axon number
Previous analysis of axon number in the median nerve of humans demonstrated a reduction with age (Brown, 1972). Here we examined the effect of deleting myostatin on the age-related influence on axon number. Our results revealed that the radial nerve of 12-month-old wild-type mice showed a significant 18.5 ± 3.1% reduction in axon number compared to 6-month-old animals. In contrast, the mstn−/− mice showed a non-significant decrease of 3.4 ± 2.3% during this period (Fig. 1B). Similarly, the ischiatic nerve of mstn+/+ 12-month-old mice showed a significant reduction in axon number of 8.2 ± 2.7% compared to a non-significant reduction of 3.7 ± 0.8% in mstn−/− mice (Fig. 1C). Interestingly, analysis of 24-month-old mstn−/− showed a highly significant reduction in the total number of axons (22.2 ± 2.9% and 11.1 ± 1.4% for radial and ischiatic nerves, respectively) compared to 6-month-old mstn−/− mice (Fig. 1D). Our results demonstrate that myostatin deletion does not cause a significant reduction in the number of the axons at 12 months of age compared to the wild type. However, this was not permanent, as nerve fibre depletion became evident by 24 months.
Hypertrophy followed by age-related hypotrophy in myostatin null axons
Previous studies have demonstrated that myostatin deletion induces a shift from small to large fast myofibres (Girgenrath et al., 2005; Elashry et al., 2009). Therefore, we examined whether the muscle fibre size shift induced by myostatin loss is accompanied by changes in the CSA of the nerve fibres. Our results for the radial and ischiatic nerves from 6-month-old mstn−/− animals showed increases in the CSA of the individual nerve fibres compared to tissue from age-matched mstn+/+ controls (Fig. 2A,D, also Fig. 2G and I; quantification in Table 1).
Table 1.
Average cross-sectional area (CSA) of whole nerve fibre, axon and myelin sheath from 6- and 12-month-old mstn+/+ and 6-, 12- and 24-month-old mstn−/− mice. All data are shown as mean ± SEM.
| Radial | Ischiatic | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mstn+/+ | Mstn−/− | Mstn+/+ | Mstn−/− | ||||||
| Motor unit CSA (μm)2 | 6 months | 12 months | 6 months | 12 months | 24 months | 6 months | 12 months | 6 months | 12 months |
| Nerve fibre | 40.4 ± 1.4 | 27.1 ± 0.7 | 48.9 ± 1.2 | 40.6 ± 1.1* | 22.3 ± 0.5§# | 29.8 ± 0.7 | 26.8 ± 0.6 | 48.2 ± 0.9 | 31 ± 0.6* |
| Axon | 18.8 ± 0.8 | 10.9 ± 0.3 | 20.1 ± 0.6 | 17.1 ± 0.5* | 9.5 ± 0.3§# | 10.2 ± 0.3 | 9.7 ± 0.3 | 18.5 ± 0.5 | 11.8 ± 0.3* |
| Myelin sheath | 21.7 ± 0.7 | 16.2 ± 0.3 | 28.9 ± 0.7 | 23.5 ± 0.6* | 12.8 ± 0.3§# | 19.6 ± 0.4 | 17.1 ± 0.4 | 29.7 ± 0.5 | 19.2 ± 0.3* |
Radial nerve – nerve fibre: significant difference (P < 0.001) from *6-month-old mstn−/−, §6-month-old mstn−/− and # 12-month-old mstn−/− mice; axon: significant difference (P < 0.001) from *6-month-old mstn−/−, §6-month-old mstn−/− and #12-month-old mstn−/− mice; myelin sheath: significant difference (P < 0.001) from *6-month-old mstn−/−, §6-month-old mstn−/− and # 12-month-old mstn−/− mice.
Ischiatic nerve – nerve fibre: *significant difference (P < 0.001) from 6-month-old mstn−/−mice; axon: *significant difference (P < 0.001) from 6-month-old mstn−/−; myelin sheath: *significant difference (P < 0.001) from 6-month-old mstn−/−.
We then addressed the question of whether the CSA increase resulted from expansion of the axon or its surrounding myelin sheath. Our analysis of the 6-month-old radial and ischiatic nerves of mstn−/− animals revealed significant increases in the CSA of both the axon and myelin sheath (Fig. 2B,C,E,F, compare G and I). At 6 months of age, the axonal expansion was greater in the ischiatic nerve than in the radial nerve (Fig. 2B,E).
Radial and ischiatic nerves from both mstn−/− and mstn+/+ genotypes exhibited age-related hypotrophy in the CSA of individual fibres by 12 months (Fig. 2A,D). The degree of hypotrophy depended on the identity of the nerve. The radial nerve from the mstn−/− mice showed less hypertrophy compared to age-matched mstn+/+ mice (Fig. 2A). The opposite was found for the ischiatic nerve (Fig. 2D). Detailed analysis of the axon and myelin sheath characteristics revealed some interesting findings. The radial nerve of mstn+/+ mice showed greater age-related hypotrophy in both the axon and myelin sheath compared to mstn−/− mice (Fig 2B,C). In contrast, the ischiatic nerve of the mstn−/− animals displayed a greater reduction in the diameter of both axon and myelin sheath compared to mstn+/+animals (Fig. 2E,F, also compare G with H and I with J).
The CSA of motor neurons is directly related to the size of the motor unit. During aging, loss of (large-diameter) fast motor neurons leads to muscle fibres being re-innervated by (small-diameter) slow motor neurons. We therefore assessed whether the size of nerve fibres changed over time. We grouped nerve fibres into three categories – small (0–20 μm2), medium (20–60 μm2) and large (more than 60 μm2) – to evaluate the percentage of each population present at a given time. (All data are shown as percentage ± SEM in Table 2). Our analysis for the radial nerve showed that the mstn−/− mice contained fewer small-sized nerve fibres but more large-sized nerve fibres compared to the mstn+/+ animals (Fig. 3A). A similar situation was found at 12 months, with the major difference being that the proportion of small nerves had increased with age for both genotypes. Following this trend, similar significant changes in the percentage of large and small nerve fibres with both genotype and age were revealed following examination of the ischiatic nerve (Fig. 3B). These results suggest that myostatin deletion induced a nerve fibre shift towards large-sized units as compared to age-matched wild types. On the other hand, both genotypes gained more small-sized units at the expense of large-sized units during aging. To confirm and extend these results, we examined the radial nerve from 24-month-old mstn−/− mice and compared it with younger mutant animals. One-way anova revealed that the radial nerve from 24-month-old mstn−/− mice showed a significant increase in the percentage of small-sized axons compared to 6- and 12-month-old mice and a significant decrease in the percentage of large-sized axons, with no significant difference in the percentage of medium-sized axons (Fig. 3C, compare D–F, and Tables 1 and 2). Our results suggest that myostatin deletion reduces the rate of age-related nerve fibre hypotrophy but does not in any way halt this process. The mstn−/− nerve does undergo significant hypotrophy by 24 months (Fig. 3F). Unfortunately, comparisons with wild types were not possible due to the premature death of age-matched litter mates.
Table 2.
Average relative percentage of small, medium and large nerve fibres (μm2) from 6- and 12-month-old mstn+/+ and 6-, 12- and 24-month-old mstn−/− mice. All data are shown as mean ± SEM.
| Radial | Ischiatic | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| mstn+/+ | mstn−/− | mstn+/+ | mstn−/− | ||||||
| Motor unit percentage (%) | 6 m | 12 m | 6 m | 12 m | 24 m | 6 m | 12 m | 6 m | 12 m |
| Small (0-20 μm2) | 38.2 ± 8.9 | 46.5 ± 3.1 | 25.2 ± 2.1 | 37.1 ± 5.5 | 59.6 ± 5.9*§ | 38.1 ± 2.3 | 49.8 ± 3.5 | 6.2 ± 1.1 | 34.9 ± 3.0* |
| Medium (20–60 μm2) | 40.5 ± 2.6 | 44.4 ± 2.6 | 41.6 ± 1.1 | 36.3 ± 1.1 | 35.5 ± 4.5 | 53.0 ± 2.7 | 40.3 ± 1.1 | 67.8 ± 4.7 | 58.8 ± 2.4 |
| Large (> 60 μm2) | 21.3 ± 9 | 9.2 ± 3.5 | 33.2 ± 2.7 | 26.6 ± 6.3 | 4.9 ± 1.8*§ | 8.8 ± 2.3 | 9.9 ± 3.7 | 26.0 ± 5.3 | 8.8 ± 1.6* |
Radial nerve – small: *significant difference from 6-month-old mstn−/− mice (P = 0.007); §significant difference (P = 0.04) from 12-month-old mstn−/− mice; medium: no significant changes; large: *significant difference from 6-month-old mstn−/− mice (P = 0.008), §12-month-old mstn−/− mice (P = 0.02).
Ischiatic nerve – small: *significant difference from 6-month-old mstn−/− mice (P = 0.001); medium: no significant changes; large: *significant difference from 6-month-old mstn−/− mice (P = 0.02).
Fig. 3.

Myostatin deletion causes a shift from small to large nerve fibres. Average radial and ischiatic nerve fibres were categorized into small (0–20 μm2), medium (20–60 μm2) and large (>60 μm2). (A) Mstn−/− (light and dark green) mice showed a tendency towards a reduction in the percentage of small nerve fibres and a significant increase in the percentage of large nerve fibres in the radial nerve compared to mstn+/+ (pink and red) (P = 0.07 and P < 0.03, respectively). A significant interaction between genotype and age was apparent for changes in medium-sized nerve fibres (P = 0.05). (B) mstn−/− ischiatic nerves (light and dark green) showed significant increases in the percentage of medium and large nerve fibres, respectively, compared with mstn+/+ nerves (P < 0.001 and P = 0.05, respectively), with a tendency towards a reduction in small nerve fibres (P = 0.06). In 12-month-old animals, significant genotype-independent changes were seen in the percentage of small nerve fibres during aging (P < 0.02). A significant interaction between genotype and age was apparent for changes in small and large nerve fibres (P < 0.001 and P < 0.05, respectively). (C) Average percentage of small, medium and large radial nerve axons from 6-, 12- and 24-month-old mstn−/− mice. The 24-month-old radial nerves (black) harbour significant increases in small nerve fibres and decreases in large nerve fibres. Error bars displayed as mean ± SEM. (D–F) Representative images of the radial nerve from mstn−/− animals at 6- (D), 12- (E) and 24-months of age (F). Scale bar: 20 μm.
Hyperplasia of muscle spindles in the myostatin null mice
It is well established that myostatin deletion increases skeletal muscle mass through varying degrees of hyperplasia and hypertrophy (McPherron et al., 1997; Elashry et al., 2009). However, it is not clear whether the hyperplasia of extrafusal fibres is accompanied by an increase in the number of muscle spindles. Previous studies have shown that postural muscles contain a high proportion of muscle spindles. Therefore, to analyse the effect of myostatin loss on muscle spindle number, we carried out a study on the flexor digitorum superficialis and the flexor digitorum brevis as representatives of postural muscles of the fore- and hindlimbs, respectively. We counted every muscle spindle in these two muscles using histological (Fig. 4A) and immunohistochemical techniques (Fig. 4A’–A’’), based either on the presence of nuclear bag/chain structures (Fig 4A’) or the expression of MHC within intrafusal fibres (contractile apparatus) at both polar ends of each muscle spindle (Fig. 4A”). Our results show that most of the spindles were distributed in the mid-belly region of the muscle, with only a few located at the periphery. Mstn−/− muscles contained significantly increased numbers of the muscle spindles in both muscles (increases of 26 and 41% for the FDS and FDB muscles, respectively, P<0.05) (Fig. 4B). Analysis of the number of extrafusal fibres revealed that mstn−/− mice had 16 ± 1% and 69 ± 3% more muscle fibres in their FDS and FDB muscles, respectively.
Fig. 4.
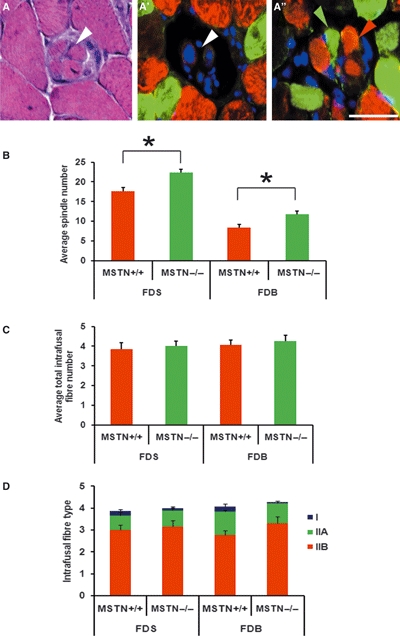
Myostatin deletion causes an increase in the number of muscle spindles but no changes in number of the intrafusal fibres or MHC-expressing fibres. (A) Haematoxylin and eosin staining. (A’–A’’) Double immunofluorescent labelling (right) against MHC IIa (green) and MHC IIb (red) stained with 4’,6-diamidino-2-phenylindole (DAPI, blue) showing the equatorial nuclear region (A’, white arrowhead) and the myosin heavy chain region at the polar end showing type IIa (A’’, green arrowhead) and type IIb (A’’, red arrowhead). Scale bar: 50 μm. (B) Average number of muscle spindles in the FDS and FDB muscles. Significant increases in muscle spindle number were observed in muscles from both mstn−/− (green bars) and age-matched mstn+/+ mice (red bars) (*P < 0.05). (C) Average total number of intrafusal fibres in muscle spindles from the FDS and FDB of mstn+/+ (red bars) and mstn−/− (green bars) mice. No significant differences were present. (D) The average number of MHC type I (blue), IIa (green) and type IIb (red) intrafusal fibres revealed no significant change in distribution in the mstn−/− compared to mstn+/+ mice.
Next, we investigated whether myostatin deletion increases the number of intrafusal fibres within each spindle. The average number of intrafusal fibres varied from 4.1 ± 0 and 4.26 ± 0 for the FDB and 3.8 ± 0 and 4 ± 0 for the FDS in the mstn+/+ and mstn−/− mice, respectively. Surprisingly, we failed to find any significant differences in the number of intrafusal fibres of both muscles in the absence of myostatin when compared to the age-matched mstn+/+ control (Fig. 4C). These studies show that myostatin deletion results in an increase in the number of muscle spindles but not in an increase in the number of intrafusal myofibres present within each of the muscle spindles.
Myostatin deletion has no effect on the fibre-type properties of intrafusal fibres
We determined the molecular properties of the intrafusal fibres to establish whether the deletion of myostatin induces a fibre-type conversion similar to that observed in the extrafusal fibres (Elashry et al., 2009). Interestingly, muscle spindles of mstn−/− animals showed no significant difference in the fibre-type composition in either the FDS or the FDB muscles, compared to age-matched mstn+/+ controls (Fig. 4D, see also Fig. 5A–D).
Fig. 5.
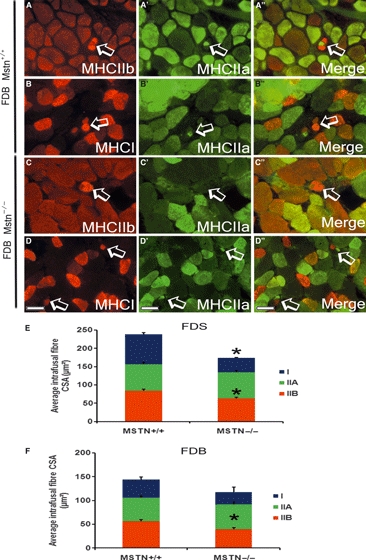
Myostatin deletion caused a decrease in the CSA of the intrafusal fibres but no changes in the histochemical properties of the spindle. (A-D) Double immunofluorescent staining for MHC IIa and IIb (A–A’’ and C–C’’) or IIa and I (B–B’’ and D–D’’) on transverse section of FDB muscle from mstn+/+ (A–B’’) and mstn−/− (C–D’’) highlighting intrafusal fibres (arrows). Scale bar: 50 μm. Quantification of the average CSA of MHC type I, IIa and IIb positive intrafusal fibres from the FDS (E) and FDB (F). (E) The average CSA of type I and type IIb intrafusal fibres was significantly smaller in the FDS muscle in the mstn−/− than in age-matched mstn+/+ mice (*P = 0.001). (F) The FDB muscle only displays a significant reduction in the average CSA of type IIb intrafusal fibres compared to mstn+/+ counterpart (*P = 0.005). All data are shown as mean ± SEM.
Intrafusal fibre hypotrophy in myostatin null mice
Our results demonstrated that myostatin inhibition had no effect on the total number and MHC profile of the intrafusal fibres. Lastly, we examined whether myostatin deletion had any effect on the regulation of the CSA of the intrafusal fibres. Surprisingly, the mstn−/− FDS muscle intrafusal fibres showed a significant reduction in the CSA of type IIb and type I intrafusal fibres (Fig. 5E) compared to age-matched mstn+/+ mice. Similarly, there was a significant reduction in the CSA of type IIb myofibres in the FDB compared to age-matched mstn+/+ mice (Fig. 5F) but not in type I fibres. These data suggest that the mechanisms regulating intrafusal fibre-type development and hypertrophy differ from those that control these processes in extrafusal fibres.
Discussion
In this study we report the effect of a genetic manipulation that causes an increase in skeletal muscle fibre number on axonal numbers and sizes in two major limb nerves. We show that following the deletion of myostatin, a potent inhibitor of skeletal muscle development, the number and size of axons in both the radial and ischiatic nerves of the mouse increase compared to wild-type litter mates. Furthermore, we show that the age-related decrease in the number of axons is attenuated in the mstn−/− compared to mstn+/+ animals. Finally, we show that the increase in extrafusal fibres that results in the absence of myostatin is matched by an increase in the number of muscle spindles.
Investigations going back over 100 years have documented the link between motor neuron number and the number of muscle fibres. The general consensus is that the developing vertebrate embryo generates many more motor neuron cells than are required, a finding that has been interpreted as a fail-safe mechanism that insures that all muscle fibres are innervated.
Three mechanisms are used during prenatal and post-natal life to control the number of motor neurons. During early embryogenesis, the motor neuron population is restricted to the ventral portion of the neural tube following its induction by signals originating from the floor plate. Recent work suggests that any motor neurons that have been incorrectly induced along the dorsal ventral axis of the neural tube are subsequently eliminated (Oppenheim et al., 1999). The second phase of motor neuron number control occurs later in life but still at the prenatal stages when motor neurons compete for trophic factors on recently differentiated myofibres. Motor neurons unable to secure a source of trophic factors are thereafter eliminated. Finally, motor neuron number is also regulated after birth. It is worth noting that in invertebrates, neural activity is required at an earlier stage of muscle development and is responsible for promoting myoblast proliferation and subsequent patterning (Fernandes & Keshishian, 2005).
Classical experiments carried out by Hollyday & Hamburger (1976) and Pittman & Oppenheim (1979) demonstrated that increasing the number of potential motor neuron targets or applying neuromuscular blockading chemicals greatly increases the number of surviving motor neurons by preventing normal cell death, a process termed hypothanasia (Hollyday & Hamburger, 1976). These and other studies led to the development of the trophic factor access hypothesis that large numbers of motor neurons are initially generated, and that these could at early stages of development access trophic factors produced over extended regions of the muscle fibres (reviewed by Banks & Chamberlain, 2005 and Landmesser, 1992). Trophic factors, possibly including IGF-1, glial derived neurotrophic factor and CNTF, are retrogradely transported from the muscle to the motor neuron cell body (Bartlett et al., 1998; Reynolds et al., 2000). However, muscle contraction activity thereafter leads to the remodelling (constriction) of the neuromuscular junction(s) and concentrates the production of trophic factors in this region.
Classical studies examined the increase in axon number only up to later stages of prenatal development and therefore did not address whether sufficient trophic factors would be present to maintain them into adult life. These studies also did not address whether mechanisms that control the number of motor neurons in adult life would correct the extra cells that survived the prenatal cull. Here we show that deletion of myostatin results in a sustained increase in axon numbers that extends into adult life. We suggest that the additional fibres that are generated in the absence of myostatin are able to produce sufficient trophic factors to sustain neurons not only at early stages but also into adulthood. According to the trophic factor access hypothesis, muscle fibres are initially poly-innervated and then the remodelling of the neuro-muscular junction results in all but one axon terminal maintaining contact with the muscle fibre. We suggest that the large increase in muscle fibres during both primary and secondary myogenesis (Matsakas et al., in press) may lead to fewer poly-innervated myofibres, as there are more target cells available to the developing motor neurons. Work is ongoing to investigate this line of thinking.
An important finding from our analysis of the two nerves examined was that the deletion of myostatin delays the decrease in axon number and size seen in the wild-type mice at 12 months of age. Aging has long been known to be associated with the progressive loss of motor neurons. Fast motor neurons are preferentially lost, resulting in the loss of type II muscle fibres. These subsequently are re-innervated by surviving slow motor neurons, which ultimately leads to a change in the muscle profile towards an oxidative phenotype (Lexell et al., 1988; Lexell, 1997; Degens, 2007). Our finding that the loss of axons is delayed in the myostatin null could be explained by the fact that the muscle fibres in the mutant are considerably larger than in the wild type and therefore may produce more trophic factors. Indeed, additional productions of trophic factors have been demonstrated to promote motor neuron survival (Dobrowolny et al., 2005). These could counter the detrimental effects of aging taking place in the fast motor neuron, thereby delaying axon degeneration. However, it should be noted that the number and size of axons do eventually decrease, and the extra production of trophic factors are unable to prevent the detrimental effects of aging in the long term. Interestingly, even though the large-diameter axons were lost in the mstn−/− animals, the muscle seems to retain fast fibres (Elashry et al., 2009). These results would suggest that the genetic deletion of myostatin prevents the fibres from responding to the change in innervation that takes place during aging.
It is of interest that although both the radial and ischiatic nerves of the mstn−/− mice were resistant to age-related axonal loss, the ischiatic axons from the null mice displayed a greater degree of hypotrophy compared with the radial axons. At present we do not understand the mechanism responsible for this difference. However, we propose a possible explanation based on the muscles that each nerve innervates. The radial nerve mainly innervates the triceps and extensor muscles in the forearm. In contrast, the ischiatic nerve innervates large muscles in the back of the leg and shank. We suggest that these large muscles contain a greater proportion of fibres that have undergone extensive hypertrophy following deletion of myostatin and that, during ageing, they undergo a greater reduction in size compared with those in the forelimb. A possible consequence of this hypotrophy is the decreased production of factors maintaining axon anabolic processes.
Our study also addresses the development of the muscle spindles in a model that induces fibre hyperplasia and hypertrophy. We found that there was an increase in the number of muscle spindles that develop in both the FDS and FDB muscles examined. Our detailed analysis of muscle spindles of the myostatin null mice led to three unexpected findings. First, we show that the intrafusal fibres within the spindles from mstn−/− mice had not undergone hyperplasia, even though the extrafusal fibres had increased in number. Secondly, we found that the intrafusal fibres from mstn−/− mice had undergone hypotrophy compared to the intrafusal fibres of mstn+/+ mice. Thirdly, we show that the intrafusal fibres from mstn−/− animals did not show the fibre-type switch towards type IIb that is seen in extrafusal fibres.
Much progress has been made regarding our understanding of fibre-type development. Key molecules regulating this process (MEF2C and MEF2D) were recently identified by Olson's group (Potthoff et al., 2007). Overexpression of either molecule leads to the expression of slow MHC and the deletion of these genes leads to loss of slow fibres and development of fast fibres (Potthoff et al., 2007). Interestingly, myostatin has been shown positively to regulate the expression of these genes (Hennebry et al., 2009). Therefore the loss of myostatin would lead to the lack of MEF2 expression, allowing a fast fibre default of the muscle fibres. However, this does not seem to be the case for muscle spindle fibres and we suggest either that other mechanisms are used in these cells or that the cells are able to over-ride the loss of myostatin. Investigations are currently underway to identify the mechanisms regulating fibre development in muscle spindles.
In summary, our work shows that increasing the number of muscle fibres leads to an increase in axon number. Furthermore, we demonstrate that the axons also increase in diameter and this possibly reflects the fast fibre composition of the muscle. Interestingly, we show that, in the absence of myostatin, there is a delay in the loss of axons. We suggest that this could be due to the enlarged muscle producing elevated levels of neurotrophic factors. This could be of clinical importance, as a number of diseases involve motor neuron loss, e.g. amyotrophic lateral sclerosis (ALS), a fatal disease in humans. Motor neuron loss in animal models of ALS can be delayed through the overexpression of IGF-1. Recent studies have shown that inhibition of myostatin results in beneficial outcomes in rodent models of ALS, promoting studies of this molecule for therapeutic uses (Holzbaur et al., 2006; Morrison et al., 2009).
Acknowledgments
We would like to thank the Egyptian Ministry of Higher Education, the Wellcome Trust (078649), and University of Reading for generous funding, which allowed this work to be carried out.
References
- Banks GB, Chamberlain JS. Relevance of motoneuron specification and programmed cell death in embryos to therapy of ALS. Birth Defects Res C Embryo Today. 2005;75:294–304. doi: 10.1002/bdrc.20051. [DOI] [PubMed] [Google Scholar]
- Bartlett SE, Reynolds AJ, Hendry IA. Retrograde axonal transport of neurotrophins: differences between neuronal populations and implications for motor neuron disease. Immunol Cell Biol. 1998;76:419–423. doi: 10.1046/j.1440-1711.1998.00767.x. [DOI] [PubMed] [Google Scholar]
- Brown WF. A method for estimating the number of motor units in thenar muscles and the changes in motor unit count with ageing. J Neurol Neurosurg Psychiatry. 1972;35:845–852. doi: 10.1136/jnnp.35.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeli E, Reznick AZ. The physiology and biochemistry of skeletal muscle atrophy as a function of age. Proc Soc Exp Biol Med. 1994;206:103–113. doi: 10.3181/00379727-206-43727. [DOI] [PubMed] [Google Scholar]
- Degens H. Age related skeletal muscle dysfunction: causes and mechanisms. J Musculoskelet Neuronal Interact. 2007;7:246–252. [PubMed] [Google Scholar]
- Dobrowolny G, Giacinti C, Pelosi L, et al. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J Cell Biol. 2005;168:193–199. doi: 10.1083/jcb.200407021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elashry MI, Otto A, Matsakas A, et al. Morphology and myofiber composition of skeletal musculature of the forelimb in young and aged wild type and myostatin null mice. Rejuvenation Res. 2009;12:269–281. doi: 10.1089/rej.2009.0870. [DOI] [PubMed] [Google Scholar]
- Fernandes JJ, Keshishian H. Motoneurons regulate myoblast proliferation and patterning in Drosophila. Dev Biol. 2005;15:493–505. doi: 10.1016/j.ydbio.2004.09.038. [DOI] [PubMed] [Google Scholar]
- Forger NG, Prevette D, deLapeyriere O, et al. Cardiotrophin-like cytokine/cytokine-like factor 1 is an essential trophic factor for lumbar and facial motoneurons in vivo. J Neurosci. 2003;23:8854–8858. doi: 10.1523/JNEUROSCI.23-26-08854.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girgenrath S, Song K, Whittemore LA. Loss of myostatin expression alters fiber-type distribution and expression of myosin heavy chain isoforms in slow- and fast-type skeletal muscle. Muscle Nerve. 2005;31:34–40. doi: 10.1002/mus.20175. [DOI] [PubMed] [Google Scholar]
- Gray H, Williams PL, Bannister LH. Gray's Anatomy: The Anatomical Basis of Medicine and Surgery. 38th edn. New York: Churchill Livingstone; 1995. [Google Scholar]
- Grieshammer U, Lewandoski M, Prevette D, et al. Muscle-specific cell ablation conditional upon Cre-mediated DNA recombination in transgenic mice leads to massive spinal and cranial motoneuron loss. Dev Biol. 1998;197:234–247. doi: 10.1006/dbio.1997.8859. [DOI] [PubMed] [Google Scholar]
- Habgood MD, Hopkins WG, Slack JR. Muscle size and motor unit survival in mice. J Physiol. 1984;356:303–314. doi: 10.1113/jphysiol.1984.sp015466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V. The effects of wing bud extirpation on the development of the central nervous systerm in chick embryos. J Exp Zool. 1934;68:449–494. [Google Scholar]
- Hennebry A, Berry C, Siriett V, et al. Myostatin regulates fiber-type composition of skeletal muscle by regulating MEF2 and MyoD gene expression. Am J Physiol Cell Physiol. 2009;296:C525–C534. doi: 10.1152/ajpcell.00259.2007. [DOI] [PubMed] [Google Scholar]
- Hollyday M, Hamburger V. Reduction of the naturally occurring motor neuron loss by enlargement of the periphery. J Comp Neurol. 1976;170:311–320. doi: 10.1002/cne.901700304. [DOI] [PubMed] [Google Scholar]
- Holzbaur EL, Howland DS, Weber N, et al. Myostatin inhibition slows muscle atrophy in rodent models of amyotrophic lateral sclerosis. Neurobiol Dis. 2006;23:697–707. doi: 10.1016/j.nbd.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Illanes O, Henry J, Skerritt G. Light and electron microscopy studies of the ulnar, saphenous, and caudal cutaneous sural nerves of the dog. Am J Anat. 1990;187:158–164. doi: 10.1002/aja.1001870204. [DOI] [PubMed] [Google Scholar]
- Lance-Jones C. Motoneuron cell death in the developing lumbar spinal cord of the mouse. Brain Res. 1982;256:473–479. doi: 10.1016/0165-3806(82)90192-4. [DOI] [PubMed] [Google Scholar]
- Landmesser L. The relationship of intramuscular nerve branching and synaptogenesis to motoneuron survival. J Neurobiol. 1992;23:1131–1139. doi: 10.1002/neu.480230906. [DOI] [PubMed] [Google Scholar]
- Lexell J. Evidence for nervous system degeneration with advancing age. J Nutr. 1997;127:1011S–1013S. doi: 10.1093/jn/127.5.1011S. [DOI] [PubMed] [Google Scholar]
- Lexell J, Taylor CC, Sjostrom M. What is the cause of the ageing atrophy? Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15- to 83-year-old men. J Neurol Sci. 1988;84:275–294. doi: 10.1016/0022-510x(88)90132-3. [DOI] [PubMed] [Google Scholar]
- Matsakas A, Otto A, Elashry MI, et al. Altered primary and secondary myogenesis in the myostatin-null mouse. Rejuvenation Res. doi: 10.1089/rej.2010.1065. in press (in press) [DOI] [PubMed] [Google Scholar]
- Maier A. Development and regeneration of muscle spindles in mammals and birds. Int J Dev Biol. 1997;41:1–17. [PubMed] [Google Scholar]
- McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- Morrison BM, Lachey JL, Warsing LC, et al. A soluble activin type IIB receptor improves function in a mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2009;217:258–268. doi: 10.1016/j.expneurol.2009.02.017. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Homma S, Marti E, et al. Modulation of early but not later stages of programmed cell death in embryonic avian spinal cord by sonic hedgehog. Mol Cell Neurosci. 1999;13:348–361. doi: 10.1006/mcne.1999.0755. [DOI] [PubMed] [Google Scholar]
- Phelan KA, Hollyday M. Embryonic development and survival of brachial motoneurons projecting to muscleless chick wings. J Comp Neurol. 1991;311:313–320. doi: 10.1002/cne.903110302. [DOI] [PubMed] [Google Scholar]
- Pittman R, Oppenheim RW. Cell death of motoneurons in the chick embryo spinal cord. IV. Evidence that a functional neuromuscular interaction is involved in the regulation of naturally occurring cell death and the stabilization of synapses. J Comp Neurol. 1979;187:425–446. doi: 10.1002/cne.901870210. [DOI] [PubMed] [Google Scholar]
- Potthoff MJ, Wu H, Arnold MA, et al. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J Clin Invest. 2007;117:2459–2467. doi: 10.1172/JCI31960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu SF, Zhuang HX, Marsh DJ, et al. Time-dependent alteration of insulin-like growth factor gene expression during nerve regeneration in regions of muscle enriched with neuromuscular junctions. Brain Res Mol Brain Res. 1999;63:207–216. doi: 10.1016/s0169-328x(98)00250-2. [DOI] [PubMed] [Google Scholar]
- Reynolds AJ, Bartlett SE, Hendry IA. Molecular mechanisms regulating the retrograde axonal transport of neurotrophins. Brain Res Brain Res Rev. 2000;33:169–178. doi: 10.1016/s0165-0173(00)00028-x. [DOI] [PubMed] [Google Scholar]
- Sendtner M, Pei G, Beck M, et al. Developmental motoneuron cell death and neurotrophic factors. Cell Tissue Res. 2000;301:71–84. doi: 10.1007/s004410000217. [DOI] [PubMed] [Google Scholar]
- Shorey M. The effect of the destruction of peripheral areas on the differentiation of the neuroblasts. J Exp Zool. 1909;7:25–64. [Google Scholar]
- Steinberg DA. Scientific neurology and the history of the clinical examination of selected motor cranial nerves. Semin Neurol. 2002;22:349–356. doi: 10.1055/s-2002-36756. [DOI] [PubMed] [Google Scholar]
- Strelau J, Strzelczyk A, Rusu P, et al. Progressive postnatal motoneuron loss in mice lacking GDF-15. J Neurosci. 2009;29:13640–13648. doi: 10.1523/JNEUROSCI.1133-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Landmesser LT. Cell death of lumbosacral motoneurons in chick, quail, and chick-quail chimera embryos: a test of the quantitative matching hypothesis of neuronal cell death. J Neurosci. 1986;6:2889–2899. doi: 10.1523/JNEUROSCI.06-10-02889.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]