

Abstract
Our previous studies have shown that the nonsteroidal anti-inflammatory drug indomethacin exhibits antileukemic activity in vitro and can inhibit the aldo-keto reductase AKR1C3, which we identified as a novel target in acute myeloid leukemia. However, the antileukemic actions of indomethacin are likely to be complex and extend beyond inhibition of either AKR1C3 or cycloxygenases. To further understand the antileukemic activity of indomethacin we have used untargeted nuclear magnetic resonance-based metabolic analysis to characterize the responses of KG1a and K562 cell lines in both normal culture conditions and in hypoxia, which better represents the tumor environment in vivo. Hypoxia induced dramatic metabolic changes in untreated KG1a and K562, including adaptation of both phospholipid and glycolytic metabolism. Despite these changes, both cell lines sustained relatively unaltered mitochondrial respiration. The administration of indomethacin induced similar metabolic responses regardless of the oxygen level in the environment. Notable exceptions included metabolites associated with de novo fatty acid synthesis and choline phospholipid metabolism. Collectively, these results suggest that leukemia cells have the inherent ability to tolerate changes in oxygen tension while maintaining an unaltered mitochondrial respiration. However, the administration of indomethacin significantly increased oxidative stress in both KG1a and K562, inducing mitochondrial dysfunction, regardless of the oxygenation conditions. These findings emphasize the particular pertinence of the tricarboxylic acid cycle to the survival of cancer cells and may explain why some antileukemic drugs have been discovered and developed successfully despite the use of culture conditions that do not reflect the hypoxic environment of cancer cells in vivo.
Several studies have highlighted potential roles for the aldo-keto reductase 1C3 (AKR1C3) in the etiology1−5 and pathophysiology of cancer,6,7 fostering interest in generating selective AKR1C3 inhibitors as potential chemopreventive and chemotherapeutic agents.8−11 Our interest in the role of AKR1C3 in acute myeloid leukemia (AML) arose from studies of the in vitro antileukemic activity of the nonsteroidal inflammatory drug (NSAID) indomethacin, which we discovered to include inhibition of AKR1C3.1,6,11 We later published crystal structures of indomethacin bound to AKR1C3, and consequently drug development studies have used indomethacin as a starting point for AKR1C3 inhibitor design.8−10 The antileukemic actions of indomethacin are likely to be complex and extend beyond inhibition of either AKR1C3 or cycloxygenases. Nuclear magnetic resonance (NMR) spectroscopy is a commonly used analytical method that provides unique insight into the composition of small metabolites, that is, the metabolome, of cellular model systems as well as human and animal biofluids and tissues.12−15 We have therefore used this approach here to investigate the metabolic effects of indomethacin treatment on two myeloid leukemia cell lines (K562 and KG1a).
Importantly, our previous studies of the actions of indomethacin against myeloid leukemia cells used conventional culture conditions that do not reflect the hypoxic conditions present in the malignant bone marrow. Evidence exists that reduced oxygen tension affects cellular metabolism and the microenvironment, including pH level.16−20 For example, glycolysis is enhanced in hypoxia via the increased activity of glucose transporters and glycolytic enzymes, ultimately inducing an increased lactate production and decreased pH levels.16,21 These and other modulations in metabolite concentrations induced by hypoxia can affect the response to pharmacological treatment.(17) Therefore here we utilize NMR-based metabolic profiling to study the impact of indomethacin treatment on the metabolism of K562 and KG1a myeloid leukemia cell lines with the aims of gaining further mechanistic insight into the antileukemic activities of this drug as well as the effects of the local environment on the outcome of treatment. To achieve the latter we investigated the effects of treatment both in standard culture conditions and in hypoxic conditions, which better mimics the tumor microenvironment in vivo. Furthermore, we utilized two cell lines that apoptose in response to the drug in order to examine the generality of any metabolic responses detected. Our findings demonstrate that despite dramatic changes in the metabolome of myeloid cells grown under conventional or hypoxic culture conditions the tricarboxylic acid (TCA) cycle is relatively preserved. However, in the presence of indomethacin, the TCA cycle becomes commonly deregulated independently of the prevailing oxygen tension.
Results and Discussion
Metabolic Changes Induced by Hypoxic Environment
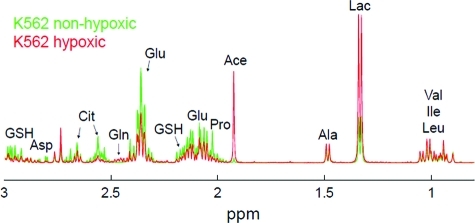
1H 2-dimensional J-resolved (J-RES) NMR spectra of extracts of KG1a and K562 AML cell lines grown under either hypoxic or normal culture conditions were recorded. We previously showed that the metabolic profiles of these two AML cell lines, when grown in normal culture conditions, differ considerably.(14) Here we demonstrate (as shown, for KG1a cells, in the NMR spectra in 1, panel a) that hypoxia induced marked changes in the metabolic profiles of K562 and KG1a cell lines. Moreover, regardless of the constitutive metabolic differences between these two cell lines, the hypoxia-induced changes in the metabolic profiles were strikingly similar between KG1a and K562. Principal component analysis (PCA) was performed on the projected J-RES NMR data sets of the two untreated cell lines grown in both hypoxic and normal conditions; the resulting PCA scores plot (Supplementary Figure 1) highlights remarkable differences between the metabolomes of K562 and KG1a cells (almost 70% of the variability is captured by the first principal component). The metabolic differences between the hypoxic and normal culture environments, accounted for on PC2, comprise about a third of the variation described along PC1. The metabolic variability within each of the 4 groups was minor relative to the changes induced by cell type and oxygen conditions in the culture.
Figure 1.

Metabolic differences induced by treatment with indomethacin in acute myeloid leukemia cell lines grown under nonhypoxic or hypoxic conditions. a) 1H projected J-resolved NMR spectra acquired on the polar fraction of KG1a cell extracts grown either in nonhypoxic (green lines, showing all 12 replicate spectra) or in hypoxic (∼1% oxygen; red line, showing all 6 replicate spectra) conditions. b) Loadings plot along PC2 obtained from the principal component analysis of the 1H NMR spectra acquired on the intracellular extracts of KG1a and K562 acute myeloid leukemia cell lines treated with indomethacin and grown in nonhypoxic or in hypoxic conditions. c) Scores plot obtained from the principal component analysis of the 1H NMR data sets of untreated (green symbols) and indomethacin-treated (red symbols) KG1a and K562 cells grown in hypoxic and nonhypoxic conditions. d) Average concentrations of selected TCA cycle intermediates and reduced glutathione in untreated or indomethacin-treated KG1a and K562 cells in nonhypoxic (average of 12 samples) or in hypoxic (average of 6 samples) conditions. e) Average concentrations of metabolites of the choline pathway in untreated or indomethacin-treated KG1a and K562 cells in nonhypoxic (average of 12 samples) or in hypoxic (average of 6 samples) conditions. f) Treatment with indomethacin induces the formation of reactive oxygen species in AML cells. Percent of ROS positive cells in untreated and indomethacin-treated KG1a and K562 AML cells grown and treated either in nonhypoxic or in hypoxic conditions, measured using flow cytometry. (Phe, phenylalanine; Tyr, tyrosine; His, histidine; Fum, fumarate; UDP, uridine diphosphate; Lac, lactate; Ala, alanine; Cre, creatine; PCre, phosphocreatine; Gly, glycine; Tau, taurine; m-Ino, myo-inositol; Cho, choline; PCho, phosphocholine; GPCho, glycerophosphocholine; Asp, aspartate; Asn, asparagine; Suc, succinate; Glu, glutamate; Gln, glutamine; GSH, glutathione; Cit, citrate; Pro, proline; Ace, acetate; Val, valine; Ile, isoleucine; Leu, leucine; *: p < 0.01; **: p < 0.0001).
Multiple univariate analyses (t tests) followed by a correction for multiple hypothesis testing (false discovery rate, FDR(22)) at the 1% level was performed on the J-RES NMR data sets after noise exclusion (resulting in about 18,000 data points per data set); specifically the hypoxic and normal culture condition data sets for each cell line were compared. This confirmed that the vast majority of the detected metabolite peaks differed significantly between the conditions of growth. The effect of the hypoxic environment on the metabolic profiles of K562 and KG1a AML cells causes a significant increase in the concentrations of branched amino acids (valine, leucine, and isoleucine), lactate, alanine, creatine, phosphocreatine, glycerol-3-phosphate, glycerophosphocholine, myo-inositol, uridine-5′-diphospho-N-acetyl-glucosamine (UDP-GlcNAc), uridine-5′-diphospho-N-acetyl-galactosamine (UDP-GalNAc), and phenylalanine. In contrast, proline, phosphocholine, citrate, glutamate, glutathione, and aspartate are significantly decreased in cells grown in hypoxic conditions (Supplementary Figure 2). Moreover, in both cell lines, fumarate, malate, and succinate are not significantly different between cells grown in hypoxic versus nonhypoxic conditions. Among all the detected metabolites common to both cell lines, only the concentrations of sarcosine and glycine change in opposite directions, i.e., in cells grown under hypoxic conditions. Both metabolites have increased concentrations in K562 cells and decreased levels in KG1a cells. Moreover, glutamine is present in detectable amounts only in K562 cells. To assess the extent of these changes in a more quantitative manner, 1H 1D NMR spectra were collected with a long repetition time to allow for full longitudinal relaxation of the protons. The concentrations of GSH and several other metabolites involved in or related to the TCA cycle and to choline metabolism for cells grown in either hypoxic or nonhypoxic conditions are reported in Supplementary Figure 3.
Regardless of the constitutive differences between the two AML cell lines, the metabolic alterations induced by the hypoxic environment are strikingly similar in KG1a and K562 cells. These results suggest that both the AML cell lines adopt a common adaptive mechanism when exposed to an environment with low oxygen tension. In agreement with previous studies performed on cell model systems, we observe an enhancement of the glycolytic activity (as confirmed by an intracellular accumulation of alanine and lactate) induced by the hypoxic environment.(18) This is likely the outcome of the overexpression of glucose transporters (particularly GLUT1 and GLUT3) and an increased activity of the glycolytic enzymes in hypoxia.23,24 The increased expression of hypoxia-inducible factor (HIF) in hypoxic tumors has been shown to trigger the expression of several glycolytic enzymes, favoring the accumulation of lactate and carbonic acids resulting in an increased acidity in the intracellular environment.17,19,25
Interestingly, although glycolysis appears to be enhanced in cells grown in hypoxia, the concentrations of metabolites within the TCA cycle are, except for citrate, not significantly altered. These observations indicate that adaptive mechanisms conserved between the cell types serve to protect mitochondrial respiration and thereby promote cell survival. The stability of the TCA cycle under reduced oxygen tension conditions is made yet more noteworthy by the observed hypoxia-induced changes in other pathways. Hypoxia altered several metabolites associated with the biosynthesis and the catabolism of phospholipids and other lipids (e.g., citrate as a precursor of de novo fatty acid synthesis) in both cell lines. The accumulation of phosphocreatine, a metabolite recognized to function as a phosphagen and hence energy reservoir for cells, is indicative of an adaptive response to an enhanced energy metabolism in hypoxic cells.(18) The hypoxia-induced decrease in phosphocholine and increase in glycerophosphocholine concentrations have been attributed to a reduction of phosphatidylcholine turnover,18,26 opposite to what has been reported for prostate cancer cell lines in hypoxia.(20) Similarly, the accumulation of myo-inositol in AML cells under hypoxic conditions might be related to the phosphatidylcholine turnover. In fact, altered requirements of myo-inositol, an isomer of glucose having several important functions in mammalian cells (e.g., as an osmolyte), have been associated with the modulation of phospholipid levels.27,28 Increased concentrations of metabolites of the hexosamine pathway can be attributed with the up-regulation of glucose metabolism affecting the synthesis of glycolytic intermediates.29−31 Metabolites in the hexosamine pathway, such as UDP-GlcNAc and UDP-GalNAc, are important precursors for the formation of N-glycans, and their elevation has been related to response of several forms of cellular stress including hypoxia.(31)
The relative stability of the TCA cycle between different states of oxygenation, in the context of other quite large changes in the metabolome, is interesting to consider since in other systems hypoxia tends to down-regulate the TCA cycle.(32) Hemopoietic stem cells (HSC) and progenitor cells are able to leave their hypoxic niches in the bone marrow, enter into the normoxic peripheral circulation, and rehome to the bone marrow. This happens at a very low frequency but is exploited in transplant medicine by the mobilization of high numbers of HSCs into the periphery of donors using cytokine stimuli. These mobilized stem cells then, when introduced to the recipient, again home from the normoxic periphery into hypoxic niches in the patients’ marrow. Thus the ability to tolerate exchanges of oxygen tensions is an inherent ability of HSCs and progenitor cells. This ability is shared by AML blasts, which again proliferate in the bone marrow but also accumulate in the periphery. Our data indicate that despite prolonged culture in artificial conditions, KG1a and K562 cells have retained this plasticity and provide novel insight into the molecular mechanisms that permit this behavior.
Metabolic Changes Induced by a 24-h Treatment with Indomethacin
Tissue culture studies using cell lines have been widely used to study potential cancer treatments in vitro. Despite numerous successes this approach is often criticized as being flawed because conventional culture conditions do not reflect the hypoxic environment of tumor cells. Here we addressed the metabolic consequences of indomethacin treatment on two AML cell lines under both normal and arguably more realistic hypoxic conditions. PCA was performed on the NMR data sets of the AML cell lines, with and without indomethacin treatment, grown under both hypoxic and nonhypoxic conditions (loadings and scores plots included in 1, panels b and c). The resulting PCA scores plot (PC1 vs PC2; 1, panel c) indicates that the separation between the indomethacin-treated and untreated samples (within each cell line and oxygen level) is much smaller than the separation caused by the genetic variability between cell types or by the oxygen tension. The extent of the differences induced by the drug treatment can be better highlighted by performing a PCA on each subset of NMR data limited to one cell line and one oxygenation condition. The four scores plots, comparing the control and indomethacin treatment data sets for KG1a and K562 cells grown under hypoxic and nonhypoxic conditions, are shown in Supplementary Figure 4. A clear separation is observed between the solvent control and indomethacin treated KG1a and K562 cells grown in both hypoxic and nonhypoxic conditions.
The analysis of the associated PCA loadings plots (data not shown explicitly) as well as the results of univariate analyses (t tests with FDR correction at 5% significance), coupled with a visual interpretation of the NMR spectra, are summarized in the biochemical pathways depicted in Supplementary Figure 5 (treated cells under nonhypoxic conditions) and 2 (treated cells under hypoxia). Both common and disparate treatment-induced changes between the two cell lines and the conditions of growth can be highlighted. For example, glutathione, creatine, and α-ketoglutarate are all significantly decreased after drug treatment in both cell lines and in both conditions of growth. Concurrently, succinate, fumarate, choline, glycerol-3-phosphate, alanine, UDP-GlcNAc, and UDP-GalNAc all significantly accumulate in both cell lines regardless of the oxygen level. However, changes in citrate concentration follow opposite trends for cells grown in hypoxic or in nonhypoxic conditions (in the hypoxic cells, citrate significantly accumulated in the drug treated vs untreated samples; in the nonhypoxic counterpart a significant depletion of citrate in the treated samples was observed). Similarly, the concentration changes of some metabolites involved in choline metabolism are common to both the cell lines but differ when hypoxia and nonhypoxia data sets are compared. In fact, cells treated with indomethacin in a nonhypoxic environment show an accumulation of glycerophosphocholine and a decrease of phosphocholine, whereas the administration of the drug under hypoxic conditions produces the opposite effect. Some of these observations, in particular for metabolites involved in the TCA cycle or choline metabolism, were quantified from the NMR spectra in 1, panels d and e.
Figure 2.

Metabolic differences induced by treatment with indomethacin in acute myeloid leukemia cell lines grown in hypoxic conditions. Schematic representation of the metabolic pathways showing the most relevant metabolic changes induced by treatment with indomethacin and common to both KG1a and K562 cell lines grown under hypoxic conditions. Metabolites in green/red have significantly increased/decreased concentrations in both KG1a and K562 cell lines treated with indomethacin and grown in a hypoxic environment; metabolites in black are detected/identified in the NMR spectra but change differently or non-significantly in the two cell lines; metabolites in gray were not detected in the NMR spectra.
The observed indomethacin-driven depletion of α-ketoglutarate and associated rise in succinate mirrored observations made in our previous study where we demonstrated that combining medroxyprogesterone acetate (MPA) with bezafibrate (Bez), in normal culture conditions, generated reactive oxygen species (ROS).(33) Following earlier observations showing that ROS directly converted α-ketoglutarate into succinate(34) and treatment of KG1a cells with H2O2 recapitulated α-ketoglutarate depletion and succinate accumulation, we concluded that the ROS generated by combined MPA and Bez is the mechanism that causes the derangement of the TCA cycle.(14) In the current study we therefore measured induction of ROS upon treatment with indomethacin (1, panel f). The drug treatment induced significant increases in ROS in both cell lines. Strikingly, despite the cells being able to protect the TCA cycle under conditions of low oxygen availability, indomethacin deregulated the balance of TCA metabolites in both cell lines and in both normal and hypoxic culture conditions. Moreover, all of the observed responses of TCA intermediates (with the exception of citrate) to indomethacin were consistent in both culture conditions. The significant depletion of α-ketoglutarate and the concurrent accumulation of succinate and fumarate suggest a partial truncation of the TCA cycle induced by the generation of ROS triggered by the indomethacin treatment. The indomethacin-induced generation of ROS also relates to the observation that reduced glutathione is depleted in the AML cells after treatment at different oxygen levels. Similar observations were previously reported by de Groot and colleagues.(35) The striking, oxygen-dependent behavior of citrate seems to indicate that the administration of indomethacin in combination with hypoxia counters the effect of hypoxia alone and points to an accumulation of citrate in the cytosol due to a hindered synthesis of de novo fatty acids, cholesterol, and isoprenoids through the conversion of citrate to acetyl-CoA.(36) The accumulation of a pool of citrate outside of the TCA cycle therefore colors the interpretation of the indomethacin-induced effects of this intermediate with the mitochondria.
Not all indomethacin-induced changes were as consistent as those observed in the TCA cycle. Treatment of cells in normal culture conditions resulted in alterations of choline phospholipid metabolism that are in general agreement with previous reports.37,38 However, the administration of indomethacin to cells under hypoxia induced disparate alterations to these metabolites. Similar to the changes in citrate concentration these opposite trends suggest that the anti-inflammatory effects of indomethacin on the inflammatory pathways depend on the oxygen level in the culture environment. In colorectal tumor cells and in prostate cancer cell lines, HIF-1 has been shown to up-regulate COX2.(39) Thus, it can be argued that in leukemia cell lines treatment with indomethacin interferes with the effect of hypoxia on the enzymes regulating the choline phospholipid metabolism.
Implications for Drug Development
From a therapeutic perspective the key observation made here is that the administration of indomethacin significantly increased oxidative stress via generation of ROS in both KG1a and K562 leukemia cells, inducing mitochondrial dysfunction regardless of the oxygenation conditions. These findings may emphasize the particular pertinence of the TCA cycle to the survival of cancer cells and may explain why some antileukemic drugs have been discovered and developed successfully despite the use of culture conditions that do not reflect the hypoxic environment of cancer cells in vivo. It is also noteworthy that indomethacin alone induced ROS, an observation not seen in our earlier study using the less AKR1C3-selective inhibitor MPA.(14) This finding lends support to the approach being used by others to develop novel AKR1C3 inhibitors based on the structure of indomethacin.8−10 Finally, since a variety of NSAIDs inhibit AKR1C3,11,40−43 future studies should address whether the chemopreventive actions of NSAIDs reported in a number of cancers44−47 include deregulation of the TCA cycle in premalignant cells.
Methods
Cell Culture Conditions
KG1a and K562 AML cell lines were maintained in exponential proliferation in RPMI 1640 medium (Gibco-Invitrogen) with 10% fetal bovine serum (FBS, Gibco-Invitrogen) and 100 units mL−1 of penicillin and 100 μg mL−1 of streptomycin (Gibco-Invitrogen). The cells were cultured in a humidified chamber at 37 °C and with 5% CO2 (nonhypoxic condition). For experiments performed in hypoxic conditions, the cells were moved to a hypoxic chamber (humidified at 37 °C, with 1% O2 and 5% CO2) and accustomed to this environment for 24 h prior to treatment.
Cell Treatments
Cells (5 × 107) were treated with either solvent control (ethanol) or 20 μM indomethacin. For each treatment, 12 replicate samples were prepared for the nonhypoxic growth condition and 6 replicate samples for the hypoxic one. After 24 h the cells were washed with PBS (Lonza Group Ltd.) and harvested by centrifugation. The cell pellets were immediately frozen in liquid nitrogen and stored at −80 °C.
NMR Sample Preparation
The extraction of polar intracellular metabolites from cell pellets was performed using a modified Bligh−Dyer procedure.(48) Samples were dried overnight in a centrifugal vacuum concentrator. The dried polar extracts were redissolved in 90% H2O/10% D2O (GOSS Scientific Instruments Ltd.) prepared as 100 mM phosphate buffer (pH 7.0), containing 0.5 mM sodium 3-(trimethylsilyl)propionate-2,2,3,3-d4 (TMSP, Cambridge Isotope Laboratories) as internal reference.
NMR Experiments and Data Processing
A 500 MHz Bruker spectrometer equipped with a cryogenically cooled probe was used for 2D 1H J-RES(49) and 1D 1H NMR data acquisition. In both cases the water resonance was suppressed using excitation sculpting.(50)
1D 1H NMR Data for Quantitative Determination of Metabolites
1D spectra were acquired with a long relaxation delay of 15 s and a 30° flip angle to guarantee near complete longitudinal relaxation. 1D spectra were acquired with a spectral width of 5 kHz and 256 transients.
2D 1H J-RES NMR
2D J-RES spectra were collected using a double spin echo sequence with 16 transients per increment and 32 increments. Strong coupling artifacts were suppressed by phase cycling.(51) Prior to Fourier transformation, 2D J-RES spectra were multiplied by a combined sine-bell/exponential window function in the direct dimension and by a sine bell function in the incremented dimension.(52) Skyline projections (of the nontilted and nonsymmetricized 2D spectra) were calculated and then aligned to TMSP at 0.0 ppm. Selected signals arising from residual solvents and TMSP were excluded. Spectra were normalized according to the probabilistic quotient method(53) and binned at 0.0015 ppm. A generalized-log transformation was applied prior to conducting the multivariate statistical analysis.(54) Multivariate statistical analysis (PCA) of the projected J-RES NMR data was carried out using PLS toolbox (Version 4.1; eigenvector Research).
Multiple univariate analyses (t tests) were performed on all the data points of 1H 1D NMR spectra after the exclusion of noise (3 times the noise level calculated according to ref (55)). The results were then false discovery rate (FDR) corrected to significance levels of 1% or 5%.
All the NMR data sets were processed using NMRLab(56) in the MATLAB programming environment (The MathWorks, Inc.). NMR resonances of metabolites were assigned and quantified using the Chenomx NMR Suite (version 5.0; Chenomx Inc.). The concentrations of metabolites are reported as mean values ± standard deviation. The reported statistical significance is based on a t test.
Assessment of ROS Formation
Carboxy-H2DCFDA (Molecular Probes, Invitrogen) was added to the cells during the last 30 min of the 24-h treatment period. Following incubation, cells were washed with PBS and analyzed by flow cytometry.
Acknowledgments
This work was supported by grants from Leukaemia Research (LRF, U.K.) and a Marie Curie Transfer of Knowledge (ToK) award (MOTET). We also thank the Wellcome Trust for supporting the HWB·NMR facility in Birmingham, and Chenomx Inc. for use of their NMR metabolomics software. We acknowledge HWB·NMR staff, in particular C. Ludwig, for assistance with NMR pulse sequences and data sets.
#. Author Contributions
#These authors contributed equally to this work.
¶. Author Contributions
¶These authors contributed equally as senior authors.
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Birtwistle J.; Hayden R. E.; Khanim F. L.; Green R. M.; Pearce C.; Davies N. J.; Wake N.; Schrewe H.; Ride J. P.; Chipman J. K.; Bunce C. M. (2009) The aldo-keto reductase AKR1C3 contributes to 7,12-dimethylbenz(a)anthracene-3,4-dihydrodiol mediated oxidative DNA damage in myeloid cells: Implications for leukemogenesis. Mutat. Res. 662, 67–74. [DOI] [PubMed] [Google Scholar]
- Courter L.; Pereira C.; Baird W. (2007) Diesel exhaust influences carcinogenic PAH-induced genotoxicity and gene expression in human breast epithelial cells in culture. Mutat. Res. 625, 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa J.; Malats N.; García-Closas M.; Real F.; Silverman D.; Kogevinas M.; Chanock S.; Welch R.; Dosemeci M.; Lan Q.; Tardón A.; Serra C.; Carrato A.; García-Closas R.; Castaño-Vinyals G.; Rothman N. (2008) Bladder cancer risk and genetic variation in AKR1C3 and other metabolizing genes. Carcinogenesis 29, 1955–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Q.; Mumford J.; Shen M.; Demarini D.; Bonner M.; He X.; Yeager M.; Welch R.; Chanock S.; Tian L.; Chapman R.; Zheng T.; Keohavong P.; Caporaso N.; Rothman N. (2004) Oxidative damage-related genes AKR1C3 and OGG1 modulate risks for lung cancer due to exposure to PAH-rich coal combustion emissions. Carcinogenesis 25, 2177–2181. [DOI] [PubMed] [Google Scholar]
- Palackal N.; Lee S.; Harvey R.; Blair I.; Penning T. (2002) Activation of polycyclic aromatic hydrocarbon trans-dihydrodiol proximate carcinogens by human aldo-keto reductase (AKR1C) enzymes and their functional overexpression in human lung carcinoma (A549) cells. J. Biol. Chem. 277, 24799–24808. [DOI] [PubMed] [Google Scholar]
- Desmond J. C.; Mountford J. C.; Drayson M. T.; Walker E. A.; Hewison M.; Ride J. P.; Luong Q. T.; Hayden R. E.; Vanin E. F.; Bunce C. M. (2003) The aldo-keto reductase AKR1C3 is a novel suppressor of cell differentiation that provides a plausible target for the non-cyclooxygenase-dependent antineoplastic actions of nonsteroidal anti-inflammatory drugs. Cancer Res. 63, 505–512. [PubMed] [Google Scholar]
- Penning T.; Byrns M. (2009) Steroid hormone transforming aldo-keto reductases and cancer. Ann. N.Y. Acad. Sci. 1155, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrns M.; Steckelbroeck S.; Penning T. (2008) An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3α-HSD, type 5 17β-HSD, and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochem. Pharmacol. 75, 484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobec S.; Brozic P.; Rizner T. (2005) Nonsteroidal anti-inflammatory drugs and their analogues as inhibitors of aldo-keto reductase AKR1C3: new lead compounds for the development of anticancer agents. Bioorg. Med. Chem. Lett. 15, 5170–5175. [DOI] [PubMed] [Google Scholar]
- Lovering A.; Ride J.; Bunce C.; Desmond J.; Cummings S.; White S. (2004) Crystal structures of prostaglandin D(2) 11-ketoreductase (AKR1C3) in complex with the nonsteroidal anti-inflammatory drugs flufenamic acid and indomethacin. Cancer Res. 64, 1802–1810. [DOI] [PubMed] [Google Scholar]
- Bunce C. M.; Mountford J. C.; French P. J.; Mole D. J.; Durham J.; Michell R. H.; Brown G. (1996) Potentiation of myeloid differentiation by anti-inflammatory agents, by steroids and by retinoic acid involves a single intracellular target, probably an enzyme of the aldoketoreductase family. Biochim. Biophys. Acta 1311, 189–198. [DOI] [PubMed] [Google Scholar]
- Blaise B. J.; Giacomotto J.; Elena B.; Dumas M. E.; Toullhoat P.; Segalat L.; Emsley L. (2007) Metabotyping of Caenorhabditis elegans reveals latent phenotypes. Proc. Natl. Acad. Sci. U.S.A. 104, 19808–19812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanska H. M.; Tiziani S.; Howe R. C.; Gunther U. L.; Guizar Z.; Lalani E. N. (2009) Nuclear magnetic resonance detects phosphoinositide 3-kinase/Akt-independent traits common to pluripotent murine embryonic stem cells and their malignant counterparts. Neoplasia 11, 1301–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiziani S.; Lodi A.; Khanim F. L.; Viant M. R.; Bunce C. M.; Günther U. L. (2009) Metabolomic profiling of drug responses in acute myeloid leukaemia cell lines. PLoS ONE 4, e4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiziani S.; Lopes V.; Gunther U. L. (2009) Early stage diagnosis of oral cancer using H-1 NMR-based metabolomics. Neoplasia 11, 269–U269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A. L. (2002) Hypoxia − A key regulatory factor in tumour growth. Nat. Rev. Cancer 2, 38–47. [DOI] [PubMed] [Google Scholar]
- Pouyssegur J.; Dayan F.; Mazure N. M. (2006) Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441, 437–443. [DOI] [PubMed] [Google Scholar]
- Ackerstaff E.; Artemov D.; Gillies R. J.; Bhujwalla Z. M. (2007) Hypoxia and the presence of human vascular endothelial cells affect prostate cancer cell invasion and metabolism. Neoplasia 9, 1138–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denko N. C. (2008) Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 8, 705–713. [DOI] [PubMed] [Google Scholar]
- Glunde K.; Shah T.; Winnard P. T.; Raman V.; Takagi T.; Vesuna F.; Artemov D.; Bhujwalla Z. M. (2008) Hypoxia regulates choline kinase expression through hypoxia-inducible factor-1α signaling in a human prostate cancer model. Cancer Res. 68, 172–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Sanchez R.; Rodriguez-Enriquez S.; Marin-Hernandez A.; Saavedra E. (2007) Energy metabolism in tumor cells. FEBS J. 274, 1393–1418. [DOI] [PubMed] [Google Scholar]
- Benjamini Y.; Hochberg Y. (1995) Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 57, 289–300. [Google Scholar]
- Mazurek S.; Boschek C. B.; Hugo F.; Eigenbrodt E. (2005) Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol. 15, 300–308. [DOI] [PubMed] [Google Scholar]
- Vizan P.; Mazurek S.; Cascante M. (2008) Robust metabolic adaptation underlying tumor progression. Metabolomics 4, 1–12. [Google Scholar]
- Cardone R. A.; Casavola V.; Reshkin S. J. (2005) The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat. Rev. Cancer 5, 786–795. [DOI] [PubMed] [Google Scholar]
- Podo F. (1999) Tumour phospholipid metabolism. NMR Biomed. 12, 413–439. [DOI] [PubMed] [Google Scholar]
- Ferretti A.; D’Ascenzo S.; Knijn A.; Iorio E.; Dolo V.; Pavan A.; Podo F. (2002) Detection of polyol accumulation in a new ovarian carcinoma cell line, CABA I: a H-1 NMR study. Br. J. Cancer 86, 1180–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knijn A.; Brisdelli F.; Ferretti A.; Iorio E.; Marcheggiani D.; Bozzi A. (2005) Metabolic alterations in K562 cells exposed to taxol and tyrphostin AG957: H-1 NMR and biochemical studies. Cell Biol. Int. 29, 890–897. [DOI] [PubMed] [Google Scholar]
- Lau K. S.; Partridge E. A.; Grigorian A.; Silvescu C. I.; Reinhold V. N.; Demetriou M.; Dennis J. W. (2007) Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 129, 123–134. [DOI] [PubMed] [Google Scholar]
- Mendelsohn R.; Cheung P.; Berger L.; Partridge E.; Lau K.; Datti A.; Pawling J.; Dennis J. W. (2007) Complex N-glycan and metabolic control in tumor cells. Cancer Res. 67, 9771–9780. [DOI] [PubMed] [Google Scholar]
- Zachara N. E.; Hart G. W. (2004) O-GlcNAc a sensor of cellular state: the role of nucleocytoplasmic glycosylation in modulating cellular function in response to nutrition and stress. Biochim. Biophys. Acta, Gen. Subj. 1673, 13–28. [DOI] [PubMed] [Google Scholar]
- Brahimi-Horn M.; Chiche J.; Pouysségur J. (2007) Hypoxia signalling controls metabolic demand. Curr. Opin. Cell Biol. 19, 223–229. [DOI] [PubMed] [Google Scholar]
- Khanim F.; Hayden R.; Birtwistle J.; Lodi A.; Tiziani S.; Davies N.; Ride J.; Viant M.; Günther U.; Mountford J.; Schrewe H.; Green R.; Murray R.; Drayson M.; Bunce C. (2009) Combined bezafibrate and medroxyprogesterone acetate: Potential novel therapy for acute myeloid leukaemia. PLoS ONE 4, e8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedotcheva N.; Sokolov A.; Kondrashova M. (2006) Nonenzymatic formation of succinate in mitochondria under oxidative stress. Free Radical Biol. Med. 41, 56–64. [DOI] [PubMed] [Google Scholar]
- de Groot D. J. A.; van der Deen M.; Le T. K. P.; Regeling A.; de Jong S.; de Vries E. G. E. (2007) Indomethacin induces apoptosis via a MRPI-dependent mechanism in doxorubicin-resistant small-cell lung cancer cells overexpressing MRPI. Br. J. Cancer 97, 1077–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen O. E.; Kalhan S. C.; Hanson R. W. (2002) The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 277, 30409–30412. [DOI] [PubMed] [Google Scholar]
- Ackerstaff E.; Gimi B.; Artemov D.; Bhujwalla Z. M. (2007) Anti-inflammatory agent indomethacin reduces invasion and alters metabolism in a human breast cancer cell line. Neoplasia 9, 222–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan K.; Mori N.; Artemov D.; Bhujwalla Z. M. (2002) Exposure of human breast cancer cells to the anti-inflammatory agent indomethacin alters choline phospholipid metabolites and Nm23 expression. Neoplasia 4, 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidi A.; Qualtrough D.; Williams A. C.; Paraskeva C. (2006) Direct transcriptional up-regulation of cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes colorectal tumor cell survival and enhances HIF-1 transcriptional activity during hypoxia. Cancer Res. 66, 6683–6691. [DOI] [PubMed] [Google Scholar]
- Lin H. K.; Jez J. M.; Schlegel B. P.; Peehl D. M.; Pachter J. A.; Penning T. M. (1997) Expression and characterization of recombinant type 2 3α-hydroxysteroid dehydrogenase (HSD) from human prostate: demonstration of bifunctional 3α/17β-HSD activity and cellular distribution. Mol. Endocrinol. 11, 1971–1984. [DOI] [PubMed] [Google Scholar]
- Matsuura K.; Shiraishi H.; Hara A.; Sato K.; Deyashiki Y.; Ninomiya M.; Sakai S. (1998) Identification of a principal mRNA species for human 3α-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D2 11-ketoreductase activity. J. Biochem. 124, 940–946. [DOI] [PubMed] [Google Scholar]
- Pawlowski J.; Huizinga M.; Penning T. M. (1991) Isolation and partial characterization of a full-length cDNA clone for 3α-hydroxysteroid dehydrogenase: a potential target enzyme for nonsteroidal anti-inflammatory drugs. Agents Actions 34, 289–293. [DOI] [PubMed] [Google Scholar]
- Penning T. M.; Burczynski M. E.; Jez J. M.; Lin H. K.; Ma H.; Moore M.; Ratnam K.; Palackal N. (2001) Structure-function aspects and inhibitor design of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3). Mol. Cell. Endocrinol. 171, 137–149. [DOI] [PubMed] [Google Scholar]
- Gupta R. A.; Dubois R. N. (2001) Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat. Rev. Cancer 1, 11–21. [DOI] [PubMed] [Google Scholar]
- Stack E.; DuBois R. N. (2001) Role of cyclooxygenase inhibitors for the prevention of colorectal cancer. Gastroenterol. Clin. North Am. 30, 1001–1010. [DOI] [PubMed] [Google Scholar]
- Wechter W. J.; Kantoci D.; Murray E. D. J.; Quiggle D. D.; Leipold D. D.; Gibson K. M.; McCracken J. D. (1997) R-flurbiprofen chemoprevention and treatment of intestinal adenomas in the APC(Min)/+ mouse model: implications for prophylaxis and treatment of colon cancer. Cancer Res. 57, 4316–4324. [PubMed] [Google Scholar]
- Zhang X.; Morham S. G.; Langenbach R.; Young D. A. (1999) Malignant transformation and antineoplastic actions of nonsteroidal anti-inflammatory drugs (NSAIDs) on cyclooxygenase-null embryo fibroblasts. J. Exp. Med. 190, 451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H. F.; Southam A. D.; Hines A.; Viant M. R. (2008) High-throughput tissue extraction protocol for NMR and MS-based metabolomics. Anal. Biochem. 372, 204–212. [DOI] [PubMed] [Google Scholar]
- Aue W.; Karhan J.; Ernst R. (1976) Homonuclear broad-band decoupling and 2-dimensional J-resolved NMR spectroscopy. J. Chem. Phys. 64, 4226–4227. [Google Scholar]
- Hwang T. L.; Shaka A. J. (1995) Water suppression that works - Excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J. Magn. Reson. Ser. A 112, 275–279. [Google Scholar]
- Thrippleton M. J.; Edden R. A. E.; Keeler J. (2005) Suppression of strong coupling artefacts in J-spectra. J. Magn. Reson. 174, 97–109. [DOI] [PubMed] [Google Scholar]
- Tiziani S.; Lodi A.; Ludwig C.; Parsons H. M.; Viant M. R. (2008) Effects of the application of different window functions and projection methods on processing of H-1 J-resolved nuclear magnetic resonance spectra for metabolomics. Anal. Chim. Acta 610, 80–88. [DOI] [PubMed] [Google Scholar]
- Dieterle F.; Ross A.; Schlotterbeck G.; Senn H. (2006) Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in H-1 NMR metabonomics. Anal. Chem. 78, 4281–4290. [DOI] [PubMed] [Google Scholar]
- Parsons H. M.; Ludwig C.; Günther U. L.; Viant M. R. (2007) Improved classification accuracy in 1-and 2-dimensional NMR metabolomics data using the variance stabilising generalised logarithm transformation. BMC Bioinf. 8, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golotvin S.; Williams A. (2000) Improved baseline recognition and modeling of FT NMR spectra. J. Magn. Reson. 146, 122–125. [DOI] [PubMed] [Google Scholar]
- Günther U. L.; Ludwig C.; Ruterjans H. (2000) NMRLAB - Advanced NMR data processing in MATLAB. J. Magn. Reson. 145, 201–208. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.