

Abstract
We have studied the gene expression pattern of invasive primary mammary tumor cells using a unique in vivo invasion assay that isolates the invasive tumor cells by chemotaxis. One of the genes upregulated in the invasive tumor cells is Mena, an actin binding protein involved in the regulation of cell motility. There are multiple known splice variants of Mena accounted for by four alternatively included exons, +, ++, +++ and 11a. Using the in vivo invasion assay in rats and mice with mammary tumors we observed that two isoforms of Mena, ++ and +++, are upregulated in the invasive tumor cells and one isoform, 11a, is downregulated. The Mena isoform switching pattern described here may provide a new biomarker for the presence of metastatic cancer cells and for prognosis.
Keywords: Breast cancer, Metastasis, Biomarker, Mena, Splice variant
Introduction
Invasion of tumor cells into surrounding tissue and intravasation into blood and lymphatic vessels is implicated in the progression of metastatic mammary cancer. This multistep process involves a number of phenotypic changes that occur sequentially and give rise to an invasive tumor cell capable of enhanced EGF stimulated motility and invasion [1]. In our effort to identify these individual events and to understand the molecular pathways underlying these phenotypic changes, we have developed animal models as well as a unique invasion assay that allows the isolation of invasive cells directly from the primary tumor and separates them from the average primary tumor cells (APTC) [2]. Isolation of the invasive tumor cells and subsequent gene expression analysis resulted in the identification of a specific gene expression signature in invasive tumor cells from mammary tumors of rats and mice called the “invasion signature” [3-5]. In several studies, a number of genes were identified that must be coordinately up or downregulated in the invasive tumor cells in order for their invasion to lead to metastasis [3, 6].
One of the key upregulated genes in invasive mammary tumor cells encodes the actin regulatory protein Mena, which we hypothesize to be a central switch point in the regulation of the pathways encoded by the invasion signature. Mena is a member of the Ena/VASP family, proteins that control the geometry of assembling F-actin networks and play a role in cell migration in a number of cell types and organisms [7]. They antagonize capping of actin filaments by capping proteins at their barbed ends and also reduce branching density through an unknown mechanism [8, 9]. The anti-capping activity of Mena has been proposed to amplify the barbed end output of the cofilin and Arp2/3 complex pathways, particularly in response to EGF, which is sufficient to increase the metastatic potential of mammary tumors [1, 7, 10]. Ena/VASP proteins are also constituents of the adherence junctions necessary to seal membranes in an epithelial sheet and control actin organization on cadherin adhesion contact [11]. The stability of cadherens junctions is frequently perturbed in invasive cancer cells and Mena may play a role in this transition to invasion.
Ena/VASP proteins share a conserved domain structure including the N-terminal EVH1 (Ena/VASP Homology) domain, that plays an essential role in intracellular protein localization by interacting with FP4 motifs found in proteins such as zyxin and vinculin [12]. The LIM3 domain of the tumor suppressor Tes binds specifically to Mena EVH1 and competes with FP4-containing proteins for interaction with Mena [13]. The central proline-rich region mediates interaction with proteins containing SH3 and WW domains and with the actin monomer binding protein profilin [8]. The C-terminal EVH2 domain binds to G and F-actin and contains a coiled-coil that mediates formation of stable tetramers [14]. The interaction of the EVH2 domain with the growing ends of the actin filaments is essential for targeting the Ena/VASP to lamellipodia [15].
Upregulation of Mena mRNA expression in both mouse and rat invasive mammary cancer cells [3, 4] is consistent with the observed increased expression of Mena protein in human breast cancer tissues and cancer cell lines [16-18]. These observations, and the proposed central role of Mena in regulating invasion and metastasis [1], prompted us to look deeper into this expression pattern and the pattern of expression of Mena splice variants in particular. Previously, mouse and human Mena homologs have been cloned, sequenced and a number of splice variants have been identified [8, 19]. Recently it has been shown that splice variants can work very efficiently as cancer biomarkers [20, 21]. Our study goal was to identify any known or new splice variants of Mena that are up or downregulated specifically in invasive mammary cancer cells. The value of such information would be in the design of new molecular probes (either nucleic acid or protein) to identify metastatic tumor cells in vivo.
Methods
Isolation of invasive tumor cells using the in vivo invasion assay and fluorescence-activated cell sorting of tumor cells at different stages of the metastasis
All procedures involving rats and mice were conducted in accordance with the National Institutes of Health regulations concerning the use and care of experimental animals, and approved by the Albert Einstein College of Medicine animal use committee. We used MTLn3-derived mammary tumors in rats [3] and MDA-MB-231 derived tumors raised in SCID mice along with the PyMT driven mouse mammary cancer transgenic model and the in vivo invasion assay has been described previously [3, 4] to study the gene expression pattern of invasive subpopulations of carcinoma cells within live primary tumors. A schematic diagram showing details of the in vivo invasion assay along with a photograph of the setup and some fluorescent images that explain the function of the assay is included as supplementary Fig. 2 [2, 22, 23]. For human cell line, a total of 2 × 106 MDA-MB-231-GFP cells per animal were resuspended in sterile PBS with 20% collagen I (BD Biosciences, Franklin Lakes, NJ). Orthotopic xenografts were produced in severe combined immunodeficiency mice (SCID) (NCI, Frederick, MD) by injecting the cell suspension into the lower left mammary fat pad. All experiments were performed on tumors that were 1.2-1.5 cm in diameter.
In the in vivo invasion assay cells can only enter the needles by active migration since a block is used to prevent passive collection of cells and tissue during insertion of the needle into the tissue. The block is removed and cell migration can be observed using 2-photon microscopy and has been demonstrated to be required for cell collection [2-4] (supplementary Fig. 2). One tenth of the volume from each needle was used to determine the number of cells collected. Collected cells were a mixture of carcinoma cells (75%) and macrophages (25%) in case of the rodent models, and carcinoma cells (95%) and macrophages and other cells (5%) for the human MDA-MB-231 cells. From the remaining 9/10 volume from the microneedle, macrophages were removed by magnetic separation using CD11b beads (Mitenyl Biotech, USA), and RNA was extracted from purified carcinoma cells as described before [3]. To isolate the average primary tumor cells (APTCs) and lung metastasis, a small piece of tumor or lung were minced, and filtered twice through nylon filters to obtain single cell suspension. To isolate the circulating tumor cells from blood, right auricular puncture was performed in anesthetized animals; red blood cells were lysed using ammonium chloride lysis buffer. Fluorescence-activated cell sorting was performed based on their green fluorescent protein (GFP) expression in tumor cells. GFP-positive tumor cells were collected into a tube and lysed directly for RNA extraction. All the procedures were done on ice or at 4°C. The same procedures for extraction of RNA and cDNA synthesis and QRT-PCR as described before [5] were used for all the three models.
Controls for invasion specific gene expression pattern
During the collection of invasive cells using the in vivo invasion assay, the invasive cells are subjected to either EGF or CSF1 as chemoattractant, and matrigel in the needle. To detect an effect of these agents on gene expression, cell lines used to prepare tumors and tumor cells FACs sorted from primary tumors were placed in collection needles and subjected to matrigel, and EGF stimulation. QRT-PCR was used to determine if the expression of key genes of the invasion signature were altered. No gene expression changes resembling the invasion signature were observed (supplementary Fig. 1) and these results are consistent with results obtained by us previously [3]. Similar results were obtained using CSF1 [3]. Another step in the isolation of invasive tumor cells is the separation of tumor cells from accompanying macrophages using magnetic beads coated with anti-macrophage cell surface antibodies as described previously [3]. No effect on gene expression was detected using the method described above as a result of using antibody beads directed against invasive cells to separate cell types (supplementary Fig. 1A). These results indicate that only the environment within the primary tumor generates the pattern of gene expression of invasive cells termed the invasion signature.
Cell line and cell culture
MTLn3 rat adenocarcinoma cells were cultured in alpha MEM with 5% FBS as described previously [5]. MDA-MB-231 cell line was propagated in DMEM with 10% FBS. The cells were maintained at 37°C in a humidified atmosphere in 5% CO2 in air. All cells used from PyMT tumors were spontaneously generated by the oncogene in vivo and were not cell lines.
RT-PCR and QRT-PCR
RT-PCR and QRT-PCR was performed using primers mentioned in the supplementary Table 1. QRT-PCR was performed using SyBr Green kit, ABI 7300 sequence detector and data analysis performed using ABI Sequence Detection Software (Applied Biosystems Foster City, CA).
RACE, cloning and sequencing
RACE was performed using Clontech kit (sequences given in supplementary Table 1 and primer design strategy is given in Fig. 2) and cloned using Invitrogen kit following manufacturer's protocol. Briefly, PCR was performed using two internal primers for the ++ and +++ sequences (supplementary Table 1) and an oligo dT primer for the 5′ and poly G primer for the 3′ ends. The PCR products were cloned into pCR-TOPO vector and transformed into chemically competent cells. Clones were sequenced using M13 primers. Sequence alignment was performed using DNASTAR software.
Fig. 2.

Strategy for primer design for each of the Mena exons and Smart RACE along with Nucleotide and protein sequences of the ++ and +++ isoforms. a The position of the primers and the strategy for primer design along with both the 3′ and 5′ RACE strategies are shown in this figure. Primer numbers refer to those in Supplementary Table 1. b Sequence alignment for ++ and +++ exons expressed in invasive tumor cells are identical to the published mouse sequences. The ++ exon nucleotide and inferred amino acid sequences are aligned in 1 and the +++ exon nucleotide and inferred amino acid sequences are aligned in 2
Results and discussion
Identification of specific isoforms of Mena, that are up or downregulated in invasive mammary cancer cells, was measured in three models: MTLn3 rat adenocarcinoma allografts, human breast cancer cell line MDA-MB-231 xenografts generated in SCID mice and PyMT mouse transgenic mammary tumors. In all the models, tumor cells were labeled with GFP. We reported previously that overall Mena expression at the mRNA level is upregulated 3-4 fold in the invasive cancer cells in both types of rodent tumors [3]. From the rat MTLn3, human MDA-MB-231 xenografts and mouse PyMT transgenic models, we collected and separated the invasive cells using the in vivo invasion assay, and the average primary tumor cells (APTCs) by FACS sorting.
To determine if any of the steps required to collect the invasive tumor cell population might lead to the pattern of gene expression we call the invasion signature, we analyzed control cells that were cultured within a needle in the presence of matrigel and EGF. These controls demonstrate that the expression of the invasion signature is not induced by any of the steps or reagents used for cell collection in this study (supplementary Fig. 1). These results indicate that only the tumor microenvironment induces the pattern of expression seen in the invasion signature of which Mena is a part.
RT-PCR analysis of the invasive tumor cells using primers that detect all Mena isoforms showed the expected Mena upregulation. Amplicons containing the Mena ++ and Mena +++ exons were upregulated while amplicons containing Mena 11a were downregulated specifically in the invasive tumor cell population as compared to the APTC. QRT-PCR studies confirmed the RT-PCR finding and showed upregulation of both ++ and +++ exons and the downregulation of the 11a exon specifically in the invasive tumor cells isolated from both mammary tumor models (Fig. 1; Fig. 2a shows the locations of the primers utilized in this study). An amplicon specific for the + exon was also detected in APTC and invasive tumor cells. Similar levels of a splice variant that did not contain either the ++ or the +++ was detected in both invasive tumor cells and APTC. This indicates that the increased expression of pan mena as observed in Figs. 1 and 3 are mainly due to the splice variants ++ and +++.
Fig. 1.

The expression of transcripts containing Mena ++ and Mena +++ exons are upregulated while those containing Mena 11a are downregulated specifically in the invasive tumor cell population as compared to the APTC. Quantification of Mena isoforms by QRT-PCR in MTLn3 rat allograft model (a), and PyMT mouse transgenic model (b). The levels of transcript observed using pan Mena primers (Mena), and ++ and +++ primers are increased while that observed using 11a primers is greatly reduced in both animal tumor models. *11a Message was undetectable in the PyMT mouse transgenic invasive tumor cells. The error bars show standard errors of mean (SEM) performed on three biological repeats and three technical repeats
Fig. 3.
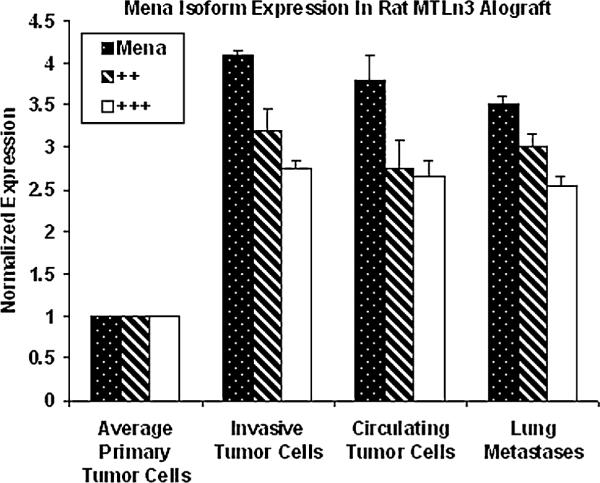
Expression of the isoforms of Mena characteristic of invasive tumor cells are stably expressed in different stages of metastasis in rat adenocarcinoma (MTLn3) cells. The expression status of Mena ++ and +++ isoforms was determined by QRT-PCR. The isoforms characteristic of invasive tumor cells show 3.5-4 fold over expression in the invasive tumor cells, circulating tumor cells in blood and in tumor cells growing as lung metastasis. The stability of elevated expression during progression suggests that these isoforms of Mena may be markers for the presence of metastatic tumor cells in several sites. The error bars show standard errors of mean (SEM) performed on three biological repeats and three technical repeats
These results indicate that there is a switch in expression from Mena11a to Mena ++ and +++ variants when tumor cells become invasive in vivo. However, it was unclear if the exons are present in a single transcript or on separate transcripts. This information is crucial for the design of molecular probes that might be designed and used for diagnostic purposes. To address this question, we cloned and sequenced ++ and +++ bearing transcripts from the invasive PyMT mouse transgenic tumors. RACE analysis was selected as we wanted to identify transcripts that definitely contained the ++ and +++ exons. The consensus results from sequencing at least 10 clones for each transcript demonstrated a 100% match with the published mouse sequences and indicated that the ++ and +++ exons are in separate transcripts (Fig. 2b). It has been previously shown that the ++ and +++ regions of Mena have the same sequence in human and mice [19].
These results demonstrate that there is an increase in expression of both the Mena ++ and +++ variants as separate transcripts when tumor cells become invasive in vivo. To determine if the increased expression of these two variants persists during the progression of metastasis, we compared the expression pattern of invasive tumor cells to circulating tumor cells and those that had formed metastatic tumors of the lung in rats with MTLn3-derived tumors. Figure 3 shows that both the ++ and +++ splice variants of Mena remained upregulated in circulating tumor cells in blood and the tumor cells that had formed metastases in the lung. Similar results were obtained in PyMT transgenic mice where a 2-3 fold upregulation of expression of both the ++ and +++ exons in lung metastases was observed. However, the 11a splice variant was not detected in the lung metastasis indicating at least an eight fold down regulation (Fig. 3).
In order to determine the efficacy of this isoform switch as a potential for metastatic biomarker we extended this study in human cell line MDA-MB-231 and observed that Mena was upregulated by 6-8 fold in the invasive tumor cells. In particular, the splice variant ++ was found to be upregulated 2-3 fold compared to APTCs. The splice variant 11a, which was detected in the APTC, was reduced to undetectable levels in the invasive tumor cells indicating at least an eight fold down regulation. The splice variant +++ which was detected in the MDA-MB-231 cell line was undetectable in the invasive cells due to technical reasons resulting from the small number of invasive tumor cells collected from MDA-MB-231 derived tumors. Together these data are consistent with those obtained in rat and mouse mammary tumors.
Several conclusions can be drawn from the studies reported here. First, invasive carinoma cells from three different types of primary mammary tumors exhibited significant upregulation of Mena expression when compared to APTCs. These results are consistent with observations in human breast cancers and cancer cell lines where Mena protein expression is up [12, 15]. Second, we found that Mena isoforms containing the 11a exon were downregulated in the invasive tumor cell population. This is in agreement with our recent results in human pancreatic cancer indicating that hMena+11a isoform is exclusively expressed in the pancreatic cancer cell lines that are Ecadherin positive and not found in E-cadherin negative pancreatic cancer cells [24] Interestingly, however, cancer cell lines treated with EGF in culture upregulate and hyperphosphorylate Mena variants containing the 11a exon [17]. Therefore it appears likely that the microenvironment of the tumor generates patterns of expression of variants that cannot be mimicked by in vitro manipulations. This is consistent with control experiments in our study which indicate that the invasion signature cannot be induced in vitro. Third, we report for the first time a selective upregulation of Mena variants containing a 4 amino acid region (++) and a 19 amino acid region (+++) along with the downregulation of a 21 amino acid region (11a) in invasive tumor cells.
While the existence of Mena splice variants have been reported previously [7, 17, 19], relatively little is known about the functional significance of these variants. The observed isoform switching likely reflects critical functional differences between the Mena variants. Expression of the Mena isoform containing the sequence encoded by +++ confers a striking increase in the ability of cells to invade into a 3D collagen gel and potentiates motility responses elicited by EGF [10]. Given the selective inclusion of the +++ exon in invasive cells observed in this study and that this isoform has a potent effect of increasing invasion, we propose to denote it as “INV” and Mena isoforms containing this exon as MenaINV (Fig. 4). It will be of great interest to understand how the Mena isoforms differ functionally. The INV exon is included just after the EVH1 domain; it is therefore possibility that INV may influence the function of Mena EVH1. Interestingly, Mena EVH1 (but not VASP or EVL) was recently shown to bind to the TES tumor suppressor [21]. Experiments are underway to determine whether MenaINV also interacts with TES. The 11a exon is included within the EVH2 domain adjacent to the known F-actin binding motif. F-actin binding is critical for almost all known Ena/VASP functions including localization to the tips of lamellipods and the ability drive to filopod and lamellipod formation and extension [7, 15]. In vitro, F-actin binding is required for the anti-capping activity of Ena/VASP and is disrupted by phosphorylation at nearby sites [8]. Since 11a is phosphorylated [17], it is possible that inclusion of this exon provides an additional site of negative regulation within Mena 11a.
Fig. 4.

Domain organization of Mena and location of alternatelyincluded exons. A schematic of Mena is shown. The EVH1 domain binds to proteins that contain a motif with the consensus: (D/E) FPPPPX(D/E)(D/E); an exception to this rule is TES, which binds to Mena EVH1 through a Lim domain [19]. EVH1-interactions are required for subcellular targeting of Mena and to bring it into signaling complexes. The invasion-specific exon “INV” is included in between the EVH1 domain and an extended “LERER” repeat region of unknown function. “PRO” denotes a proline-rich region that contains binding sites for the actin-monomer binding protein profilin as well as several SH3- and WW-domains. The EVH2 domain contains binding sites for G-actin (“G”), F-actin (“FAB”) and has a coiled-coil (“CC”) that forms tetramers. The “11a” exon which is present in Mena within primary tumors and downregulated in invasive cells is included adjacent to the FAB site
Recently it has been shown that splice variants can work very efficiently as cancer biomarkers [20, 21]. Analysis of the relative levels of MenaINV, Mena++, Mena 11a in tumor tissue, may have the potential as a new ratiometric prognostic marker for metastasis; a finding of INV or ++ up and 11a downregulation may predict metastatic disease. Since the upregulation of expression of the INV and ++ exons observed here is a stable change in invasive and metastatic mammary tumor cells, probes specifically directed at these exons would be powerful diagnostic and prognostic markers for the presence of metastatic cells and therefore the potential of metastatic disease at the primary site, and in the circulating population of tumor cells. Further work will be required to evaluate these predictions.
Gene expression changes in the invasive cells could result from two mechanisms. One is that stable genetic and epigenetic changes occur in the cancer cell as it progressively acquires favorable mutations. Secondly the progressing tumor induces transient changes in gene expression of cancer cells that are responsive to the microenvironment and are limited to the cell in transit. To understand why the preferred expression of Mena variants containing the INV or ++ exons occurs throughout progression will require further study to determine if the selection for these variants is a consequence of epigenetic changes occurring in response to the microenvironment of the tumor or if these are stable changes resulting from a genetic mutation carried in the invasive subpopulation of cancer cells into the lung metastases.
Supplementary Material
Acknowledgements
The authors wish to acknowledge Andrew Freidman for technical help, Drs Erik Sahai and Jeffrey Segall for their help in discussions. U.P. was supported by the Anna Fuller Molecular Oncology Fund and the Ludwig Center for Molecular Oncology. This work is supported by NIH CA 100324 (SG and JC) and CA113395 (JC), NIH grant # GM58801 and ICBP grant # 1-U54-CA112967 for FBG, Lega Italiana per la Lotta Contro i Tumori and Associazione Italiana per la Ricerca sul Cancro (AIRC) for PN. SG is the recipient of the Young Investigator Award from Breast Cancer Alliance Inc.
References
- 1.Condeelis J, Singer RH, Segall JE. The great escape: when cancer cells hijack the genes for chemotaxis and motility. Annu Rev Cell Dev Biol. 2005;21:695–718. doi: 10.1146/annurev.cellbio.21.122303.120306. doi:10.1146/annurev.cellbio.21. 122303.120306. [DOI] [PubMed] [Google Scholar]
- 2.Wyckoff J, Wang W, Lin EY, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64(19):7022–7029. doi: 10.1158/0008-5472.CAN-04-1449. doi:10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- 3.Wang W, Goswami S, Lapidus K, et al. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 2004;64(23):8585–8594. doi: 10.1158/0008-5472.CAN-04-1136. doi:10.1158/0008-5472.CAN-04-1136. [DOI] [PubMed] [Google Scholar]
- 4.Wang W, Wyckoff JB, Goswami S, et al. Coordinated regulation of pathways for enhanced cell motility and chemotaxis is conserved in rat and mouse mammary tumors. Cancer Res. 2007;67(8):3505–3511. doi: 10.1158/0008-5472.CAN-06-3714. doi:10.1158/0008-5472.CAN-06-3714. [DOI] [PubMed] [Google Scholar]
- 5.Goswami S, Wang W, Wyckoff JB, Condeelis JS. Breast cancer cells isolated by chemotaxis from primary tumors show increased survival and resistance to chemotherapy. Cancer Res. 2004;64(21):7664–7667. doi: 10.1158/0008-5472.CAN-04-2027. doi:10.1158/0008-5472.CAN-04-2027. [DOI] [PubMed] [Google Scholar]
- 6.Wang W, Mouneimne G, Sidani M, et al. The activity status of cofilin is directly related to invasion, intravasation, and metastasis of mammary tumors. J Cell Biol. 2006;173(3):395–404. doi: 10.1083/jcb.200510115. doi: 10.1083/jcb.200510115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gertler FB, Niebuhr K, Reinhard M, Wehland J, Soriano P. Mena, a relative of VASP and Drosophila Enabled, is implicated in the control of microfilament dynamics. Cell. 1996;87:227–239. doi: 10.1016/s0092-8674(00)81341-0. doi: 10.1016/S0092-8674(00)81341-0. [DOI] [PubMed] [Google Scholar]
- 8.Barzik M, Kotova TI, Higgs HN, et al. Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. J Biol Chem. 2005;280(31):28653–28662. doi: 10.1074/jbc.M503957200. doi: 10.1074/jbc.M503957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bear JE, Svitkina TM, Krause M, et al. Antagonism between Ena/VASP Proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109:509–521. doi: 10.1016/s0092-8674(02)00731-6. doi:10.1016/S0092-8674 (02)00731-6. [DOI] [PubMed] [Google Scholar]
- 10.Philippar U, Roussos ET, Oser M, Yamaguchi H, Kim H, Giampieri S, Wang Y, Goswami S, Wyckoff JB, Sahai E, Condeelis JS, Gertler FB. A Mena invasion isoform potentiates EGFinduced carcinoma cell invasion and metastasis. Dev Cell. 2008 doi: 10.1016/j.devcel.2008.09.003. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott JA, Shewan AM, den Elzen NR, et al. Ena/VASP proteins can regulate distinct modes of actin organization at cadherin-adhesive contacts. Mol Biol Cell. 2006;17(3):1085–1095. doi: 10.1091/mbc.E05-07-0644. doi: 10.1091/mbc.E05-07-0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prehoda KE, Lee DJ, Lim WA. Structure of the enabled/VASP homology 1 domain-peptide complex: a key component in the spatial control of actin assembly. Cell. 1999;97(4):471–480. doi: 10.1016/s0092-8674(00)80757-6. doi: 10.1016/S0092-8674(00)80757-6. [DOI] [PubMed] [Google Scholar]
- 13.Boeda B, et al. Tes, a specific Mena interacting protein, breaks the rules for EVH1 binding. Mol Cell. 2007;28:1071–1082. doi: 10.1016/j.molcel.2007.10.033. doi: 10.1016/j.molcel.2007.10.033. [DOI] [PubMed] [Google Scholar]
- 14.Kuhnel K, Jarchau T, Wolf E, et al. The VASP tetramerization domain is a right-handed coiled coil based on a 15-residue repeat. Proc Natl Acad Sci USA. 2004;101(49):17027–17032. doi: 10.1073/pnas.0403069101. doi: 10.1073/pnas.0403069101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loureiro JJ, Rubinson DA, Bear JE, et al. Critical roles of phosphorylation and actin binding motifs, but not the central proline-rich region, for Ena/vasodilator-stimulated phosphoprotein (VASP) function during cell migration. Mol Biol Cell. 2002;13(7):2533–2546. doi: 10.1091/mbc.E01-10-0102. doi:10.1091/mbc.E01-10-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Modugno F, Bronzi G, Scanlan MJ, et al. Human Mena protein, a serex-defined antigen overexpressed in breast cancer eliciting both humoral and CD8 + T-cell immune response. Int J Cancer. 2004;109(6):909–918. doi: 10.1002/ijc.20094. doi:10.1002/ijc.20094. [DOI] [PubMed] [Google Scholar]
- 17.Di Modugno F, DeMonte L, Balsamo M, et al. Molecular cloning of hMena (ENAH) and its splice-variant hMena + 11a. Epidermal growth factor (EGF) increases their expression and stimulates hMena + 11a phosphorylation in breast cancer cell lines. Cancer Res. 2007;67:2657–2665. doi: 10.1158/0008-5472.CAN-06-1997. doi:10.1158/0008-5472.CAN-06-1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Modugno F, Mottolese M, Di Benedetto A, et al. The cytoskeleton regulatory protein hMena (ENAH) is overexpressed in human benign breast lesions with high risk of transformation and human epidermal growth factor receptor-2-positive/hormonal receptor-negative tumors. Clin Cancer Res. 2006;12(5):1470–1478. doi: 10.1158/1078-0432.CCR-05-2027. doi: 10.1158/1078-0432.CCR-05-2027. [DOI] [PubMed] [Google Scholar]
- 19.Urbanelli L, Massini C, Emiliani C, et al. Characterization of human Enah gene. Biochim Biophys Acta. 2006;1759(1-2):99–107. doi: 10.1016/j.bbaexp.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 20.Brinkman BM. Splice variants as cancer biomarkers. Clin Biochem. 2004;37(7):584–594. doi: 10.1016/j.clinbiochem.2004.05.015. doi:10.1016/j.clinbiochem.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 21.Venables JP. Unbalanced alternative splicing and its significance in cancer. BioEssays. 2006;28(4):378–386. doi: 10.1002/bies.20390. doi:10.1002/bies. 20390. [DOI] [PubMed] [Google Scholar]
- 22.Wyckoff J, Segall J, Condeelis J. The collection of the motile population of cells from a living tumor. Cancer Res. 2000;60:5401–5404. [PubMed] [Google Scholar]
- 23.Wyckoff J, Segall JE, Condeelis J. Single-cell imaging in animal tumors in vivo. In: Spector D, Goldman R, editors. Live cell imaging, a laboratory manual, chap 22. Cold Spring Harbor Laboratory Press; 2005. pp. 409–422. [Google Scholar]
- 24.Pino MS, Balsamo M, Di Modugno F, et al. Human Mena + 11a isoform serves as a marker of epithelial phenotype and sensitivity to epidermal growth factor receptor inhibition in human pancreatic cancer cell lines. Clin Cancer Res. 2008;14(15):4943–4950. doi: 10.1158/1078-0432.CCR-08-0436. doi:10.1158/1078-0432.CCR-08-0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.