

Abstract
Enterotoxigenic Escherichia coli (ETEC) is a common cause of acute diarrhea in resource-poor settings. We report that some ETEC strains elicit a reduction in trans-epithelial electrical resistance (TER) in polarized T84 epithelial cell monolayers. The effect was irreversible up to 48 hours after a three-hour infection and was observed with heat-labile enterotoxin (LT)–producing strains, but not with heat-stable enterotoxin (ST)–producing strains. Using purified LT, a mutant with reduced ADP-ribosylating activity, and the LT-B subunit alone, we demonstrate that TER reduction requires a functional enterotoxin. Treatment of monolayers with LT or LT-producing strains of ETEC increases paracellular permeability to fluorescein isothiocyanate–dextran. Our data suggest that LT-producing ETEC strains may induce intestinal barrier dysfunction.
Introduction
Enterotoxigenic E. coli (ETEC) is among the most common causes of diarrhea among infants and children worldwide.1–3 The current paradigm of ETEC pathogenesis suggests that the organism colonizes the small intestine by virtue of proteinaceous colonization factors, previously called colonization factor antigens, followed by the elaboration of heat-stable (ST) enterotoxin and/or heat-labile enterotoxin (LT).4 Among populations of infants and children in resource-poor areas, infections with ST-producing strains are more strongly associated with illness.3 Strains producing only LT are often not associated with diarrhea in such settings,5,6 although first encounters with the pathogen likely result in diarrhea.3 Importantly, however, colonization with LT-ETEC in the absence of symptoms commonly occurs among those previously exposed to LT, and possibly among some with no immunologic experience.6 The large number of asymptomatic colonization events makes it important to understand the potential ill effects of ETEC exposure, other than fluid and ion secretion.
LT is an A1:B5 ADP-ribosylating exotoxin closely related to cholera toxin, and the two toxins share a similar mechanism of action.4 The B subunits of LT bind with high affinity to cellular GM1 ganglioside and possibly other membrane receptors. Upon binding, the B pentamer directs endocytosis of the catalytic A-subunit, which enters the cytoplasm by retrograde transport via the endoplasmic reticulum complex. Once in the cytoplasm, the A subunit mediates ADP-ribosylation of the Gsα signaling protein, which leads to activation of adenylate cyclase, followed by accumulation of cAMP, and ultimately activation of the cystic fibrosis transmembrane conductance regulator chloride channel on the enterocyte surface. These events ultimately induce secretion of chloride and fluid.
LT may confer other pathogenic effects. The toxin has a long history as a potent immune adjuvant, yet a role for this effect in the setting of natural infection has not yet been demonstrated. More recently, LT has also been shown to be a potential colonization factor for ETEC srains,7 and it is possible that still more contributions may be uncovered.
Many enteric pathogens induce disruption of the intestinal barrier,8,9 an effect that may be detrimental to the host via loss of absorptive gradients and/or leakage of fluid, electrolytes, and proteins. Previous studies have shown that infection of T84 cells with Vibrio cholerae induces barrier disruption,10 and application of LT to T84 monolayers induces electrophysiologic abnormalities,11 which could include enhanced permeability. To study the effects of ETEC on the epithelial barrier, we used the polarized T84 adenocarcinoma cell line, which forms functional tight junctions and has been used extensively to study the effects of bacterial infection on transepithelial electrical resistance (TER).12 We show that human ETEC strains induce intestinal barrier dysfunction, and implicate the LT toxin in this effect; the effect is dependent on LT ADP-ribosylating activity. Additionally, we show that LT induces morphologic changes in cell shape, which may be related to the barrier effects. These observations may have implications for patients colonized with ETEC strains.
Materials and Methods
Bacterial strains and toxins.
Bacterial stocks were stored at –80°C in L-broth containing 30% glycerol. To prepare bacteria for infection, a colony from a freshly streaked plate was grown overnight in L-broth at 37°C under static conditions. The next day, bacteria were subcultured into Dulbecco's minimal essential medium (DMEM)–high glucose (Invitrogen, Carlsbad, CA), grown statically for 3-4 hours, and normalized to a mean ± SD optical density at 600 nm of 0.30 ± 0.20. Two hundred microliters were added to each well of the culture dish at time of infection.
Purified LT, LT mutant R129G, and the purified LT B subunit were prepared as described.13 Heat inactivation of the toxin was accomplished by heating at 65°C for 45 minutes.
Cell culture studies.
Human colonic T84 intestinal epithelial cells (ATCC CCL-248) were routinely maintained in DMEM/F-12 medium (Invitrogen) containing 10% fetal bovine serum (Sigma, St. Louis, MO), 50 U/mL of penicillin, and 50 mg/mL of streptomycin and were polarized as described.12 To generate polarized monolayers, T84 cells between passages 8 and 20 were seeded at a density of 3 × 105 cells/mL on collagen-coated 12 mm polycarbonate Transwell® permeable supports with 0.4-μm pores (Costar; Corning, Ithaca, NY) and grown for 10–14 days, during which time cells were fed daily. Monolayers were used for experiments when TER reached levels > 1,000 ohms/cm2. Infections were carried out as described.14 Briefly, T84 cells were washed with Hanks' balanced salt solution (Invitrogen) three times and equilibrated in DMEM/F-12 medium containing 1% methyl-α-D-mannopyranoside for 30 minutes, after which 200 μL of bacterial inoculum or toxin preparation was added to the apical compartment of the Transwell inserts. After three hours, the apical compartments of the wells were washed three times with Hanks' balanced salt solution, and medium was replaced with DMEM/F-12 containing 100 μg/mL of gentamicin. Gentamicin was also added to the basolateral compartment and TER was measured by using EVOM with STX2 chopstick electrodes (World Precision Instruments, Sarasota, FL).
Paracellular permeability assay.
Infections were performed as described above and followed for a period of 18 hours post-infection, at which time the basolateral medium was exchanged for either P buffer (10 mM HEPES, pH 7.4, 1 mM sodium pyruvate, 10 mM glucose, 3 mM CaCl2, 145 mM NaCl) or P/EGTA buffer as a positive control (10 mM HEPES, 1 mM sodium pyruvate, 10 mM glucose, 145 mM NaCl, 2 mM EGTA). P-buffer or P/EGTA buffer containing dextran conjugated to fluorescein isothiocyanate (FITC-dextran; Sigma) or BSA conjugated to FITC (FITC-BSA; Sigma) at 10 mg/mL was added to the apical side. After six hours of incubation with the FITC conjugates, the basolateral medium was removed and the intensity of light emission at 518 nm in the basolateral media was measured in triplicate on a Cary Eclipse Fluorometer (Varian Inc. Scientific Instruments, Lexington, MA).
Immunofluorescent assay.
At 24 hours post-infection, T84 cell monolayers on Transwell membranes were rinsed three times with phosphate-buffered saline (PBS) and fixed in ice-cold ethanol at –20°C for 20 minutes. After one rinse with PBS, cells were permeabilized in 1% Triton X-100/PBS for 10 minutes at room temperature and blocked for 30 minutes in 5% normal goat serum (NGS) for 30 minutes. The membranes were then incubated for one hour at room temperature with mouse anti-zonula occludens-1 (ZO-1) antibodies (Invitrogen) diluted in 1% NGS/PBS. After three washes in PBS, phalloidin-568 and goat anti-mouse 488 (Invitrogen) diluted in 1% NGS/PBS were added to the cells and incubated for one hour at room temperature in the dark. Antibodies were diluted in 1% NGS/PBS and added to the cells for one hour at room temperature. Cells were washed three times with PBS, once with distilled water, and mounted by using Vectashield® Hard set™ mounting medium (Vector Laboratories, Burlingame, CA). Images chosen were representative of the entire micrograph data set. Images were captured by using an LSM510 META laser scanning confocal microscope (Zeiss, Thornwood, NY) in the LSM510 software package (Zeiss). Figures were assembled in Adobe Photoshop 7.0 (Adobe, San Jose, CA).
Statistical analysis.
Experiments were performed three times with each sample in triplicate. Means were compared by the Mann-Whitney test with significance set at P < 0.05. Data are reported as mean ± SEM or 95% confidence intervals where noted.
Results
Effect of ETEC on TER of polarized T84 monolayers.
In separate experiments, we infected polarized T84 cell monolayers with four ETEC isolates, H10407 (LT/ST+), E2528C1 (LT+), 1AB7A (LT+), or A338C5 (ST+) for three hours, after which time the cells were washed free of bacteria and TER was monitored for an additional 21 hours. At 24 hours (21 hours post-infection) all LT-producing strains caused significant decreases in TER compared with uninfected cells or cells infected with A338C5 (H10407; P = 0.0079, E2528C1 and 1AB7A; P = 0.0357, by Mann-Whitney test) (Figure 1A and B). The deterioration of TER persisted for up to 48 hours after infection (Figure 1C). The 24-hour time point was used for further characterization of the effect.
Figure 1.
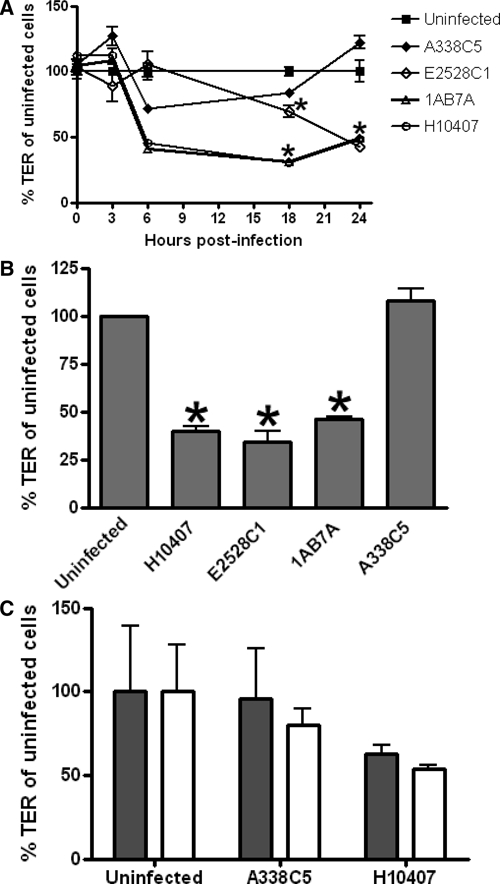
Reduction in trans-epithelial electrical resistance (TER) of polarized T84 monolayers by enterotoxigenic Escherichia coli. A, Fully polarized T84 monolayers mounted in Transwell cell culture inserts were infected with ETEC strains H10407, E2528C1, 1AB7A, or A338C5 for three hours. Monolayers were then washed, and TER was measured by using an Ohm meter at time points up to 21 hours post-infection. Graphs show mean ± SEM. Asterisk indicates significant decreases in TER at 18 hours and 21 hours post-infection compared with A338C5 and uninfected cells (by Mann-Whitney test). B, % TER of uninfected cells at 24 hours. Asterisk indicates significance by the Mann-Whitney test (P < 0.05). C, Persistence of effects of ETEC on TER for at least 48 hr after infection. Cells were infected for three hours. The TER was then measured at 21 hours and 48 hours post-infection. No significant difference in TER was observed between the 21-hour (gray bar) and 48-hour (white bar) time points (mean and 95% confidence intervals are shown).
Effect of purified LT on TER.
All ETEC strains we used produced the LT enterotoxin except strain A338C5, which produced only ST; A338C5 was the only strain that did not affect TER. To ascertain whether LT affected barrier resistance, we tested the ability of purified LT to induce reduction of TER in the polarized T84 model. The toxin was applied to the monolayer for three hours, after which time the apical chamber was replaced with toxin-free buffer. At a concentration of 10 μg/mL, LT induced a significant reduction in TER beginning as soon as 6 hours after intoxication (P = 0.0079 at 24 hr post-infection, by Mann-Whitney test) to a degree comparable to that observed with LT-producing strains of ETEC (Figure 2A). Heat-inactivated LT failed to cause a decrease in TER.
Figure 2.
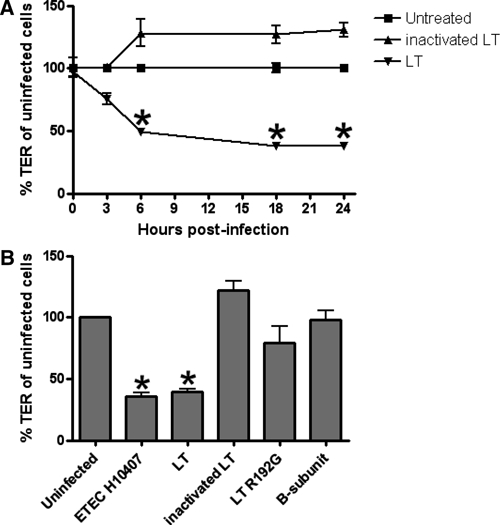
Reduction in trans-epithelial electrical resistance (TER) in T84 cells by purified catalytically active heat-labile enterotoxin (LT). A, Polarized T84 monolayers were treated with LT or with heat-inactivated LT for 3 hours, washed, then monitored for TER over the next 21 hours. Three independent experiments were performed and results were pooled for comparison; graphs show mean ± SEM. Asterisks indicate significance by the Mann-Whitney test (P < 0.05). B, Cells were treated with LT, heat-inactivated LT, the mutant R192G, or the B-subunit for three hours, washed to remove extracellular toxin, then monitored for TER.
We investigated whether the ADP-ribosylating activity of LT was required for the effect of the toxin on epithelial barrier permeability. To address this question, we tested the effects of the purified LT-B subunit, and an A subunit mutant (R192G) that induces 1,000-fold less cAMP in vitro than native LT.15 Only the A1:B5 holotoxin elicited a reduction in TER (P = 0.0079 at 24 hours, by Mann-Whitney test) (Figure 2B).
Effect of LT treatment of T84 cells on paracellular permeability.
Because monolayer electrophysiology obeys Ohm's law, it is possible that alterations in TER may be secondary to modulation of transepithelial current. To assess directly the permeability of the monolayer, we tested the ability of FITC-dextran (molecular mass = 4 kD) to traverse the paracellular pathway after application of ETEC or LT to polarized T84 monolayers. Infections were performed as above and followed for an additional 18 hours at which time the basolateral medium was replaced with P-buffer and the apical medium was replaced with P-buffer containing 10 mg/mL of FITC-dextran. Treatment with ETEC A338C5, inactivated LT, R192G, or the B-subunit alone did not result in significant penetration of FITC-dextran into the lower chamber (Figure 3A). In contrast, treatment with either purified LT (P = 0.0095) or the LT-producing strains E2528C1 (P = 0.0411) or H10407 (P = 0.0022) resulted in significant increases in the concentration of FITC-dextran in the basolateral compartment, consistent with penetration of the marker through the paracellular pathway. To ascertain the size limits of the permeability defect, we assessed monolayer permeability to FITC-BSA (67 kD); this larger marker was not found at levels higher than the negative control with any of the treatments (Figure 4B), suggesting that the size of the barrier defect was between 4 kD and 67 kD.
Figure 3.
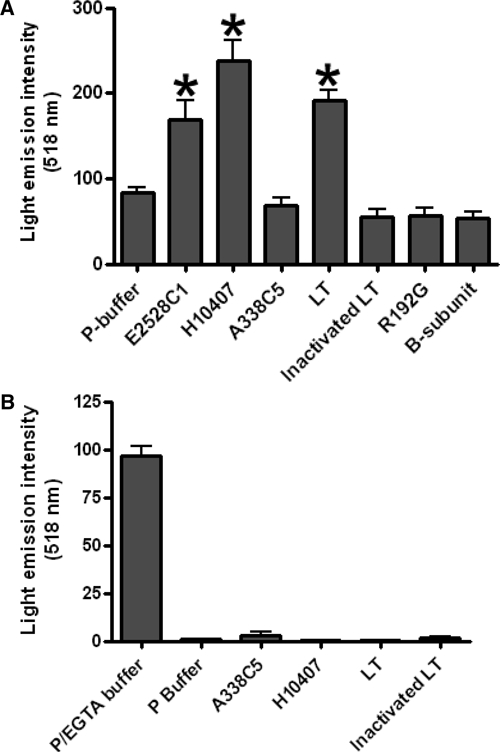
Increased paracellular permeability of T84 monolayers to fluorescein isothiocyanate (FITC)–labeled markers by heat-labile enterotoxin (LT)–producing enterotoxigenic Escherichia coli (ETEC) and purified catalytically active LT. A, Cells were treated for three hours, washed, and incubated. At 18-hours post-infection, FITC-dextran was added to the apical side of each well. Six hours later, fluorescence in the basolateral compartment was measured by using a fluorimeter. Values are the mean ± SEM. The Mann-Whitney test was used to determine significance. B, Penetration of FITC–bovine serum albumin through monolayers treated with ETEC strains or LT. Detection of the marker was not significantly above the negative control for any of the treatments (mean and 95% confidence intervals are shown).
Figure 4.
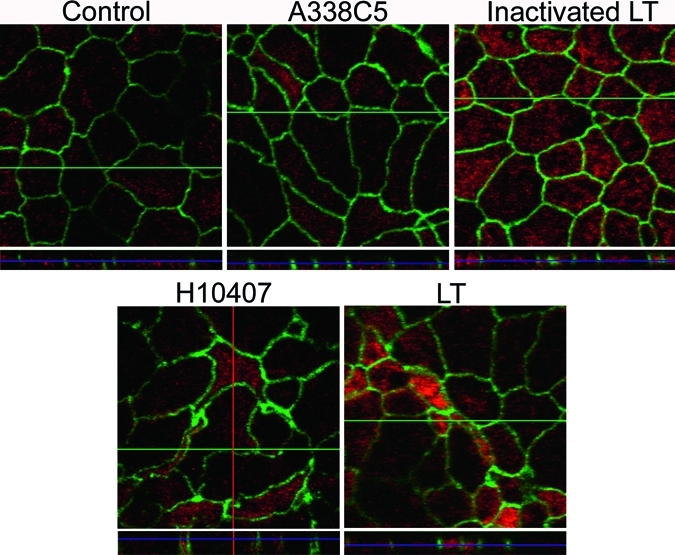
Morphologic changes in T84 monolayers by heat-labile enterotoxin (LT)–producing H10407 and purified LT. After a three-hour exposure to bacteria or the toxin, polarized T84 monolayers were washed and incubated for 21 hours. Cells were then stained for zonula occludens-1 (ZO-1) and phalloidin-568 as described in the Materials and Methods. Z-stacking was used to visualize the vertical axis of the corresponding images.
Effet of infection with ETEC H10407 or treatment with LT on morphology of polarized T84 cells.
We hypothesized that exposure of polarized T84 monolayers to LT-producing ETEC or purified functional LT would result in disruption of tight junctions, and that these alterations would be apparent by immunofluorescence microscopy of tight junction proteins. To address this hypothesis, we performed immunofluorescent assays on cells infected with A338C5, H10407, LT, or inactivated LT by using antibodies against tight junction protein zonula occludens-1 (ZO-1); filamentous actin was visualized simultaneously. We found that untreated monolayers, monolayers infected with A338C5, or those treated with inactivated LT were morphologically normal, exhibiting the expected chicken-wire pattern of apposing cell membranes (Figure 4). In contrast, infection with LT-producing H10407 or purified LT caused distortion of cell morphology, including elongation and irregular shape of some cells. Affected monolayers also exhibited many small cells, which stained normally for actin. In all cases ZO-1 remained localized to the tight junctions.
Discussion
Diarrheal diseases continue to exact a heavy toll of mortality in underdeveloped countries, particularly among children less than five years of age.16 There remains, however, uncertainty regarding the importance of more subtle physiologic effects of colonization by enteric pathogens.17 Adverse effects caused by asymptomatic carriage are suggested by diverse sources.1 Among farm animals, reduction of subclinical infections is one mechanism by which antibiotics are thought to improve growth of livestock.18 Given that most exposures to enteric pathogens among infants and children in developing countries are likely not to result in frank diarrheal illness,3 it is imperative to consider thoroughly the possible adverse consequences of frequent subclinical infection.
Intestinal barrier function is important to intestinal health. The epithelial fence, maintained by a firm tight junction complex between adjacent cells, serves as a barrier against invasion by pathogenic and non-pathogenic intestinal microbiota, limits exposure to non-self antigens, and secures the gradients that drive intestinal absorption and secretion. Perturbation of the barrier is associated with a myriad of diseases, both mucosal and systemic. Prominent among them for resource-poor settings is the incompletely understood entity called tropical or environmental enteropathy.19,20 This syndrome is characterized by loss of intestinal barrier function, bacterial overgrowth, malabsorption, and other abnormalities in gut function. Whereas the full pathophysiologic picture of environmental enteropathy is unclear, it virtually always occurs in populations frequently exposed to enteric pathogens that induce barrier dysfunction. Our data suggest for the first time that human ETEC could constitute one such deleterious exposure.
The mechanism of the effects we observe is not entirely apparent. Our data suggest that the effects require intact ADP-ribosylating activity of the A subunit. However, there is no clear connection between activation of the Gsα signaling complex and tight junction integrity. Lycke and others have reported enhance gut permeability in mice fed cholera toxin, an effect that also required intact ADP-ribosylating activity.21 Maintenance of barrier function is a dynamic, controlled process that involves several signal transduction pathways. One potential mechanism for the effects we observe could involve the prostaglandin signaling pathway, specifically the production of prostaglandin E2, which has been linked to the effects of cholera toxin and to barrier dysfunction.22–24 The interface between LT-dependent ADP ribosylation and tight junction signaling is the subject of ongoing work in our laboratory.
Our data suggest that the effects of LT on epithelial resistance are caused by enhanced permeability to low molecular mass molecules (< 67 kD). This observation suggests that the effect is unlikely to include enhanced permeability to invasion by enteric pathogens. However, loss of the absorptive gradient and leakage of interstitial fluid could accompany low molecular weight barrier dysfunction. The full implications of these observations await observations in animal models or in clinical studies.
ACKNOWLEDGMENT
We thank the Institute of Human Virology Center for Fluorescence Spectroscopy at the University of Maryland School of Medicine, a National Institutes of Health–supported facility, for use of the fluorescent plate reader.
Footnotes
Financial support: This study was supported by grant AI33096 from the National Institutes of Health to Joseph P. Nataro.
Authors' addresses: Roderick B. Kreisberg, Division of Gastroenterology and Hepatology, Department of Medicine, University of Maryland School of Medicine, Baltimore, MD, E-mail: rkrei001@yahoo.com. Jill Harper, Maura C. Strauman, and Mark Marohn, Department of Microbiology and Immunology, University of Maryland School of Medicine, Baltimore, MD, E-mails: jharper@medicine.mumaryland.edu, mstrauman@wistar.org, and mmaro001@umaryland.edu. John D. Clements, Department of Microbiology and Immunology, Tulane University Health Sciences Center, New Orleans, LA, E-mail: jclemen@tulane.edu. James P. Nataro, Department of Pediatrics, University of Virginia School of Medicine, Charlottesville, VA, E-mail: jpn2r@virginia.edu.
References
- 1.Gupta SK, Keck J, Ram PK, Crump JA, Miller MA, Mintz ED. Part III. Analysis of data gaps pertaining to enterotoxigenic Escherichia coli infections in low and medium human development index countries, 1984–2005. Epidemiol Infect. 2008;136:721–738. doi: 10.1017/S095026880700934X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Okoh AI, Osode AN. Enterotoxigenic Escherichia coli (ETEC): a recurring decimal in infants' and travelers' diarrhea. Rev Environ Health. 2008;23:135–148. doi: 10.1515/reveh.2008.23.2.135. [DOI] [PubMed] [Google Scholar]
- 3.Valentiner-Branth P, Steinsland H, Fischer TK, Perch M, Scheutz F, Dias F, Aaby P, Molbak K, Sommerfelt H. Cohort study of Guinean children: incidence, pathogenicity, conferred protection, and attributable risk for enteropathogens during the first 2 years of life. J Clin Microbiol. 2003;41:4238–4245. doi: 10.1128/JCM.41.9.4238-4245.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fleckenstein JM, Hardwidge PR, Munson GP, Rasko DA, Sommerfelt H, Steinsland H. Molecular mechanisms of enterotoxigenic Escherichia coli infection. Microbes Infect. 2010;12:89–98. doi: 10.1016/j.micinf.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nataro JP, Kaper JB. Diarrheagenic Escherichia coli. Clin Microbiol Rev. 1998;11:142–201. doi: 10.1128/cmr.11.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steinsland H, Valentiner-Branth P, Gjessing HK, Aaby P, Molbak K, Sommerfelt H. Protection from natural infections with enterotoxigenic Escherichia coli: longitudinal study. Lancet. 2003;362:286–291. doi: 10.1016/S0140-6736(03)13971-2. [DOI] [PubMed] [Google Scholar]
- 7.Johnson AM, Kaushik RS, Francis DH, Fleckenstein JM, Hardwidge PR. Heat-labile enterotoxin promotes Escherichia coli adherence to intestinal epithelial cells. J Bacteriol. 2009;191:178–186. doi: 10.1128/JB.00822-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berkes J, Viswanathan VK, Savkovic SD, Hecht G. Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation. Gut. 2003;52:439–451. doi: 10.1136/gut.52.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guttman JA, Finlay BB. Tight junctions as targets of infectious agents. Biochim Biophys Acta. 2009;1788:832–841. doi: 10.1016/j.bbamem.2008.10.028. [DOI] [PubMed] [Google Scholar]
- 10.Fullner KJ, Lencer WI, Mekalanos JJ. Vibrio cholerae-induced cellular responses of polarized T84 intestinal epithelial cells are dependent on production of cholera toxin and the RTX toxin. Infect Immun. 2001;69:6310–6317. doi: 10.1128/IAI.69.10.6310-6317.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lencer WI, Constable C, Moe S, Rufo PA, Wolf A, Jobling MG, Ruston SP, Madara JL, Holmes RK, Hirst TR. Proteolytic activation of cholera toxin and Escherichia coli labile toxin by entry into host epithelial cells. Signal transduction by a protease-resistant toxin variant. J Biol Chem. 1997;272:15562–15568. doi: 10.1074/jbc.272.24.15562. [DOI] [PubMed] [Google Scholar]
- 12.Nataro JP, Hicks S, Phillips AD, Vial PA, Sears CL. T84 cells in culture as a model for enteroaggregative Escherichia coli pathogenesis. Infect Immun. 1996;64:4761–4768. doi: 10.1128/iai.64.11.4761-4768.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chong C, Friberg M, Clements JD. LT(R192G), a non-toxic mutant of the heat-labile enterotoxin of Escherichia coli, elicits enhanced humoral and cellular immune responses associated with protection against lethal oral challenge with Salmonella spp. Vaccine. 1998;16:732–740. doi: 10.1016/s0264-410x(97)00255-7. [DOI] [PubMed] [Google Scholar]
- 14.Harrington SM, Strauman MC, Abe CM, Nataro JP. Aggregative adherence fimbriae contribute to the inflammatory response of epithelial cells infected with enteroaggregative Escherichia coli. Cell Microbiol. 2005;7:1565–1578. doi: 10.1111/j.1462-5822.2005.00588.x. [DOI] [PubMed] [Google Scholar]
- 15.Dickinson BL, Clements JD. Dissociation of Escherichia coli heat-labile enterotoxin adjuvanticity from ADP-ribosyltransferase activity. Infect Immun. 1995;63:1617–1623. doi: 10.1128/iai.63.5.1617-1623.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Black RE, Cousens S, Johnson HL, Lawn JE, Rudan I, Bassani DG, Jha P, Campbell H, Walker CF, Cibulskis R, Eisele T, Liu L, Mathers C. Global, regional, and national causes of child mortality in 2008: a systematic analysis. Lancet. 2010;375:1969–1987. doi: 10.1016/S0140-6736(10)60549-1. [DOI] [PubMed] [Google Scholar]
- 17.Prentice AM, Darboe MK. Growth and host-pathogen interactions. Nestle Nutr Workshop Ser Pediatr Program. 2008;61:197–210. doi: 10.1159/000113495. [DOI] [PubMed] [Google Scholar]
- 18.Cromwell GL. Why and how antibiotics are used in swine production. Anim Biotechnol. 2002;13:7–27. doi: 10.1081/ABIO-120005767. [DOI] [PubMed] [Google Scholar]
- 19.Humphrey JH. Child undernutrition, tropical enteropathy, toilets, and handwashing. Lancet. 2009;374:1032–1035. doi: 10.1016/S0140-6736(09)60950-8. [DOI] [PubMed] [Google Scholar]
- 20.Ramakrishna BS, Venkataraman S, Mukhopadhya A. Tropical malabsorption. Postgrad Med J. 2006;82:779–787. doi: 10.1136/pgmj.2006.048579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lycke N, Karlsson U, Sjolander A, Magnusson KE. The adjuvant action of cholera toxin is associated with an increased intestinal permeability for luminal antigens. Scand J Immunol. 1991;33:691–698. doi: 10.1111/j.1365-3083.1991.tb02542.x. [DOI] [PubMed] [Google Scholar]
- 22.Beltinger J, del Buono J, Skelly MM, Thornley J, Spiller RC, Stack WA, Hawkey CJ. Disruption of colonic barrier function and induction of mediator release by strains of Campylobacter jejuni that invade epithelial cells. World J Gastroenterol. 2008;14:7345–7352. doi: 10.3748/wjg.14.7345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bagley KC, Abdelwahab SF, Tuskan RG, Lewis GK. Cholera toxin indirectly activates human monocyte-derived dendritic cells in vitro through the production of soluble factors, including prostaglandin E(2) and nitric oxide. Clinical Vaccine Immunol. 2006;13:106–115. doi: 10.1128/CVI.13.1.106-115.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodriguez-Lagunas MJ, Martin-Venegas R, Moreno JJ, Ferrer R. PGE2 promotes Ca2+-mediated epithelial barrier disruption through EP1 and EP4 receptors in Caco-2 cell monlayers. Am J Physiol Cell Physiol. 2010;299:C324–C334. doi: 10.1152/ajpcell.00397.2009. [DOI] [PubMed] [Google Scholar]