

Abstract
Excessive generation of nitric oxide radical (NO•) in neuroinflammation, excitotoxicity and during age-related neurodegenerative disorders entails the localized and concerted increase in nitric oxide synthase(s) expression in glial cells and neurons. The aim of the present study was to assess the biological significance of the impact of NO• on the cell’s thiol status with emphasis on S-glutathionylation of targeted proteins. Exposure of primary cortical neurons or astrocytes to increasing flow rates of NO• (0.061–0.25 μM/s) resulted in the following. (i) A decrease in GSH (glutathione) in neurons accompanied by formation of GSNO (S-nitrosoglutathione) and GSSG (glutathione disulfide); neurons were far more sensitive to NO• exposure than astrocytes. (ii) A dose-dependent oxidation of the cellular redox status: the neuron’s redox potential increased ~ 42 mV and that of astrocytes ~ 23 mV. A good correlation was observed between cell viability and the cellular redox potential. The higher susceptibility of neurons to NO• can be partly explained by a reduced capacity to recover GSH through lower activities of GSNO and GSSG reductases. (iii) S-glutathionylation of a small subset of proteins, among them GAPDH (glyceraldehyde-3-phosphate dehydrogenase), the S-glutathionylation of which resulted in inhibition of enzyme activity. The quantitative analyses of changes in the cell’s thiol potential upon NO• exposure and their consequences for S-glutathionylation are discussed in terms of the distinct redox environment of astrocytes and neurons.
Keywords: astrocyte, energy metabolism, neurodegeneration, neuron, nitric oxide, S-glutathionylation
INTRODUCTION
Excessive generation of NO• in neuroinflammation, excitotoxicity, and during aging and age-related neurodegeneration entails the up-regulation of the inducible (iNOS) and neuronal (nNOS) isoforms of nitric oxide synthase, enzymes that catalyse the five-electron oxidation of the guanidine group of L-arginine with the subsequent release of nitric oxide radical (NO•) [1,2]. The diverse biological effects of NO• and NO•-derived species can be attributed in part to their ability to regulate the activity of target proteins through modifications of redox-sensitive thiols. S-nitrosylation of reactive thiol cysteine residues has been proposed as a signalling mechanism for NO• under physiological and pathological circumstances [3]. Several proteins have been shown to be S-nitrosylated during exposure to NO• [4,5]; however, the nature of this modification is labile and its stability is often affected by the presence of thiols [6], largely by GSH (glutathione), the most abundant non-protein thiol, ubiquitously present in most cells at ~ 1–10 mM concentrations [7]. The reaction of GSH with S-nitrosylated proteins by a transnitrosation mechanism (eqn 1) [3,6] yields GSNO (nitrosoglutathione); that by a S-thiolation mechanism (eqn 2) yields a protein mixed disulfide or S-glutathionylated protein and nitroxyl anion (NO−); the rapid consumption of NO− exerts a kinetic control on this reaction [8].
| (1) |
| (2) |
As to which modification is likely to be the consequential end point is determined largely by the pKa of the thiolate moiety, an example of the nuances of NO•-mediated regulation of protein function. The product of the S-thiolation mechanism (eqn 2), an S-glutathionylated protein, can also be formed via thiol–disulfide exchange (eqn 3) and sulfenic acid intermediates (eqn 4) [9].
| (3) |
| (4) |
Regardless of the molecular mechanisms, S-glutathionylation has been shown to be a regulatory device for mitochondrial and cytosolic proteins, such as aconitase, adenine nucleotide transporter, GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and PTEN (phosphatase and tensin homologue deleted on chromosome 10) [3,9–12]. Hence, S-glutathionylation is an important regulator of protein function in response to several stressors or stimuli in many cellular and disease contexts [3,7,9]. Although S-glutathionylation is often assumed to be a consequence of increased oxidative stress, recent evidence points to S-glutathionylation of proteins as a consequence of nitrosative stress as well [11,13].
The increased generation of NO• and the presence of S-glutathionylated proteins in neurodegenerative disorders (such as Alzheimer’s disease) has incited further clarification of the role of protein S-nitrosylation and S-glutathionylation in neuronal dysfunction [7,9]. The selective loss of neurons in neurodegeneration points to a differential susceptibility between neurons and their surrounding glial cells in the disease process. As such, astrocytes have shown to be more resistant to NO• toxicity than neurons and this has been attributed in part to the glutathione system [14,15]. Within this context, the redox buffering capacity of the cell is likely to influence S-glutathionylation of proteins inasmuch as protein mixed disulfide formation can be viewed as a shift of the cellular redox environment towards a more oxidized state [16].
With the aid of a sensitive HPLC method for detecting intracellular formation of GSNO and GSSG, the present study was aimed at assessing (i) whether or not S-glutathionylation of redox-sensitive proteins is a consequence of increased NO• levels (mediated by GSNO and GSSG formation), and (ii) the role of NO• modulated S-glutathionylation of proteins in neuronal cell injury.
MATERIALS AND METHODS
Chemicals and biochemicals
MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide], BCNU [1,3-bis(2-chloroethyl)-1-nitrosourea], protease inhibitor cocktail (catalogue number P8340), PMSF, NP-40 (Nonidet P40), L-glutamine, L-glutamate, o-metaphosphoric acid, NEM (N-ethylmalemide), ammonium sulfamate, acetonitrile, sodium monobasic phosphate, 1-octanesulfanic acid, o-phosphoric acid, NADH, NADPH, GSNO, GSSG and GAPDH were purchased from Sigma Chemical Co. NBM (neurobasal medium), B-27 supplements and penicillin/streptomycin were purchased from Invitrogen.
Animals
Fischer 344 rats (Simonsen Laboratories) were housed under an automated 12 h light/12 h dark cycle with food and water available ad libibum and were starved overnight before being killed. All experimental procedures were subject to the approval of the Institutional Animal Care and Use Committee (IAUC) of the University of Southern California, Los Angeles, CA, U.S.A.
Primary cortical neurons and astrocytes
Primary cortical neurons were isolated using the methods described previously [17,18]. Briefly, cortical neurons were isolated from the fetuses of timed pregnant (E16–E18, where E is embryonic day) Fischer 344 rats, plated at a density of ~ 106 cells/well in 0.1 % polyethyleneimine-coated six-well plates and seeded in NBM supplemented with B-27, 25 units/ml penicillin, 25 μg/ml streptomycin, 25 μM L-glutamate and 0.5 mM L-glutamine at 37 % in a humidified 5 % CO2 atmospher for the first 3 days and thereafter, maintained in NBM without L-glutamate. All experiments were carried out on 10–14-day-old primary cortical neurons. Rat primary astrocytes were isolated from neonatal Fischer 344 rats (2–4 days old) and maintained in DMEM (Dulbecco’s modified Eagle’s medium)/Ham’s F12 (Cell Culture Core Facility, University of Southern California) supplemented with 10 %(v/v) heat-inactivated fetal bovine serum (Gemini Bio-Products), 50 units/ml penicillin and 50 μg/ml streptomycin until the cultures reached an age of 3 weeks; cultures were trypsinized, recounted and seeded in 0.1 % polyethyleneimine-coated six-well plates at a density of ~ 0.5 × 106 cells/well. All primary neurons and astrocytes were stained with Trypan Blue and counted before seeding into plates or dishes.
Exposure of cells to NO•
Primary cortical neurons and astrocytes were exposed to 3.4–13.8 mM DETA (diethylenetriamine)–NO, a relatively pure NO• donor that decomposes into NO• and the free amine DETA [19]. The above DETA–NO concentrations correspond to NO• release rates of 0.061–0.25 μM/s as determined by an ISO NO• electrode (World Precision Instruments). Cells were exposed to the NO• donor for 1 h to minimize the likelihood that changes in the GSH pool were due to cell death. The half-life of the donor was ~ 20 h, hence the flow of NO• release was constant under the conditions of the present study.
MTT assay
MTT (0.5 mg/ml) was dissolved in Hepes toxicity buffer and added to the wells at the end of the experiment [17]. Cells were incubated for 1.5 h at 37 °C and lysed in DMSO. MTT reduction was measured at 490 nm using a microplate spectrophotometer (Bio-Rad Laboratories). Results are expressed as percentage reductions compared with controls. All measurements were carried out in triplicate for each treatment.
Measurement of GSH, GSSG and GSNO
GSH, GSSG and GSNO was measured by HPLC with electrochemical detection as described previously [20]: cells were treated with either 5 mM NEM before lysis with 5 % o-metaphosphoric acid, or 5 % o-metaphosphoric acid with 25 mM ammonium sulfamate to prevent artefactual formation of GSNO. Cells were washed with ice-cold PBS before the addition of o-metaphosphoric acid, and total lysate was centrifuged at 12 000 g for 5 min. Supernatant was analysed for GSH, GSNO and GSSG content by injection into HPLC Agilent 1100 series (Agilent Technologies). The mobile phase used to separate GSH, GSNO and GSSG consisted of 3 % acetonitrile, 25 mM sodium monobasic phosphoric acid and 0.5 mM 1-octanesulfanic acid adjusted to pH 2.7 with o-phosphoric acid. The respective concentrations of GSH, GSNO and GSSG were calculated based on injected standards.
Cellular redox status
The cellular redox status was quantified by the Nernst equation (Ehc = Eo + 30 log([GSSG]/[GSH]2) where [GSH] and [GSSG] are molar concentrations and Eo is taken as −264 mV at pH 7.4 [21]. The Nernst potential was calculated from the molar concentration of GSH and GSSG in cell volume at the value of ~ 10 μl/mg of protein. In the experimental design of the present study whereby neurons or astrocytes are exposed to a flow of NO•, GSNO is formed. Hence, assessment of the cellular redox status by the Nernst equation may be difficult because the equation does not take into account the absolute concentration of GSNO or the GSNO/GSH ratio. Nevertheless, the formation of GSNO from GSH is reflected in the overall absolute concentration of GSH and the equation is still useful to ascertain the redox state of cells under nitrosative stress. The Nernst potential takes into the account not only the GSH/GSSG ratio, but also the absolute concentration of GSH, where any changes in the latter will change the redox potential even without significant changes in the GSH/GSSG ratio [16].
Enzyme activities
(i) Activities of GSNO and GSSG reductases. Cells were lysed in reductase buffer (20 mM Tris/HCl, pH 8, 0.5 mM EDTA, 0.1 % NP-40 and 1 mM PMSF), and sonicated three times at 20 s intervals (with 1 min of resting time on ice) to disrupt and solubilize all proteins. To detect GSNO-metabolizing activity, 1 mg/ml lysate was incubated with 100 μM GSNO in the absence or presence of 200 μM NADH. GSNO reductase activity was measured by determining the GSNO-dependent decrease in NADH absorbance at 340 nm [22]. GSSG reductase activity was measured with 1 mg/ml lysate in the presence of 100 μM GSSG and 200 μM NADPH; absorbance was measured at 340 nm (ε340 = 6.22 mM−1 · cm−1). BCNU (20 mM) was added as an inhibitor of GSSG reductase activity to ensure the specificity of the reaction. (ii) GAPDH activity in neurons was measured using the method described in [23] with some modifications: cells were lysed in GAPDH reaction buffer (100 mM Tris/HCl and 5 mM sodium arsenate, pH 8.6) with the addition of protease inhibitors and 2 mM NEM, and freeze–thawed three times. GAPDH activity was detected in the supernatant through monitoring the increase in NADH formation spectrophotometrically at 340 nm in the presence of 250 μM NAD+ and 50 mg/ml GAPDH. Purified rabbit muscle GAPDH activity was measured under the same conditions.
Western blot and immunoprecipitation
Purified rabbit GAPDH was purchased from Roche Chemicals. Monoclonal antibodies recognizing glutathionylated proteins (anti-glutathione, catalogue no. 101-A) and the agarose-conjugated form of the antibody (catalogue no. 101-A-A) were purchased from Virogen and diluted at 1:100 for immunoblotting and 1:10 for immunoprecipitation. Rabbit polyclonal GAPDH purchased from Abcam was diluted 1:1000 for immunoblotting. (i) Immunoblotting. After NO treatment, cells were washed three times in ice-cold PBS and scraped in RIPA buffer (50 mM Tris/HCl, pH 7.4, 1 % NP-40, 0.25 % sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM Na3VO4, 1 mM PMSF, 1 mM NaF, 5 mM NEM and protease inhibitor cocktail) with (non-)reducing SDS sample buffer, and separated by non-reducing SDS/PAGE (10 % gel). Using appropriate antibodies, the immunoreactive bands were visualized with an enhanced chemiluminescence reagent (Pierce Biotechnology). Western blot images were acquired using VersaDoc 1000 imaging system (Bio-Rad Laboratories). SDS/PAGE gels were also stained with Coomassie Blue to ensure equal loading. (ii) Immunoprecipitation. Cell lysates were prepared as described above and centrifuged at 10 000 g for 30 min at 4 °C with Nanosep columns with 3 kDa cut-off (VWR International) to concentrate proteins and remove endogenous GSH to prevent interference with anti-glutathione agarose-conjugated beads. Lysates were then incubated with agarose-conjugated anti-glutathione antibody diluted 1:10 for 48 h at 4 °C in the dark; immunoprecipitates were dissociated from the agarose beads by boiling in reducing sample buffer containing 100 mM DTT (dithiothreitol) (Pierce Biotechnology), immunocomplexes were separated on by non-reducing SDS/PAGE (10 % gel), and probed with the anti-GAPDH antibody and visualized by chemiluminescence as described above. Total lysate from neurons not exposed to NO• was used as a control.
Statistical significance
Results are means ± S.E.M. Statistical analysis was performed using Student’s t test for unpaired data or ANOVA. A P value of <0.05 was considered significant.
RESULTS
Exogenous NO• leads to intracellular formation of GSNO in primary cortical neurons and astrocytes
Primary cortical neurons and astrocytes were exposed to various flow rates of NO•, and the cellular content of GSH, GSSG and GSNO was assayed using a sensitive HPLC method. Astrocytes are more resilient than neurons to NO• challenges [14] and this may be partly due to differential changes in their thiol/disulfide (GSH/GSSG) status. Figure 1 shows changes in the GSH status of neurons (Figure 1A) and astrocytes (Figure 1B) upon exposure to a continuous flow of NO• for short periods. In the absence of NO•, GSH levels in neurons and astrocytes were 14.49 and 23.16 nmol/106 cells respectively. The higher GSH levels in astrocytes compared with neurons are consistent with other studies [24,25] and with the role of astrocytes as a detoxifier of oxidants in the brain [25].
Figure 1. Intracellular GSNO and GSSG formation following exposure of primary cortical neurons and astrocytes to NO•.

Primary cortical neurons or astrocytes were treated with increasing concentrations of DETA–NO for 1 h in their respective media as described in the Materials and methods section. GSH (●), GSNO (○) and GSSG ( ) levels in (A) primary cortical neurons and (B) astrocytes. n = 3.
) levels in (A) primary cortical neurons and (B) astrocytes. n = 3.
Increasing flow rates of NO• led to a dose-dependent decrease in GSH levels that were accompanied by an increase in GSNO concentrations in both primary cortical neurons and astrocytes (Figure 1). Upon exposure to higher NO• flow rates (i.e. 0.25 μM/s), GSH concentrations in neurons decreased by ~ 55 % (Figure 1A), whereas those in astrocytes decreased by ~ 14 % (Figure 1B). Exposure to lower rates of NO• delivery (0.061 μM/s) led to a ~ 35 %decrease in GSH levels in neurons, whereas those in astrocytes were hardly affected (Figure 1). This suggests that astrocytes are able to maintain their GSH concentrations more effectively than neurons during nitrosative stress. In the absence of NO•, GSSG levels in neurons and astrocytes were 0.03 ± 0.005 and 0.07 ± 0.04 nmol/106 cells respectively, increasing to 0.14 ± 0.05 and 0.18 ± 0.09 nmol/106 cells when exposed to NO• (0.25 μM/s). The decreases in GSH were accompanied by GSNO formation up to 0.23 ± 0.05 and 0.13 ± 0.008 nmol/106 cells in neurons and astrocytes respectively (when exposed to an NO• flow rate of 0.25 μM/s). Increases in GSNO were accompanied by smaller increases in GSSG. Hence this method allows for detection of a substantial pool of GSNO in both cell types.
Exposure to NO• leads to changes in the cellular redox status
The higher levels of GSH in astrocytes might afford a greater reducing capacity to buffer changes of the cellular redox status, which can be quantified by the Nernst equation (see the Materials and methods section). The cellular redox status is dependent on two factors: the GSH/GSSG ratio and the absolute concentration of GSH [7,16]. Calculation of the cellular redox potential by this equation in the presence of NO• presents some difficulties, for it does not take into account the absolute concentration of GSNO or the GSNO/GSH ratio. However, the formation of GSNO from GSH is reflected in decreases in the absolute concentration of the latter and the equation may provide a rough estimation of the cellular redox status. Figure 2 shows that exposure of neurons and astrocytes to NO• leads to a dose-dependent oxidation of the cellular redox potential, the effect being more pronounced in neurons than in astrocytes. On the basis of GSH and GSSG concentrations in the absence of NO•, the redox potentials in neurons and astrocytes were −281.29 ± 4.42 and −287.22 ± 4.67 mV respectively. These values were increased to −239.21 ± 17.9 and −272.24 ± 8.43 mV in neurons and astrocytes respectively upon exposure of NO• at a flow rate of 0.25 μM/s (Figure 2). This supports the notion of a higher susceptibility of neurons to oxidative/nitrosative stress compared with astrocytes.
Figure 2. Cellular redox status as a function of NO• flow rate.
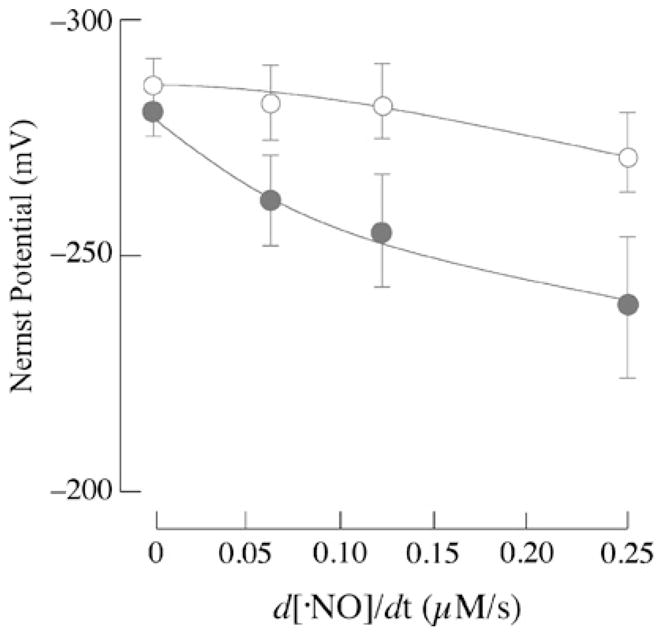
Nernst potential (mV) in neurons (●) and astrocytes (○) was calculated as described in the Materials and methods section and plotted as a function of NO• flow rate (expressed in μM/s).
Metabolism of GSNO and GSSG in primary cortical neurons and astrocytes
GSNO reductase, an evolutionarily conserved enzyme capable of metabolizing intracellular S-nitrosothiol levels, has a high affinity and specificity towards GSNO [22]. The enzyme catalyses the NADH-dependent metabolism of GSNO to GSSG and NH3 (GSNO + 2NADH + 2H+→GSNH2 + 2NAD+ + H2O; GSNH2 + GSH→GSSG + NH3) and is important in regulating cellular levels of S-nitrosothiols [22,26] and susceptibility to nitrosative stress [27]. To determine whether or not primary cortical neurons and astrocytes are able to metabolize GSNO, a fixed amount of GSNO was incubated with cellular lysates from primary cortical neurons and astrocytes in the presence or absence of NADH. Both cell types, neurons (Figure 3A) and astrocytes (Figure 3C), metabolized GSNO in an NADH-dependent manner. However, astrocytes metabolized GSNO faster (t1/2 ~ 5 min) than neurons (t1/2 ~ 10 min). Consumption of GSNO by both cell lysates was accompanied by an NADH-dependent GSSG formation (see equations above), the rate of which was higher in astrocytes than in neurons (Figures 3B and 3D).
Figure 3. GSNO metabolism in neurons and astrocytes.

Assay conditions: 1 mg/ml cell lysate supplemented with 100 μM GSNO in the absence or presence of 200 μM NADH in reductase buffer. GSNO and GSSG were determined by HPLC as described in the Materials and methods section. (●) GSNO or GSSG in the absence of NADH; (○) GSNO or GSSG in the presence of NADH. (A and B) Primary cortical neurons; (C and D) primary cortical astrocytes. n = 3; P < 0.05.
In concert with the GSNO reductase activity, reduction of GSSG by GSSG reductase regulates and maintains the GSH pool. The activities of GSNO reductase and GSSG reductase were measured in order to assess the differential ability of neurons and astrocytes to buffer significant changes in the redox potential effected by an NO• challenge. GSNO-driven NADH oxidation proceeded at rates 4-fold higher in astrocytes (8.21 nmol/min per mg of protein) than in neurons (1.88 ± 0.20 nmol/min per mg of protein) (Figure 4A). Likewise, GSSG reductase activity was 11-fold higher in astrocytes (7.41 ± 0.5 nmol/min per mg of protein) than in neurons (0.63 ± 0.14 nmol/min per mg of protein) (Figure 4B). NAD(P)H consumption in the presence of GSSG was inhibited upon pre-treatment of cells with BCNU, a GSSG reductase inhibitor (results not shown).
Figure 4. GSNO reductase and GSSG reductase activity in neurons and astrocytes.
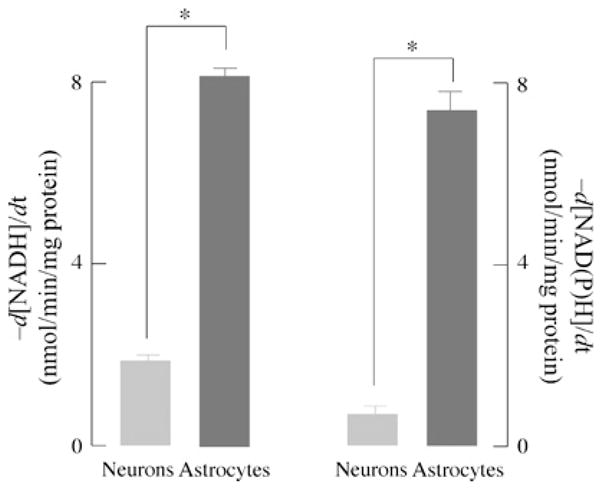
Primary cortical neurons and astrocytes were lysed in reductase buffer and GSNO/GSSG reductase activity was measured as described in the Materials and methods section. GSNO reductase activity was determined by the rate of NADH oxidation at 340 nm in 1 mg/ml cell lysate protein supplemented with 100 μM GSNO and 200 μM NADH. GSSG reductase activity was determined by the rate of NADPH oxidation at 340 nm in 1 mg/ml cell lysate protein in the presence of 100 μM GSSG and 200 μM NADPH. n = 3; *P < 0.05.
NO•, cellular redox state and cell viability
Exposure of primary cortical neurons and astrocytes to increasing flow rates of NO• resulted in a dose-dependent decrease in cellular viability in neurons (Figure 5A). NO•-induced cytotoxicity was far less pronounced in astrocytes: 49.7 ± 16.2 % of cells were viable at the highest flow rate of NO• (0.25 μM/s). Conversely, at this highest flow rate, only 9.7 ± 6.6 %of neurons were viable (Figure 5A). Addition of N-acetylcysteine, a GSH-repleting thiol, led to attenuation of cell death in neurons (results not shown). The dependence of cell viability (results shown in Figure 5A) on the cellular redox potential (expressed in mV; results shown in Figure 2) is shown in Figure 5(B): of note, cell viability diminishes dramatically within a narrow reduction potential range, 25 mV; although Figure 5(B) shows a good correlation between cell viability and the cell’s reduction potential, the reduction potential values obtained in this plot cannot be taken as correlating with apoptosis or necrosis. The redox potential, estimated from the GSH/GSSG pool, in HT29 cells was reported to increase from approx. −250 mV in differentiating cells to approx. −150 mV in apoptotic cells [21]. This discrepancy may be due, in part, to the different exposure times to NO• in experiments designed to calculate the redox potential (as a function of the concentrations of GSH, GSSG and GSNO) and cell viability. The values obtained in the present study, however, agree with previous work on Jurkat cells exposed to steady-state levels of H2O2 in which variations of the thiol redox potential were in the 5–20 mV range, depending on the extent of apoptosis [28].
Figure 5. NO•, cell viability and the cellular redox potential.

(A) Primary cortical neurons (black bars) and astrocytes (grey bars) were exposed to increasing concentrations of DETA–NO for 6 h. Viability was determined using a MTT assay. Values are expressed as percent of the viability in untreated cells. (B) Dependence of cell viability (data from A) on the cell’s reduction potential (data from Figure 2): (●) neurons, ( ) astrocytes. n = 3; P < 0.05.
) astrocytes. n = 3; P < 0.05.
Exposure of cells to NO• leads to protein glutathionylation
As shown in Figure 1, exposure of neurons and astrocytes to a flow of NO• led to the formation of GSNO and GSSG; in addition, disruption of the GSH pools may be due to a decreased synthesis of GSH, increased efflux of GSSG and/or conjugation of GSH to proteins with formation of protein mixed disulfides, i.e. S-glutathionylation. Whereas decreased GSH synthesis has been shown in neurons upon chronic exposure to NO• [15], our experimental model, entailing short exposure periods (1 h), suggests that disruption of the GSH pool is likely to be due to an increased export of GSSG [29] and/or S-glutathionylation of proteins. The latter was assessed in primary cortical neurons and astrocytes (Figure 6): exposure of neurons to increasing NO• flow rates led to a dose-dependent increase in glutathionylated proteins (Figure 6A); treatment with DTT, which reduces disulfide bonds, elicited a loss of the immunoreactive bands, thus indicating that the antibody recognized glutathionylated proteins. Diamide, a thiol-specific oxidant, was used as a positive control: supplementation of neurons with diamide led to a slew of glutathionylated proteins; conversely, NO• treatment led to glutathionylation of a small subset of proteins (only three detectable bands corresponding to approx. 37, 40 and 50 kDa). This suggests that NO•-induced protein glutathionylation proceeds by mechanisms other than thiol oxidation.
Figure 6. NO•-induced protein glutathionylation in neurons and astrocytes.

Western blot analysis of glutathionylated proteins of cellular lysate from (A) neurons and (B) astrocytes treated with increasing concentrations of DETA–NO for 1 h or 200 mM diamide for 10 min. Lysates from cells exposed to 0.25 μM/s of NO• flux for 1 h and thereafter treated with 100 mM DTT for 10 min after lysis to confirm specificity of the antibody. Glutathionylated proteins were detected with anti-glutathione antibodies (1:500). Gels were stained with Coomassie Blue to ensure equal loading. n = 3. Grey lines indicate sections of the gel where empty lanes were removed. Molecular masses are indicated in kDa.
Treatment of primary astrocytes with NO• also led to protein glutathionylation (Figure 6B); however, only one band (~ 90 kDa) increased in intensity in a dose-dependent manner. Immunoreactive bands at 40 and 37 kDa were insensitive to DTT and showed no change in intensity upon NO• treatment; this suggests some non-specific binding of the antibody, thus rendering their glutathionylated status unlikely. Treatment of astrocytes with diamide resulted in a laddering pattern, indicating that NO•-induced glutathionylation occurs in a highly specific manner, affecting a discrete number of proteins in astrocytes as well.
Modulation of GAPDH activity by NO• through glutathionylation
GAPDH, a glycolytic enzyme responsible for generating the first high-energy intermediate and pair of reducing equivalents in glycolysis, consists of four identical 37 kDa subunits, each containing four cysteine residues, two of which (Cys149 and Cys153) are located in the catalytic site. Cys149 forms a highly reactive thiolate group due to its interaction with histidine and is necessary for GAPDH activity [10]. Previous work has demonstrated that GAPDH can undergo redox-mediated post-translational modifications, such as nitrosylation, during NO• exposure [30] or glutathionylation when incubated with GSNO [10,31]. Exposure of neurons to increasing flow rates of NO• resulted in a dose-dependent decrease in GAPDH activity (Figure 7A) under experimental conditions that entailed protein glutathionylation (Figure 6A). Figure 7(B) shows GAPDH glutathionylation when neurons were exposed to an NO• flow rate of 0.12 μM/s.
Figure 7. NO•-mediated inhibition of GAPDH activity in primary cortical neurons.
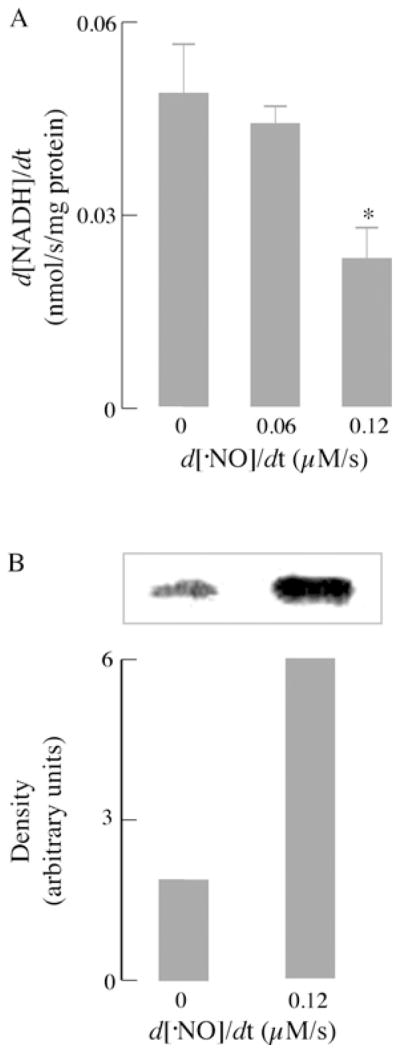
(A) GAPDH activity was measured in cell extracts by monitoring the formation of NADH in the presence of glyceraldehyde 3-phosphate (100 μg/ml) and 250 μM NAD+. Other assay conditions were as described in the Materials and methods section (n = 3; *P < 0.05). (B) Primary cortical neurons (seeded at 106 cells/well) were exposed to a flow of NO• at a rate of 0.12 μM/s for 1 h and pre-treated with 1 mM NEM for 10 min before lysis. Assay conditions were as described in the Materials and methods section.
In vitro incubation of purified rabbit GAPDH with increasing concentrations of GSSG resulted in glutathionylation of the enzyme (Figure 8A) and, consequently, inhibition of its activity (Figure 8B). Inhibition followed a sigmoidal response with the half-maximal inhibition value at ~ 1.3 mM GSSG (Figure 8B). DTT treatment rescued GSSG-elicited inhibition of GAPDH (results not shown). Maximal inhibition of GAPDH occurred at 60 % and was linear only within a certain range; this suggests that glutathionylation of GAPDH does not completely inhibit its activity and a threshold effect is present with regard to modulation of protein activity whereby the redox environment needs to be oxidized to a certain extent before GAPDH activity might be regulated through S-glutathionylation. Treatment of GAPDH incubated with 1 mM GSSG in the presence of DTT (Figure 8A) resulted in a decrease in immunoreactivity. However, there appears to be some non-specificity with the anti-glutathione antibody as it reacts with the purified GAPDH control. This may also account for some pull-down of GAPDH in neurons not treated with NO• in the immunoprecipitation experiments (Figure 7B). Careful use of concentrations, as well as different doses, was chosen to ensure detection of increased glutathionylation.
Figure 8. GSSG-dependent glutathionylation of GAPDH.

(A) Assay conditions: 10 μg of purified rabbit muscle GAPDH was incubated with increasing concentrations of GSSG in GAPDH assay buffer. Western blot analysis of glutathionylated proteins using an anti-glutathione antibody (upper panel) and semi-quantiative analysis (densitometry) (lower panel) (n = 3). (B) Inhibition of GAPDH by GSSG: 10 μg of purified rabbit muscle GAPDH was incubated with various amounts of GSSG in GAPDH assay buffer for 10 min before activity measurements were made. GAPDH activity was measured in the presence of 250 μM NAD+ and 100 μg/ml glyceraldehyde 3-phosphate. Activity was calculated as described in the Materials and methods section (n = 3; expressed as percentage inhibition compared with GAPDH activity without treatment).
DISCUSSION
Neurodegenerative diseases such as Alzheimer’s disease are characterized by the selective loss of neurons with nitrosative stress being an inherent contributor to the degenerative progression of the disease. This, together with increasing amounts of evidence supporting the central role of neuroinflammation and redox modulation of protein function in neurodegeneration, underscores the relevance of the present study to neurodegenerative diseases [1,2]. The experimental approach of the present study entailed the use of a long-acting NO• donor to mimic the exogenous generation of the steady flux of NO• encountered by neurons under neuroinflammatory conditions (in lieu of generating NO• through activation of immune cells, such as astrocytes and microglia). This approach helped us to focus specifically on the role of NO•-mediated changes responsible for cellular injury. The concentrations of NO• used in the present study are relatively high, but still reasonable when considering (i) that NOS activity in the brain is higher than in other tissues [1], (ii) that levels of 1–4 μM NO• were detected in vivo during ischaemia/reperfusion [32], and (iii) that a localized and concerted increase in NOS expression in glial cells (iNOS) and neurons (nNOS) has been observed [14,33].
One impediment to fully understand NO•-mediated redox changes is the inability to directly detect and quantify the amount of intracellular GSNO formed during NO• exposure together with changes in GSH and GSSG levels. Usually, the formation of intracellular GSNO is inferred through the formation of S-nitrosylated proteins or Hg+-mediated cleavage of low-molecular-mass thiols followed by NO• release. Although these approaches have yielded useful information, they cannot quantify the redox changes inherent in NO• exposure. The highly sensitive HPLC approach to detect intracellular formation of GSNO and GSSG enabled a quantitative evaluation of the NO•-mediated redox changes and an estimation of the cellular redox status (expressed in mV). The results demonstrate that excessive exposure to exogenous NO• can lead to perturbations of the cellular redox status of neurons as well as astrocytes through the increased formation of GSNO and GSSG and loss of GSH. Neurons were highly sensitive to NO•-mediated oxidation of the intracellular redox environment compared with astrocytes. As changes in the total intracellular GSH pool were measured 1 h after the addition of NO• and viability was measured 6 h after the addition of NO•, it may be surmised that NO•-mediated oxidation of the GSH redox status is a contributing factor to NO•-mediated neurotoxicity. This is evident by the fact that astrocytes, which have a more robust GSH redox-buffering capacity, were less susceptible to NO•-induced cell death. It has been estimated that oxidation of the redox state (Ehc,) by 30 mV leads to a 10-fold change in ratio of a protein dithiol/disulfide motif [34]; this is in agreement with results of the present study, where a 41 mV change in the Nernst potential in neurons translated to increased S-glutathionylation of proteins, whereas in astrocytes, a 23 mV change led to a smaller extent of glutathionylated proteins.
The loss of GSH observed in neurons could be due to decreased GSH synthesis and/or increased consumption of GSH through the formation of GSNO and GSSG. Although the former is likely to occur under chronic NO• exposure [15], it is unlikely under these experimental conditions, entailing brief incubation times. Instead, the loss of GSH is likely to be due to increased efflux of GSSG out of the cell, formation of GSNO and conjugation to proteins through the formation of glutathione–protein adducts, i.e. S-glutathionylation. Failure to maintain GSH levels and the higher sensitivity of neurons to NO• exposure may be partly due to the significantly lower activities of GSNO reductase and GSSG reductase; consequently, neurons have a reduced capacity to buffer NO•-induced redox changes and increased susceptibility to NO• toxicity. Accordingly, knockdown of GSNO reductase in cerebellar cells resulted in increase susceptibility to NO•-mediated neurotoxicity [27].
In this experimental model, exposure of astrocytes and neurons to a flow of NO• is expected to result in inhibition of cytochrome oxidase in both cell types [35,36]; higher levels of NO• also inhibit electron transfer at the bc1 segment of the respiratory chain in an ‘antimycin-like effect’ that results in O2•− production [37]. The diffusion-controlled reaction of O2•− with NO• to yield ONOO− [38] has been well documented to occur in mitochondria [39]. The reaction of ONOO− with GSH [40] to yield GSNO and NO• [41] may suggest that ONOO− is an important mediator in GSNO formation. However, the different susceptibility of neurons and astrocytes to either NO• or ONOO− is a function of their capacity to buffer these redox changes with their inherent GSH levels [42].
The formation of S-glutathionylated proteins in neurons and astrocytes correlated with increasing neuronal cell death. Although thiol–disulfide exchange (eqn 3) is a straightforward mechanism for S-glutathionylated protein formation, it is thermodynamically unfavourable and requires substantial decreases in the GSH/GSSG ratio [3]; during nitrosative stress, S-glutathionylation is most likely to occur through the intermediate formation of nitrosylated proteins. This is based largely on the concept that formation of ‘activated’ forms of cysteine thiols that could ‘prime’ mixed disulfide formation. The results of the present study suggest that S-glutathionylation of proteins upon NO• exposure follows S-nitrosylation (eqn 2); supporting this notion is the discrete set of proteins that are found S-glutathionylated upon exposure to NO•; this strengthens the requirements for S-nitrosylation [5] and is at variance with diamide treatment that resulted in a laddering pattern. However, it is clear that both modifications are possible if GSNO is produced in the cell, then the relative selectivity for either modification will depend largely on the affinity (determined by pKa) for and stability of the modification. It appears that this is true within the context of the cell as NO•-induced nitrosylation of proteins leads to an observable laddering pattern in the brain [4], whereas NO• under the conditions of the present study led to the glutathionylation of a small subset of proteins. It may also be reasonable to surmise that NO•-induced glutathionylation of proteins might have cellular consequences different from nitrosylation. This is exemplified by ryanodine receptor channels (RYR1) where each modification led to specific functional consequences [43]. It is therefore also important to understand the role of NO•-mediated redox signalling through S-glutathionylation of proteins [3].
The preferential glutathionylation of a small subset of proteins in neurons and astrocytes suggests a highly specific process rather than an indiscriminate consequence of redox changes during nitrosative stress. GAPDH, a glycolytic and pro-apoptotic protein [44] was found to be glutathionylated in neurons during nitrosative stress, probably as a consequence of increased GSNO and GSSG formation; glutathionylation of GAPDH led to a significant decrease in its activity, which may represent a mechanism that promotes energy failure by limiting energy substrates to mitochondria. The loss of mitochondrial energy substrates affects neurons especially, owing to their high-energy requirements for cellular function. Glutathionylation of GAPDH by GSSG followed a sigmoid curve, showing linearity within a fairly narrow range of GSSG concentrations and a maximal inhibition of 60 %, suggesting that there exists a threshold level with respect to inhibition of GAPDH by glutathionylation during oxidative or nitrosative stress. Incubation of GAPDH with its substrate, glyceraldehyde 3-phosphate, did not protect against glutathionylation-induced inhibition (results not shown); this suggests that glutathionylation by GSSG did not necessarily occur on the active-site Cys149, but at other cysteine residues, thereby facilitating an allosteric effect that mediates the decrease in affinity for the physiological substrates or promotes its apoptotic function [44]. Limitation of energy substrates to mitochondria (upon S-glutathionylation and inhibition of GAPDH) is compounded by the sensitivity of complex I proteins to S-glutathionylation and S-nitrosylation [45,46].
S-glutathionylation of proteins may also serve as a means of storing GSH and a protective mechanism for redox-sensitive cysteine residues against irreversible damage. In the case of neurons, where the activities of GSNO and GSSG reductases are marginal, preservation of the GSH moiety through S-glutathionylation might be an economical means; deglutathionylation of proteins by either direct thiol–disulfide exchange or deglutathionylation by thiol–disulfide oxidoreductases, mainly glutaredoxin, may help restore the GSH levels [3,7]. This view is reasonable considering that under nitrosative stress, neurons, with a less robust redox-buffering capacity, exhibited a greater extent of S-glutathionylation of proteins compared with astrocytes.
Acknowledgments
FUNDING
This work was supported by the National Institutes of Health [grant number 2RO1 AG016718].
Abbreviations used
- BCNU
1,3-bis(2-chloroethyl)-1-nitrosourea
- DETA
diethylenetriamine
- DTT
dithiothreitol
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GSNO
S-nitrosoglutathione
- iNOS
inducible nitric oxide synthase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide
- NBM
neurobasal medium
- NEM
N-ethylmalemide
- nNOS
neuronal nitric oxide synthase
- NP-40
Nonidet P40
Footnotes
AUTHOR CONTRIBUTION
All authors were involved in performing the experiments. The study was conceived and the paper was written by Li-Peng Yap and Enrique Cadenas.
References
- 1.Duncan AJ, Heales SJ. Nitric oxide and neurological disorders. Mol Aspects Med. 2005;26:67–96. doi: 10.1016/j.mam.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Calabrese V, Mancuso C, Calvani M, Frizzarelli E, Butterfield DA, Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007;8:766–775. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- 3.Martinez-Ruiz A, Lamas S. Signalling by NO-induced protein S-nitrosylation and S-glutathionylation: convergences and divergences. Cardiovasc Res. 2007;75:220–228. doi: 10.1016/j.cardiores.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 4.Jaffrey F, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 5.Stamler JS, Lamas S, Fang FC. Nitrosylation: the prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 6.Hogg N. The biochemistry and physiology of S-nitrosothiols. Annu Rev Pharmacol Toxicol. 2002;42:585–600. doi: 10.1146/annurev.pharmtox.42.092501.104328. [DOI] [PubMed] [Google Scholar]
- 7.Dalle-Donne I, Milzani A, Gagliano N, Colombo R, Giustarini D, Rossi R. Molecular mechanisms and potential clinical significance of S-glutathionylation. Antioxid Redox Signaling. 2008;10:445–473. doi: 10.1089/ars.2007.1716. [DOI] [PubMed] [Google Scholar]
- 8.Zeng H, Spencer NY, Hogg N. Metabolism of S-nitrosoglutathione by endothelial cells. Am J Physiol Heart Circ Physiol. 2001;281:H432–H439. doi: 10.1152/ajpheart.2001.281.1.H432. [DOI] [PubMed] [Google Scholar]
- 9.Mieyal JJ, Gallogly MM, Qanungo S, Sabens EA, Shelton MD. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid Redox Signaling. 2008;10:1941–1988. doi: 10.1089/ars.2008.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mohr S, Hallak H, de Boitte A, Lapetina EG, Brüne B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem. 1999;274:9427–9430. doi: 10.1074/jbc.274.14.9427. [DOI] [PubMed] [Google Scholar]
- 11.West MB, Hill BG, Xuan YT, Bhatnagar A. Protein glutathiolation by nitric oxide: an intracellular mechanism regulating redox protein modification. FASEB J. 2006;20:1715–1717. doi: 10.1096/fj.06-5843fje. [DOI] [PubMed] [Google Scholar]
- 12.Han D, Canali R, Garcia J, Aguilera R, Gallaher TK, Cadenas E. Sites and mechanisms of aconitase inactivation by peroxynitrite: modulation by citrate and glutathione. Biochemistry. 2005;44:11986–11996. doi: 10.1021/bi0509393. [DOI] [PubMed] [Google Scholar]
- 13.Padgett CM, Whorton AR. Cellular responses to nitric oxide: role of protein S-thiolation/dethiolation. Arch Biochem Biophys. 1998;358:232–242. doi: 10.1006/abbi.1998.0859. [DOI] [PubMed] [Google Scholar]
- 14.Bolaños JP, Almeida A. Modulation of astroglial energy metabolism by nitric oxide. Antioxid Redox Signaling. 2006;8:955–965. doi: 10.1089/ars.2006.8.955. [DOI] [PubMed] [Google Scholar]
- 15.Gegg ME, Beltran B, Salas-Pino S, Bolaños JP, Clark JB, Moncada S, Heales SJ. Differential effect of nitric oxide on glutathione metabolism and mitochondrial function in astrocytes and neurones: implications for neuroprotection/neurodegeneration? J Neurochem. 2003;86:228–237. doi: 10.1046/j.1471-4159.2003.01821.x. [DOI] [PubMed] [Google Scholar]
- 16.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radical Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 17.Zhou Q, Lam PY, Han D, Cadenas E. c-Jun N-terminal kinase regulates mitochondrial bioenergetics by modulating pyruvate dehydrogenase activity in primary cortical neurons. J Neurochem. 2008;104:325–335. doi: 10.1111/j.1471-4159.2007.04957.x. [DOI] [PubMed] [Google Scholar]
- 18.Xie Z, Wei M, Morgan TE, Fabrizio P, Han D, Finch CE, Longo VD. Peroxynitrite mediates neurotoxicity of amyloid β-peptide1-42- and lipopolysaccharide-activated microglia. J Neurosci. 2002;22:3484–3492. doi: 10.1523/JNEUROSCI.22-09-03484.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borutaite V, Brown GC. Nitric oxide induces apoptosis via hydrogen peroxide, but necrosis via energy and thiol depletion. Free Radical Biol Med. 2003;35:1457–1468. doi: 10.1016/j.freeradbiomed.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 20.Han D, Hanawa N, Saberi B, Kaplowitz N. Hydrogen peroxide and redox modulation sensitize primary mouse hepatocytes to TNF-induced apoptosis. Free Radical Biol Med. 2006;41:627–639. doi: 10.1016/j.freeradbiomed.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 21.Jones DP. Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol. 2002;348:93–112. doi: 10.1016/s0076-6879(02)48630-2. [DOI] [PubMed] [Google Scholar]
- 22.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410:490–404. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 23.Ghezzi P, Romines B, Fratelli M, Eberini I, Gianazza E, Casagrande S, Laragione T, Mengozzi M, Herzenberg LA, Herzenberg LA. Protein glutathionylation: coupling and uncoupling of glutathione to protein thiol groups in lymphocytes under oxidative stress and HIV infection. Mol Immunol. 2002;38:773–780. doi: 10.1016/s0161-5890(01)00114-6. [DOI] [PubMed] [Google Scholar]
- 24.Raps SP, Lai JC, Hertz L, Cooper AJ. Glutathione is present in high concentrations in cultured astrocytes but not in cultured neurons. Brain Res. 1989;493:398–401. doi: 10.1016/0006-8993(89)91178-5. [DOI] [PubMed] [Google Scholar]
- 25.Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- 26.Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, Stamler JS. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–628. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- 27.He J, Wang T, Wang P, Han P, Yin Q, Chen C. A novel mechanism underlying the susceptibility of neuronal cells to nitric oxide: the occurrence and regulation of protein S-nitrosylation is the checkpoint. J Neurochem. 2007;102:1863–1874. doi: 10.1111/j.1471-4159.2007.04651.x. [DOI] [PubMed] [Google Scholar]
- 28.Antunes F, Cadenas E. Cellular titration of apoptosis with steady state concentrations of H2O2: submicromolar levels of H2O2 induce apoptosis through Fenton chemistry independent of the cellular thiol state. Free Radical Biol Med. 2001;30:1008–1018. doi: 10.1016/s0891-5849(01)00493-2. [DOI] [PubMed] [Google Scholar]
- 29.Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem. 2003;384:505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- 30.Mohr S, Stamler JS, Brüne B. Posttranslational modification of glyceraldehyde-3-phosphate dehydrogenase by S-nitrosylation and subsequent NADH attachment. J Biol Chem. 1996;271:4209–4214. doi: 10.1074/jbc.271.8.4209. [DOI] [PubMed] [Google Scholar]
- 31.Klatt P, Pineda Molina E, Perez-Sala D, Lamas S. Novel application of S-nitrosoglutathione–Sepharose to identify proteins that are potential targets for S-nitrosoglutathione-induced mixed-disulphide formation. Biochem J. 2000;349:567–578. doi: 10.1042/0264-6021:3490567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malinski T, Bailey F, Zhang ZG, Chopp M. Nitric oxide measured by a porphyrinic microsensor in rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1993;13:355–358. doi: 10.1038/jcbfm.1993.48. [DOI] [PubMed] [Google Scholar]
- 33.Lam PY, Yin F, Hamilton RT, Boveris A, Cadenas E. Elevated neuronal nitric oxide synthase expression during ageing and mitochondrial energy production. Free Radical Res. 2009;43:431–439. doi: 10.1080/10715760902849813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones DP. Redefining oxidative stress. Antioxid Redox Signaling. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 35.Bolaños JP, Peuchen S, Heales SJR, Land JM, Clark JB. Nitric oxide-mediated inhibition of the mitochondrial respiratory chain in cultured astrocytes. J Neurochem. 1994;63:910–916. doi: 10.1046/j.1471-4159.1994.63030910.x. [DOI] [PubMed] [Google Scholar]
- 36.Bolaños JP, Heales SJR, Peuchen S, Barker JE, Land JM, Clark JB. Nitric oxide-mediated mitochondrial damage: a potential neuroprotective role for glutathione. Free Radical Biol Med. 1996;21:995–1001. doi: 10.1016/s0891-5849(96)00240-7. [DOI] [PubMed] [Google Scholar]
- 37.Poderoso JJ, Carreras MC, Lisdero C, Riobo N, Schopfer F, Boveris A. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys. 1996;328:85–92. doi: 10.1006/abbi.1996.0146. [DOI] [PubMed] [Google Scholar]
- 38.Kissner R, Nauser T, Bugnon P, Lye PG, Koppenol WH. Formation and properties of peroxynitrite as studied by laser flash photolysis, high-pressure stopped-flow technique, and pulse radiolysis. Chem Res Toxicol. 1997;10:1285–1292. doi: 10.1021/tx970160x. [DOI] [PubMed] [Google Scholar]
- 39.Riobó NA, Clementi E, Melani M, Boveris A, Cadenas E, Moncada S, Poderoso JJ. Nitric oxide inhibits mitochondrial NADH–ubiquinone reductase activity through the formation of peroxynitrite. Biochem J. 2001;359:139–145. doi: 10.1042/0264-6021:3590139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls: the cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 41.Moro MA, Darley-Usmar VM, Goodwin DA, Read NG, Zamora-Pino R, Feelisch M, Radomski MW, Moncada S. Paradoxical fate and biological action of peroxynitrite on human platelets. Proc Natl Acad Sci USA. 1994;91:6702–6706. doi: 10.1073/pnas.91.14.6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolaños JP, Heales SJR, Land JM, Clark JB. Effect of peroxynitrite on the mitochondrial respiratory chain: differential susceptibility of neurones and astrocytes in primary culture. J Neurochem. 1995;64:1965–1972. doi: 10.1046/j.1471-4159.1995.64051965.x. [DOI] [PubMed] [Google Scholar]
- 43.Aracena P, Sánchez G, Donoso P, Hamilton SL, Hidalgo C. S-glutathionylation decreases Mg2+ inhibition and S-nitrosylation enhances Ca2+ activation of RyR1 channels. J Biol Chem. 2003;278:42927–42935. doi: 10.1074/jbc.M306969200. [DOI] [PubMed] [Google Scholar]
- 44.Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 45.Clementi E, Brown GC, Feelish M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci USA. 1998;95:7631–7636. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taylor ER, Hurrell F, Shannon RJ, Lin TK, Hirst J, Murphy MP. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J Biol Chem. 2003;278:19603–19610. doi: 10.1074/jbc.M209359200. [DOI] [PubMed] [Google Scholar]