

Abstract
The regulated degeneration of axons or dendrites (pruning) and neuronal apoptosis are widely used during development to determine the specificity of neuronal connections. Pruning and apoptosis often share similar mechanisms; for example, developmental dendrite pruning of Drosophila class IV dendritic arborization (da) neurons is induced by local caspase activation triggered by ubiquitin-mediated degradation of the caspase inhibitor DIAP1. Here, we examined the function of Valosin-containing protein (VCP), a ubiquitin-selective AAA chaperone involved in endoplasmic reticulum-associated degradation, autophagy and neurodegenerative disease, in Drosophila da neurons. Strong VCP inhibition is cell lethal, but milder inhibition interferes with dendrite pruning and developmental apoptosis. These defects are associated with impaired caspase activation and high DIAP1 levels. In cultured cells, VCP binds to DIAP1 in a ubiquitin- and BIR domain-dependent manner and facilitates its degradation. Our results establish a new link between ubiquitin, dendrite pruning and the apoptosis machinery.
Keywords: Pruning, Apoptosis, DIAP1 (Thread), VCP (TER94), Drosophila
INTRODUCTION
Developmentally programmed neuronal cell death and developmental pruning of axons or dendrites are important processes that shape the connections in the nervous system. Pruning can occur by several mechanisms, including retraction and local degeneration of the affected processes. Local degeneration of axons or dendrites often involves caspase activation and thus shares features with apoptosis (Kuo et al., 2006; Nikolaev et al., 2009; Williams et al., 2006). In the Drosophila peripheral nervous system, class III and class IV dendritic arborization (da) neurons have stereotyped long and branched dendrites at larval stages (Grueber et al., 2002). During the pupal stage, a subset of class IV neurons prune and then regrow their dendrites (Kuo et al., 2005; Williams and Truman, 2005), whereas class III neurons undergo developmental apoptosis (Williams and Truman, 2005). Both pruning and developmental apoptosis in these neurons require caspase activation through ubiquitin-mediated degradation of the caspase inhibitor Drosophila inhibitor of apoptosis protein 1 [DIAP1; Thread (Th) – FlyBase] (Kuo et al., 2006; Williams et al., 2006; Williams and Truman, 2005).
VCP (TER94 – FlyBase; CDC-48/CDC48 in C. elegans and yeast; also known as p97 in vertebrates) is an abundant AAA ATPase that is required for the degradation of a subset of substrates of the ubiquitin-proteasome system (Jentsch and Rumpf, 2007; Schuberth and Buchberger, 2008). Its best-understood function is the degradation of misfolded proteins of the endoplasmic reticulum (ER) in the ER-associated degradation (ERAD) pathway (Ye et al., 2001), but it has also been linked to a number of other processes including nucleus reformation after mitosis (Ramadan et al., 2007), myofibril assembly (Janiesch et al., 2007) and autophagosome maturation (Ju et al., 2009; Tresse et al., 2010). Interestingly, certain autosomal dominant mutations of VCP cause a degenerative disease called inclusion body myopathy with Paget's disease of bone and frontotemporal dementia (IBMPFD), which mostly affects muscles and brain (Watts et al., 2004), thus linking VCP to brain function. It has been proposed that VCP is required for the degradation of ubiquitylated substrate proteins when they need to be extracted from the ER membrane or a tight protein complex. However, VCP is also sometimes required for the degradation of monomeric soluble proteins, such as GFP-based model substrates. A potential explanation for this observation is that these substrates are particularly strongly folded and VCP might be needed to unfold them prior to degradation (Beskow et al., 2009). However, the exact mechanism of action of VCP in many processes remains unclear owing to a lack of knowledge concerning its substrates.
We investigated neural developmental functions of VCP in Drosophila class III and class IV da neurons. We find that VCP inhibition in larval neurons induces proteotoxic stress, which, depending on the strength of the inhibition, can be cell lethal. Remarkably, intermediate levels of VCP inhibition are not lethal, but instead suppress dendrite pruning, apoptosis and caspase activation at the pupal stage. We show that these phenotypes are caused by impaired caspase activation and DIAP1 degradation. Thus, VCP-dependent DIAP1 degradation is required for proper neuronal remodeling and apoptosis.
MATERIALS AND METHODS
Fly stocks
Dendritic arborization neurons were labeled using the following GAL4 lines: ppk-GAL4 (Grueber et al., 2007), ppk-GeneSwitch [gift from Rebecca Yang (Duke University Medical Center, NC, USA) and Jay Parrish (University of Washington, WA, USA)] Gal419-12 [gift from H.-H. Lee and Y. Xiang (National Taiwan University Medical School)], GAL4109(2)80, UAS-GMA (Dutta et al., 2002; Medina et al., 2006) and ppk-CD4::tdTomato [gift from P. Soba, (University of California, San Francisco, CA, USA)]. VCP constructs (wild type, QQ dominant-negative, R152H) were cloned into pUAST (Brand and Perrimon, 1993) and injected into w1118 flies according to standard procedures. The JNK reporter line was pucA251.1F3 (Bloomington) and UAS-Xbp1-GFP (Ryoo et al., 2007) was used to assess ER stress. UAS-tau::GFP was a gift of W. Song (University of California, San Francisco, CA, USA); other lines included UAS-mCD8GFP (Lee and Luo, 1999), UAS-dVCP-IR (inverted repeat, VDRC 24354) and UAS-dcr2 (Dietzl et al., 2007), UAS-p35 (Hay et al., 1994) (Bloomington) and UAS-mCD8::PARP::Venus (Williams et al., 2006). Mutant alleles included dvcp26-8 and dvcp7-12 (Goldstein et al., 2001; Ruden et al., 2000) and th4 (Hay et al., 1995) (all from Bloomington).
Live imaging
Live imaging of da neurons in appropriately staged larvae or pupae was carried out on a Leica SP5 confocal microscope. Images were taken from neurons in abdominal segments A2-A6. In all images shown, anterior is left and dorsal is up. Fisher's exact test (GraphPad) was used for statistical comparisons.
Immunocytochemistry
Larvae or appropriately staged pupae (4 hours APF) were dissected as described (Kuo et al, 2006). Antibodies used were rat anti-VCP (1:500) (Leon and McKearin, 1999), mouse anti-ubiquitin (1:50; P4D1, Sigma), rabbit anti-β-galactosidase (1:5000; Cortex Biochem), rabbit anti-activated caspase (1:100; Trevigen), rabbit anti-cleaved PARP (1:20; Abcam 2317), rabbit anti-DIAP1 (1:1000) (Ryoo et al., 2002), rat anti-mCD8 (1:200; Invitrogen) and rabbit anti-GFP (1:3000; Jan lab). Donkey Cy2- or Rhodamine Red X-conjugated secondary antibodies (Jackson Laboratory) were used at 1:200.
Transfections, immunoprecipitation and Western blotting
FLAG-tagged forms of Drosophila VCP and HA-tagged wild-type and mutant forms of DIAP1 were in pUAST. For expression in S2 cells, plasmids were co-transfected with Actin-GAL4 using the Effectene Kit (Qiagen) according to the manufacturer's instructions. After 48 hours, cells were harvested and lysed in RIPA buffer containing 1 mM PMSF and complete protease inhibitors (Roche). Where indicated, the proteasome inhibitor MG132 (10 μM) was added 3 hours before lysis. For immunoprecipitations, cells were lysed in lysis buffer (100 mM NaCl, 50 mM Tris-HCl pH 7.5, 5 mM MgCl2, 10% glycerol, 1% Triton X-100, 1 mM PMSF, 1× complete protease inhibitors). Extracts were precleared with IgG Sepharose 6 Fast Flow beads (Pharmacia) and immunoprecipitated with mouse anti-HA beads (Sigma) for 2 hours, washed with lysis buffer and eluted with SDS loading buffer. Primary antibodies for Western blotting were mouse anti-DIAP1 (Yoo et al., 2002), mouse anti-FLAG (M2, Sigma), rat anti-HA (3F10, Roche), mouse anti-tubulin (DM1a, Sigma) and mouse anti-GAPDH (clone 71.1, Sigma).
RESULTS
Drosophila VCP is required for proper neuronal morphology and viability
In order to characterize the function of VCP in neurons, we used the GAL4/UAS system (Brand and Perrimon, 1993) to express either wild-type VCP, an ATPase-deficient VCP transgene (E375Q, E575Q, referred to as VCP QQ) that acts as a dominant-negative (Ye et al., 2001), or an RNAi construct directed against VCP (Dietzl et al., 2007). We expressed VCP RNAi or VCP QQ in class IV neurons using ppk-GAL4 and visualized them via concomitant expression of membrane-bound GFP. Overexpressed VCP was mostly localized in a characteristic perinuclear pattern in class IV neurons, suggestive of the ER, as has been observed in other cell types (e.g. Meyer et al., 2000), whereas the dominant-negative VCP QQ was localized somewhat more diffusely and also inside the nucleus (see Fig. S1 in the supplementary material). The limited sensitivity of the antibodies prevented us from assessing VCP localization in dendrites.
In third instar larvae, class IV neurons have highly branched dendrites that fully cover the larval body wall. Inhibition of VCP by VCP QQ or VCP RNAi caused a reduction in dendrite arborization, while major branches stayed intact (Fig. 1A-D). These defects in class IV neurons mostly affected higher-order branches containing filamentous actin, but not primary dendrite branches rich in microtubules, as shown with the actin marker GMA (GFP fused to the actin-binding domain of moesin) and the microtubule marker tau::GFP (see Fig. S2 in the supplementary material). We also expressed VCP RNAi or VCP QQ in class III neurons using the GAL4 driver Gal419-12. Here, the length of class III neuron dendrites seemed mildly affected by expression of VCP QQ, but spike appearance was normal (Fig. 1E-H). In order to increase the strength and penetrance of potential VCP loss-of-function phenotypes in class IV neurons, we either expressed VCP RNAi in a heterozygous dvcp7-12/+ mutant background (Goldstein et al., 2001) or used two copies of the UAS-VCP QQ transgene. Whereas the heterozygous mutations did not visibly affect the ddaC neuron (Fig. 1I), expression of VCP RNAi in vcp7-12/+ animals frequently led to a drastic reduction of the dendritic field and also induced lethality, as neurons were sometimes missing or displayed blebbing and fragmenting dendrites (drastic dendrite reduction or cell death occurred in 37% of all neurons; Fig. 1J). Similar results were seen when the copy number of VCP QQ was increased (strong defects occurred in 64% of all neurons; Fig. 1K). We also performed mosaic analysis with a repressible cell marker (MARCM) to test whether cells homozygous for the P-element insertion vcpl(2)15502 showed similar defects. At the third instar stage, the morphology of vcpl(2)15502 mutant neurons was almost wild-type, which was likely to be due to maternal contribution and protein perdurance, although these cells died shortly after the onset of the pupal phase (data not shown).
Fig. 1.

VCP inhibition affects dendrite morphology and cell viability of Drosophila larval da neurons. (A-D) VCP inhibition affects the morphology of class IV neurons. The indicated transgenes were expressed under the control of ppk-GAL4, and the class IV neuron ddaC was visualized by concomitant expression of membrane-bound GFP and imaged live at third instar. (A) Control (ppk-GAL4/+), (B) VCP RNAi (ppk-GAL4/UAS-VCP IR) and (C) VCP QQ (ppk-GAL4/UAS-VCP QQ). (D) Quantification of branching defects upon expression of VCP RNAi or VCP QQ. Data are shown as mean ± s.d., n=4; ***, P<0.0001 (Student's t-test). (E-H) No major effects of VCP inhibition on class III neuronal morphology. The indicated transgenes were expressed under Gal419-12, and the class III neurons ddaF and ddaA were imaged live at third instar. (E) Control (Gal419-12/+), (F) VCP RNAi (Gal419-12/UAS-VCP IR) and (G) VCP QQ (Gal419-12/UAS-VCP QQ). The insets show class III neuron dentrites at higher magnification for better comparison of spike density. (H) Quantification of spike numbers per 30 μm dendrite length for the indicated genotypes. Data are mean ± s.d., n=5. (I-K) Strong VCP inhibition causes severe morphological defects and lethality in class IV neurons. (I) ppk-GAL4/+, dvcp7-12/+ (heterozygous control). (J) Expression of VCP RNAi in a vcp heterozygous background (ppk-GAL4/UAS-VCP IR, dvcp7-12/+). (K) Two copies of UAS-VCP QQ (ppk-GAL4/2x UAS-VCP QQ). Arrows (J,K) indicate fragmenting dendrites. The number of dying or severely affected neurons for each genotype is indicated below each panel (***, P<0.0001; Fisher's exact test). Scale bars: 50 μm.
VCP inhibition induces several cellular stress pathways
To address the mechanism underlying cell death upon strong VCP inhibition, we next asked whether class IV neurons expressing the dominant-negative VCP QQ showed signs of proteotoxic stress. First, we asked whether we could detect signs of impaired proteasomal degradation. When we stained class IV neurons with an antibody against ubiquitin, we found that class IV neurons expressing VCP QQ showed strongly increased ubiquitin levels, indicating that ubiquitylated substrate proteins had accumulated (Fig. 2A,B). We also asked whether stress signaling pathways, such as the pro-apoptotic Jun N-terminal kinase [JNK; also known as stress-activated protein kinase (SAPK) or Basket] pathway or the unfolded protein response (UPR), were activated. To address JNK activation, we used a lacZ-containing enhancer-trap insertion in the JNK target gene puckered (puc-lacZ), which acts as a reporter for JNK activity. puc-lacZ activity was low in control class IV neurons compared with two neighboring class I neurons (ddaD and ddaE), but it was strongly increased in class IV neurons expressing VCP QQ (Fig. 2C,D). In order to assess UPR activation, we expressed an Xbp1-GFP reporter (Ryoo et al., 2007) in class IV neurons. UPR activation induces correct splicing of this construct such that GFP fluorescence becomes detectable upon ER stress. Whereas no GFP could be detected in control neurons, neurons expressing VCP QQ frequently showed GFP signals in the nucleus. Thus, consistent with previous reports (Kobayashi et al., 2002; Ye et al., 2001), VCP inhibition interferes with proteasomal protein degradation and induces JNK signaling and the UPR. Both these signaling pathways can induce apoptosis; for example, JNK signaling can induce the expression of pro-apoptotic genes such as reaper and Hid (Wrinkled – FlyBase) (McEwen and Peifer, 2005). We conclude that, depending on the strength of VCP inhibition, these stresses can eventually lead to cell death.
Fig. 2.

VCP inhibition induces proteotoxic stress in class IV neurons. (A,B) Ubiquitin staining of Drosophila third instar ppk neurons expressing different VCP transgenes. (A) The dorsal da neuron cluster of control animals and animals expressing wild-type VCP or VCP QQ in class IV neurons was stained with anti-ubiquitin antibodies (red). Genotypes from left to right: ppk-GAL4/+; ppk-GAL4/UAS-VCP wt; ppk-GAL4/UAS-VCP QQ. Dorsal da neurons were marked with anti-HRP antibodies (green); the class IV neuron ddaC is indicated by an arrow. (B) Quantification of the stainings in A. The relative pixel intensity of the ubiquitin signal of ddaC was compared with that of a neighboring class I da neuron (ddaE). Mean ± s.e.m., n=3. (C,D) VCP inhibition induces JNK activation in ppk neurons. (C) JNK activity in control ppk neurons or ppk neurons expressing VCP QQ was measured using a puc-lacZ (pucA251.1) reporter. JNK reporter activity was visualized by anti-β-galactosidase staining, and dorsal cluster da neurons were stained with anti-HRP antibodies. Genotypes: left, pucA251.1/+, ppk-GAL4/+; right, pucA251.1/+, ppk-GAL4/UAS-VCP QQ. The position of ddaC is indicated by an arrow. (D) Quantification of β-galactosidase staining shown in C. The quantification was performed as in B. Data are shown as mean ± s.d., n=3. (E) VCP inhibition induces endoplasmic reticulum (ER) stress. The UPR marker Xbp1-GFP was expressed in class IV neurons (labeled by mCD8::RFP). Wild-type VCP (left) does not induce visible GFP fluorescence (ppk-GAL4/UAS-mCD8::RFP, UAS-VCP wt, UAS-Xbp1-GFP), whereas expression of VCP QQ (right) induces a nuclear GFP signal indicative of ER stress in 70% of neurons (ppk-GAL4/UAS-mCD8::RFP, UAS-VCP QQ, UAS-Xbp1-GFP) (n=10 each). ddaC is indicated by an arrow.
VCP is required for neuronal remodeling and apoptosis
During metamorphosis, the larval dendrites of class IV neurons are pruned (i.e. severed and degraded) in a ubiquitin- and caspase-dependent process, and new, adult-type dendrites grow out later (Kuo et al., 2005; Kuo et al., 2006; Williams et al., 2006). As expression of VCP RNAi or one copy of the dominant-negative VCP QQ did not affect cell viability, we were able to examine the effects of VCP inhibition at this stage. At 18 hours after puparium formation (APF), control class IV neurons and neurons overexpressing wild-type VCP had lost all their larval dendrites [0% not severed (n=16) and 2% not severed (n=52), respectively] (Fig. 3A,C). By contrast, a high proportion of class IV neurons expressing VCP RNAi or VCP QQ retained long, attached dendrites [63% not severed (n=65) and 54% not severed (n=54), respectively] (Fig. 3B,D). In order to exclude the possibility that these defects were the consequence of the earlier dendritic morphology defects described above, we sought to inhibit VCP after dendrite morphogenesis was completed using the RU486-inducible GeneSwitch technique (Osterwalder et al., 2001). To this end, we used a ppk-CD4::tdTomato transgene to visualize neuronal morphology and a ppk-GeneSwitch (ppk-GS) transgene to induce the expression of VCP QQ. In the absence of drug, the morphology of class IV neurons appeared wild type at the third instar larval stage (data not shown) and most neurons had pruned their dendrites at 18 hours APF [defects occurred in 2/28 neurons (7%); Fig. 3E]. Addition of 100 μM RU486 24 hours before the onset of metamorphosis did not visibly change the morphology of the larval neurons (data not shown); however, a high proportion of class IV neurons in these drug-treated animals retained their dendrites at 18 hours APF [defects occurred in 24/34 neurons (70%); Fig. 3F], indicating that the phenotypes observed during metamorphosis can be disassociated from the defects at the larval stage.
Fig. 3.

VCP is required for dendrite pruning and apoptosis during Drosophila metamorphosis. Class IV neurons (ddaC) or class III neurons (ddaA and ddaF) expressing the indicated combinations of transgenes were imaged at 18 hours APF. (A-D) Effects of VCP inhibition on class IV neuron dendrite pruning. (A) Control (ppk-GAL4/+), (B) VCP RNAi (ppk-GAL4/UAS-VCP IR), (C) overexpression of wild-type VCP (ppk-GAL4/UAS-VCP wt) and (D) VCP QQ (ppk-GAL4/UAS-VCP QQ). The penetrance of pruning defects for each genotype is given under each panel (***, P<0.0001; Fisher's exact test). (E,F) Inducible VCP inhibition using ppk-GeneSwitch. Class IV neuron morphology was visualized with a ppk-CD4::tdTomato transgene (genotype ppk-GS/UAS-VCP QQ, ppk-CD4::tdTomato). VCP QQ expression was induced 24 hours before pupariation with (F) or without (E, control) 100 μM RU486. (G-K) VCP inhibition inhibits apoptosis of class III neurons during metamorphosis. (G) Control (Gal419-12/+), (H) class III neurons expressing p35 (Gal419-12/UAS-p35), (I) VCP RNAi (Gal419-12/UAS-VCP IR), (J) wild-type VCP (Gal419-12/UAS-VCP wt) and (K) VCP QQ (Gal419-12/UAS-VCP QQ). Control class III neurons or class III neurons expressing wild-type VCP have undergone apoptosis (G,J). Cell bodies of surviving class III neurons are marked by arrows (H,I,K). The number of surviving class III neurons is given below each panel (***, P<0.0001; Fisher's exact test). Scale bars: 50 μm.
We next examined the effect of VCP inhibition on class III neurons, which normally undergo developmental apoptosis soon after puparium formation such that they are completely removed at 18 hours APF (Williams and Truman, 2005). Control class III neurons or class III neurons overexpressing wild-type VCP had indeed undergone apoptosis and disappeared at this stage (0/48 surviving; Fig. 3G,J), whereas expression of the baculovirus caspase inhibitor p35 (Hay et al., 1994) caused most class III neurons to survive [49/56 (87.5%); Fig. 3H]. Interestingly, expression of VCP RNAi or VCP QQ inhibited apoptosis in a large proportion of class III neurons, allowing them to persist until 18 hours APF (Fig. 3I,K). In these experiments, the ddaF neuron was more prone to survive than ddaA, and VCP QQ was more potent in promoting survival than VCP RNAi [54/82 (66%) survived versus 18/50 (36%)]. Thus, mild VCP inhibition interferes with class IV neuron dendrite pruning and class III neuron apoptosis during the pupal stage.
VCP inhibition causes defects in caspase activation and degradation of the caspase inhibitor DIAP1
Both dendrite pruning and developmental apoptosis require caspase activity (Kuo et al., 2006; Williams et al., 2006; Williams and Truman, 2005). Caspase activity can be detected in severing class IV neuron dendrites and dying class III neurons during the early pupal stage at ~4-6 hours APF. We asked whether caspase activity and the levels of apoptotic regulators were changed in these neurons when VCP was inhibited. To visualize caspase activity, we either used a caspase reporter construct based on mammalian poly(ADP-ribose) polymerase 1 (PARP1), a known caspase substrate (Williams et al., 2006), or an antibody that recognizes cleaved mammalian and Drosophila caspases (Yu et al., 2002). At 5 hours APF, control class IV neuron dendrites showed swellings and started to fragment, and caspase activity could often be detected in fragmented dendrites with the caspase reporter (Fig. 4A,A′). When we expressed VCP QQ, at 5 hours APF class IV dendrites developed substantially fewer swellings, did not fragment and did not contain detectable caspase activity (Fig. 4B,B′). However, in class IV neurons expressing VCP QQ, we sometimes detected low levels of reporter activation around the nucleus, possibly indicating caspase activation by elevated stress levels (Fig. 4B,B′). Next, we assessed caspase activity in class III neurons at 4 hours APF. All control class III neurons examined showed high levels of active caspases in the soma, indicating that they were undergoing apoptosis (n=11; Fig. 4C,C′). By contrast, expression of VCP QQ in class III neurons strongly suppressed this activity, such that these neurons did not display caspase activity (n=12; Fig. 4D,D′).
Fig. 4.

VCP inhibition affects caspase activity during early metamorphosis. (A-B′) Caspase activity in class IV neurons was detected 4-6 hours APF with the reporter construct CD8::PARP::Venus. (A,A′) Control ddaC neuron (ppk-GAL4/UAS-CD8::Parp::Venus) and (B,B′) ddaC neuron expressing VCP QQ (ppk-GAL4/UAS-CD8::PARP::Venus, UAS-VCP QQ) (n=10 for each sample). The brackets (A,A′) indicate cleaved PARP in dendrites. (C-D′) Caspase activity in the class III neurons ddaA and ddaF (which is more dorsal) at 4 hours APF was detected with an antibody against activated caspases. (C,C′) Class III neurons overexpressing wild-type VCP (Gal419-12/UAS-VCP wt) and (D,D′) class III neurons expressing VCP QQ (Gal419-12/UAS-VCP QQ) (n=11 for each sample). Arrows (A-D′) indicate the positions of cell bodies (with weak caspase activity in or around the nucleus in B,B′). Scale bars: 50 μm in A-B′; 20 μm in C-D′.
DIAP1 is a crucial regulator of apoptosis in flies (Wang et al., 1999). High DIAP1 levels inhibit caspase activation, and DIAP1 degradation induces apoptosis (Ryoo et al., 2002; Yin and Thummel, 2004; Yoo et al., 2002) and dendrite pruning (Kuo et al., 2006; Lee et al., 2009). Although the limited resolution of the immunofluorescence experiments, especially at the pupal phase, prevented us from assessing DIAP1 levels in class IV neuron dendrites, DIAP1 levels could be easily examined in class III neurons. Consistent with the results of the caspase immunostaining, control class III neurons had very low levels of DIAP1 at 4 hours APF, as compared with adjacent cells (Fig. 5A,A′). By contrast, class III neurons expressing VCP QQ displayed much higher DIAP1 levels, which were comparable to those of adjacent cells (Fig. 5B,B′) and about four times higher than in control neurons (Fig. 5C).
Fig. 5.

Evidence that VCP affects DIAP1 during developmental apoptosis and pruning. (A-C) DIAP1 levels are upregulated in class III neurons during early metamorphosis (4 hours APF). (A,A′) DIAP1 levels in the class III neuron ddaF expressing wild-type VCP (Gal419-12/UAS-VCP wt). DIAP1 levels in the ddaF soma are lower than those in an adjacent cell (probably ddaC) and the cell body has rounded up. (B,B′) ddaF neuron expressing VCP QQ (Gal419-12/UAS-VCP QQ). DIAP1 levels are comparable to those in the adjacent cell and the ddaF soma retains its normal shape. (C) Quantification of normalized DIAP1 staining intensities in class III neurons expressing wild-type VCP or VCP QQ. Data are shown as mean ± s.d., n=5. (D-F) The DIAP1 mutant allele th4 is a suppressor of VCP during dendrite pruning. Class IV da neurons were imaged at 15 hours APF. (D) Class IV neuron in a th4/+ heterozygous background (th4/+, ppk-GAL4/+), (E) a class IV neuron expressing VCP QQ in the wild-type background (+/+, ppk-GAL4/UAS-VCP QQ) and (F) a class IV neuron expressing VCP QQ in a heterozygous DIAP1 mutant background (th4/+, ppk-GAL4/UAS-VCP QQ). The numbers of unpruned neurons are given below each panel. Scale bars: 10 μm in A-B′; 50 μm in D.
Next, we assessed a potential link between VCP and DIAP1 using a genetic approach. If impaired caspase activation through blocked DIAP1 degradation is the reason for the pruning defects upon VCP inhibition, then these defects should be reversed by lowering DIAP1 levels genetically. To this end, we expressed VCP QQ in class IV neurons either in wild type or in a heterozygous diap1 mutant background (th4/+) that should decrease the amount of DIAP1 by half. As expected, heterozygous th4 did not affect pruning (2.6% pruning defects, n=38) (Fig. 5D), but a high proportion of neurons expressing VCP QQ in the wild-type background still had attached dendrites at 15 hours APF (59%, n=19; P<0.0005) (Fig. 5E). By contrast, the majority of class IV neurons expressing VCP QQ in the th4/+ background had pruned their dendrites (12.5% still attached at 15 hours APF, n=24) (Fig. 5F), indicating that lowering DIAP1 levels suppressed the pruning defects induced by VCP inhibition. Notably, lowering DIAP1 levels also sensitizes cells to stress-induced apoptosis, and a high proportion of the VCP QQ-expressing neurons in the th4/+ background underwent apoptosis during early metamorphosis (~60% at 15 hours APF). Taken together, inhibition of VCP in class III and class IV neurons inhibits caspase activation, probably through an effect on DIAP1.
VCP is required for DIAP1 degradation in cultured Drosophila cells
Our histochemical and genetic data suggest that VCP might be involved in the degradation of DIAP1. In order to test this hypothesis further, we performed biochemical experiments. When we expressed VCP QQ in S2 cells, the levels of endogenous DIAP1 were increased (Fig. 6A). In the following experiments, we used an HA-tagged form of DIAP1 for ease of detection. When we co-expressed HA-tagged DIAP1 with VCP QQ, we observed additional HA-reactive high molecular weight species in western blots, indicative of the accumulation of poly-ubiquitylated DIAP1 (data not shown). To verify this, we co-expressed tagged DIAP1 with wild-type VCP or VCP QQ and immunoprecipitated DIAP1 under stringent buffer conditions. Anti-ubiquitin blots of the precipitates showed that poly-ubiquitylated DIAP1 did indeed accumulate upon VCP inhibition, indicating a defect in the degradation of ubiquitylated DIAP1 (Fig. 6B). In order to test whether DIAP1 and VCP interact with each other or can be found in the same protein complexes, we performed co-immunoprecipitation experiments with tagged VCP and DIAP1. In these experiments, a weak interaction between the two factors could indeed be detected (Fig. 6C). Furthermore, the DIAP1-VCP interaction was strongly enhanced with the dominant-negative VCP QQ (Fig. 6C), which acts as a substrate trap (Ye et al., 2001).
Fig. 6.
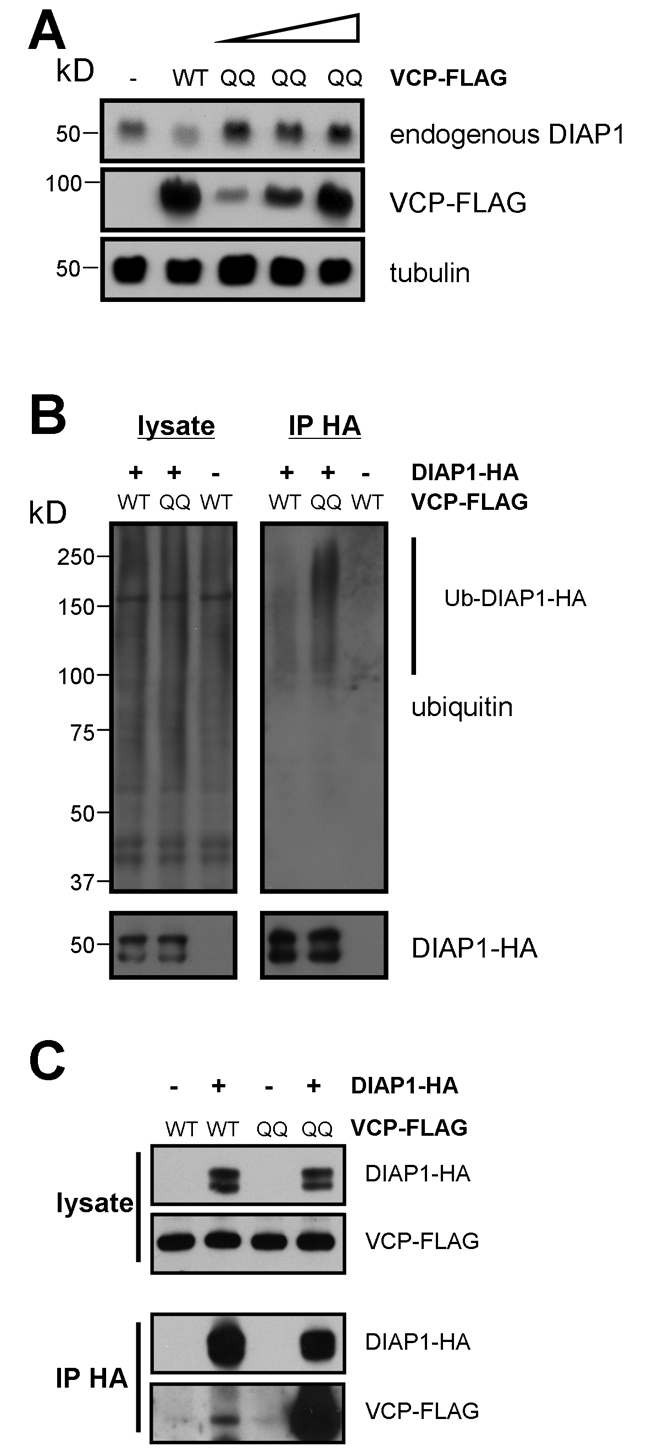
VCP is required for DIAP1 degradation in S2 cells. (A) VCP QQ expression leads to an accumulation of endogenous DIAP1. Drosophila S2 cells were transfected with wild-type VCP (0.8 μg) or increasing amounts of VCP QQ (0.2-0.8 μg), and cell extracts were probed with anti-DIAP1 antibodies. Tubulin served as a loading control. (B) Accumulation of ubiquitylated DIAP1 upon VCP inhibition. DIAP1-HA immunoprecipitates from cells expressing the indicated VCP constructs were blotted against ubiquitin. (C) Interactions between DIAP1 and VCP. DIAP1-HA and FLAG-tagged VCP constructs were expressed in S2 cells. HA immunoprecipitates (IP HA) were blotted with the indicated antibodies.
Molecular determinants of the DIAP1-VCP interaction
VCP is usually involved in ubiquitin-mediated degradation when a ubiquitylated substrate protein is part of a tight protein complex, or when it is tightly folded. To explore why VCP might be required for DIAP1 degradation, we tested whether DIAP1 forms oligomers or whether VCP QQ would change a protein interaction with a binding partner. We found that DIAP1 dimerizes (or oligomerizes) in a manner dependent on its RING domain, but only to a relatively low extent (see Fig. S3A in the supplementary material). Furthermore, the presence of VCP QQ did not alter the interaction of DIAP1 with the caspase Dronc (Nedd2-like caspase – FlyBase) (see Fig. S3B in the supplementary material), indicating that VCP might not be required to disrupt DIAP1-containing protein complexes.
Next, we asked whether specific DIAP1 domains are required for the interaction with VCP. DIAP1 has three major domains (Fig. 7A): two baculovirus inhibitor of apoptosis repeat (BIR) domains (BIR1 and BIR2 at residues 42-112 and 224-295, respectively), which are required for interactions with caspases and the RHG proteins Reaper, Hid and Grim; and a C-terminal RING finger domain that is required for the ubiquitylation of bound caspases and for the auto-ubiquitylation of DIAP1 under apoptosis-inducing conditions. We focused on the roles of the BIR1 and RING domains because both are located relatively close to the respective N- and C-termini of DIAP1 and might thus represent obstacles for proteasome-dependent unfolding. In addition, auto-ubiquitylation catalyzed by the RING domain might contribute to VCP binding. BIR and RING domains contain Zn2+ ions that are chelated by cysteine residues. We disrupted the BIR1 domain by replacing one of the coordinating cysteines with serine (C107S). This mutation lowered the levels of DIAP1, owing to increased proteasomal degradation (Fig. 7B). Interestingly, expression of VCP QQ did not increase the levels of DIAP1 C107S, indicating that the degradation of DIAP1 C107S was independent of VCP (Fig. 7B).
Fig. 7.

DIAP1 domains required for interaction with VCP. (A) Domain structure of DIAP1. The approximate positions of the mutations in C107S and C406S are indicated by asterisks. (B) Disrupton of the BIR1 domain destabilizes DIAP1. HA-tagged DIAP1 or DIAP1 C107S was expressed in Drosophila S2 cells and equal amounts of cell lysate were analyzed by SDS-PAGE. Where indicated, the proteasome inhibitor MG132 was added, or wild-type VCP or VCP QQ were co-expressed with DIAP1. The loading control was a cross-reactive band from a GAPDH antibody. (C) Effect of a RING finger mutation on the DIAP1 ubiquitylation status. HA-tagged DIAP1 or DIAP1 C406S was expressed in S2 cells together with VCP or VCP QQ, and DIAP1 was immunoprecipitated as in Fig. 6B. Immunoprecipitates were analyzed by blotting against HA and ubiquitin. (D) Effect of DIAP1 BIR1 and RING mutations on the interaction with VCP QQ. Wild-type DIAP1 or the BIR1 and RING mutants were co-transfected with VCP QQ and immunoprecipitated as in Fig. 6C. ‘2×’ indicates that twice as much BIR1 mutant immunoprecipitate was loaded to correct for the different expression levels. See also Fig. S3 in the supplementary material. (E) Model for the biphasic effects of VCP inhibition in neuronal viability, apoptosis and pruning. (F) Hypothetical mechanism for VCP-dependent DIAP1 degradation. The BIR1 domain stabilizes DIAP1 and necessitates VCP involvement to facilitate BIR1 domain unfolding and degradation. Ub, ubiquitin chain.
Next, we asked whether replacing one of the coordinating cysteines in the RING domain (C406S) would alter DIAP1 ubiquitylation. As shown above, ubiquitylated DIAP1 can be detected in S2 cells and ubiquitylated DIAP1 accumulates upon VCP inhibition. The DIAP1 C406S mutation strongly decreased the amount of ubiquitylated DIAP1, indicating that auto-ubiquitylation is responsible for a large portion of the DIAP1-ubiquitin conjugates (Fig. 7C). Residual ubiquitylation of DIAP1 C406S is consistent with the existence of several ubiquitin ligases for DIAP1 (Herman-Bachinsky et al., 2007). When we assessed the ability of these two DIAP1 mutants to interact with VCP QQ, both showed a strongly reduced interaction (Fig. 7D). These results suggest that a functional BIR1 domain and ubiquitylation are necessary for DIAP1 to be recognized as a VCP substrate.
DISCUSSION
In this study, we have analyzed the role of VCP during the development of peripheral da neurons in Drosophila. We found that VCP inhibition affects neural development and cell viability in a biphasic manner. Mild VCP inhibition causes defects in caspase activation and therefore affects apoptosis and pruning. This effect is caused by interference with the ubiquitin-dependent degradation of the caspase inhibitor DIAP1, as our data suggest that DIAP1 is a VCP substrate. By contrast, strong VCP inhibition causes severe morphological defects and cell death, probably owing to increased proteotoxic stress. A likely explanation for these seemingly contradicting phenotypes is that VCP inhibition activates pro-apoptotic signaling cascades, such as JNK signaling or the UPR. Upon strong VCP inhibition, these signals eventually override the anti-apoptotic effects through slowed DIAP1 degradation (Fig. 7E). Importantly, caspase activation can occur in the presence of DIAP1; for example, the Hid protein can displace caspases from DIAP1 and thereby activate them (Wang et al., 1999). Similar biphasic phenotypes have been reported for the ubiquitin-activating enzyme E1 (Uba1 – FlyBase): hypomorphic E1 alleles support cell viability and inhibit apoptosis via stabilization of DIAP1, whereas strong loss-of-function alleles have effects on mitosis and cell viability (Lee et al., 2008). It is interesting to speculate whether our results could be relevant for the pathogenesis of VCP-related neurodegeneration. In fact, we observed mild pruning defects in class IV neurons upon expression of a VCP disease variant (VCP R152H) (see Fig. S4 in the supplementary material). Although these defects were relatively subtle, similar defects in neuronal remodeling in humans could contribute to dementia. In addition, our results suggest that VCP mutation might induce dementia not only through stress-induced cell death but also through inhibition of other VCP-dependent neuronal processes.
Importantly, our work has identified VCP as a new regulator of DIAP1 degradation. An interesting question is how VCP might contribute to DIAP1 degradation. Our biochemical analysis suggests that an intact BIR1 domain is a major determinant for VCP binding. As our results suggest that VCP is not required to break up an interaction between DIAP1 and a binding partner, we speculate that it might be required to unfold ubiquitylated DIAP1, and specifically the BIR1 domain, prior to proteasomal degradation (Fig. 7F), as has been proposed for a GFP-based model substrate (Beskow et al., 2009). A stable BIR1 domain, in turn, might be favorable because caspase cleavage in the DIAP1 N-terminus (after amino acid 20) exposes an N-end rule degradation signal (Ditzel et al., 2003).
In other VCP-dependent pathways, such as ERAD, VCP often requires adaptor proteins to perform its functions. We also tested RNAi lines directed against the VCP interactors Ufd1-like, Npl4 (CG4673), p47 and Ufd2 (CG9934) for their effects on dendrite pruning but did not observe defects with any of these lines, indicating that VCP might not need adaptors to act on DIAP1; alternatively, other, as yet unknown adaptors might be involved.
Taken together, we have identified VCP as a new regulator of neuronal remodeling and developmental apoptosis, and we have identified DIAP1 as the relevant substrate. Our data therefore provide a new link between the ubiquitin system and apoptosis.
Supplementary Material
Acknowledgements
We thank Hsui-Hsiang Lee and Sijun Zhu for experimental advice; Anshuman Kelkar for comments on the manuscript; Rebecca Yang and Jay Parrish for ppk-GeneSwitch; Hsui-Hsiang Lee and Yang Xiang for Gal419-12; and Dennis McKearin, Darren Williams, Bruce Hay, Hermann Steller, Jay Brenman, Wei Song and Peter Soba as well as the Vienna Drosophila RNAi Center (VDRC) and the Bloomington Stock Center for other fly lines and reagents. This work was supported by a postdoctoral fellowship from the German Academic Exchange Service (DAAD) to S.R. and an NIH grant (R 37NS040929) to Y.N.J. L.Y.J. and Y.N.J. are investigators of the Howard Hughes Medical Institute. Deposited in PMC for release after 6 months.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.062703/-/DC1
References
- Beskow A., Grimberg K. B., Bott L. C., Salomons F. A., Dantuma N. P., Young P. (2009). A conserved unfoldase activity for the p97 AAA-ATPase in proteasomal degradation. J. Mol. Biol. 394, 732-746 [DOI] [PubMed] [Google Scholar]
- Brand A. H., Perrimon N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401-415 [DOI] [PubMed] [Google Scholar]
- Dietzl G., Chen D., Schnorrer F., Su K. C., Barinova Y., Fellner M., Gasser B., Kinsey K., Oppel S., Scheiblauer S., et al. (2007). A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448, 151-156 [DOI] [PubMed] [Google Scholar]
- Ditzel M., Wilson R., Tenev T., Zachariou A., Paul A., Deas E., Meier P. (2003). Degradation of DIAP1 by the N-end rule pathway is essential for regulating apoptosis. Nat. Cell Biol. 5, 467-473 [DOI] [PubMed] [Google Scholar]
- Dutta D., Bloor J. W., Ruiz-Gomez M., VijayRaghavan K., Kiehart D. P. (2002). Real-time imaging of morphogenetic movements in Drosophila using Gal4-UAS-driven expression of GFP fused to the actin-binding domain of moesin. Genesis 34, 146-151 [DOI] [PubMed] [Google Scholar]
- Goldstein E. S., Treadway S. L., Stephenson A. E., Gramstad G. D., Keilty A., Kirsch L., Imperial M., Guest S., Hudson S. G., LaBell A. A., et al. (2001). A genetic analysis of the cytological region 46C-F containing the Drosophila melanogaster homolog of the jun proto-oncogene. Mol. Genet. Genomics 266, 695-700 [DOI] [PubMed] [Google Scholar]
- Grueber W. B., Jan L. Y., Jan Y. N. (2002). Tiling of the Drosophila epidermis by multidendritic sensory neurons. Development 129, 2867-2878 [DOI] [PubMed] [Google Scholar]
- Grueber W. B., Ye B., Yang C. H., Younger S., Borden K., Jan L. Y., Jan Y. N. (2007). Projections of Drosophila multidendritic neurons in the central nervous system: links with peripheral dendrite morphology. Development 134, 55-64 [DOI] [PubMed] [Google Scholar]
- Hay B. A., Wolff T., Rubin G. M. (1994). Expression of baculovirus P35 prevents cell death in Drosophila. Development 120, 2121-2129 [DOI] [PubMed] [Google Scholar]
- Hay B. A., Wassarman D. A., Rubin G. M. (1995). Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell 83, 1253-1262 [DOI] [PubMed] [Google Scholar]
- Herman-Bachinsky Y., Ryoo H. D., Ciechanover A., Gonen H. (2007). Regulation of the Drosophila ubiquitin ligase DIAP1 is mediated via several distinct ubiquitin system pathways. Cell Death Differ. 14, 861-871 [DOI] [PubMed] [Google Scholar]
- Janiesch P. C., Kim J., Mouysset J., Barikbin R., Lochmuller H., Cassata G., Krause S., Hoppe T. (2007). The ubiquitin-selective chaperone CDC-48/p97 links myosin assembly to human myopathy. Nat. Cell Biol. 9, 379-390 [DOI] [PubMed] [Google Scholar]
- Jentsch S., Rumpf S. (2007). Cdc48 (p97): a ‘molecular gearbox’ in the ubiquitin pathway? Trends Biochem. Sci. 32, 6-11 [DOI] [PubMed] [Google Scholar]
- Ju J. S., Fuentealba R. A., Miller S. E., Jackson E., Piwnica-Worms D., Baloh R. H., Weihl C. C. (2009). Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 187, 875-888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Tanaka K., Inoue K., Kakizuka A. (2002). Functional ATPase activity of p97/valosin-containing protein (VCP) is required for the quality control of endoplasmic reticulum in neuronally differentiated mammalian PC12 cells. J. Biol. Chem. 277, 47358-47365 [DOI] [PubMed] [Google Scholar]
- Kuo C. T., Jan L. Y., Jan Y. N. (2005). Dendrite-specific remodeling of Drosophila sensory neurons requires matrix metalloproteases, ubiquitin-proteasome, and ecdysone signaling. Proc. Natl. Acad. Sci. USA 102, 15230-15235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo C. T., Zhu S., Younger S., Jan L. Y., Jan Y. N. (2006). Identification of E2/E3 ubiquitinating enzymes and caspase activity regulating Drosophila sensory neuron dendrite pruning. Neuron 51, 283-290 [DOI] [PubMed] [Google Scholar]
- Lee H. H., Jan L. Y., Jan Y. N. (2009). Drosophila IKK-related kinase Ik2 and Katanin p60-like 1 regulate dendrite pruning of sensory neuron during metamorphosis. Proc. Natl. Acad. Sci. USA 106, 6363-6368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T., Luo L. (1999). Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22, 451-461 [DOI] [PubMed] [Google Scholar]
- Lee T. V., Ding T., Chen Z., Rajendran V., Scherr H., Lackey M., Bolduc C., Bergmann A. (2008). The E1 ubiquitin-activating enzyme Uba1 in Drosophila controls apoptosis autonomously and tissue growth non-autonomously. Development 135, 43-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon A., McKearin D. (1999). Identification of TER94, an AAA ATPase protein, as a Bam-dependent component of the Drosophila fusome. Mol. Biol. Cell 10, 3825-3834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen D. G., Peifer M. (2005). Puckered, a Drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development 132, 3935-3946 [DOI] [PubMed] [Google Scholar]
- Medina P. M., Swick L. L., Andersen R., Blalock Z., Brenman J. E. (2006). A novel forward genetic screen for identifying mutations affecting larval neuronal dendrite development in Drosophila melanogaster. Genetics 172, 2325-2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H. H., Shorter J. G., Seemann J., Pappin D., Warren G. (2000). A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 19, 2181-2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev A., McLaughlin T., O'Leary D. D., Tessier-Lavigne M. (2009). APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457, 981-989 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Osterwalder T., Yoon K. S., White B. H., Keshishian H. (2001). A conditional tissue-specific transgene expression system using inducible GAL4. Proc. Natl. Acad. Sci. USA 98, 12596-12601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadan K., Bruderer R., Spiga F. M., Popp O., Baur T., Gotta M., Meyer H. H. (2007). Cdc48/p97 promotes reformation of the nucleus by extracting the kinase Aurora B from chromatin. Nature 450, 1258-1262 [DOI] [PubMed] [Google Scholar]
- Ruden D. M., Sollars V., Wang X., Mori D., Alterman M., Lu X. (2000). Membrane fusion proteins are required for oskar mRNA localization in the Drosophila egg chamber. Dev. Biol. 218, 314-325 [DOI] [PubMed] [Google Scholar]
- Ryoo H. D., Bergmann A., Gonen H., Ciechanover A., Steller H. (2002). Regulation of Drosophila IAP1 degradation and apoptosis by reaper and ubcD1. Nat. Cell Biol. 4, 432-438 [DOI] [PubMed] [Google Scholar]
- Ryoo H. D., Domingos P. M., Kang M. J., Steller H. (2007). Unfolded protein response in a Drosophila model for retinal degeneration. EMBO J. 26, 242-252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuberth C., Buchberger A. (2008). UBX domain proteins: major regulators of the AAA ATPase Cdc48/p97. Cell. Mol. Life Sci. 65, 2360-2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresse E., Salomons F. A., Vesa J., Bott L. C., Kimonis V., Yao T. P., Dantuma N. P., Taylor J. P. (2010). VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6, 217-227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. L., Hawkins C. J., Yoo S. J., Muller H. A., Hay B. A. (1999). The Drosophila caspase inhibitor DIAP1 is essential for cell survival and is negatively regulated by HID. Cell 98, 453-463 [DOI] [PubMed] [Google Scholar]
- Watts G. D., Wymer J., Kovach M. J., Mehta S. G., Mumm S., Darvish D., Pestronk A., Whyte M. P., Kimonis V. E. (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377-381 [DOI] [PubMed] [Google Scholar]
- Williams D. W., Truman J. W. (2005). Cellular mechanisms of dendrite pruning in Drosophila: insights from in vivo time-lapse of remodeling dendritic arborizing sensory neurons. Development 132, 3631-3642 [DOI] [PubMed] [Google Scholar]
- Williams D. W., Kondo S., Krzyzanowska A., Hiromi Y., Truman J. W. (2006). Local caspase activity directs engulfment of dendrites during pruning. Nat. Neurosci. 9, 1234-1236 [DOI] [PubMed] [Google Scholar]
- Ye Y., Meyer H. H., Rapoport T. A. (2001). The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414, 652-656 [DOI] [PubMed] [Google Scholar]
- Yin V. P., Thummel C. S. (2004). A balance between the diap1 death inhibitor and reaper and hid death inducers controls steroid-triggered cell death in Drosophila. Proc. Natl. Acad. Sci. USA 101, 8022-8027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S. J., Huh J. R., Muro I., Yu H., Wang L., Wang S. L., Feldman R. M., Clem R. J., Muller H. A., Hay B. A. (2002). Hid, Rpr and Grim negatively regulate DIAP1 levels through distinct mechanisms. Nat. Cell Biol. 4, 416-424 [DOI] [PubMed] [Google Scholar]
- Yu S. Y., Yoo S. J., Yang L., Zapata C., Srinivasan A., Hay B. A., Baker N. E. (2002). A pathway of signals regulating effector and initiator caspases in the developing Drosophila eye. Development 129, 3269-3278 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}