

Abstract
Peroxisome proliferators-activated receptor gamma (PPARG) ligands improve insulin sensitivity in type 2 diabetes and polycystic ovarian syndrome (PCOS). Despite clinical studies showing normalization of pituitary responsiveness to gonadotropin-releasing hormone (GnRH) in patients with PCOS, the precise role of PPARG in regulating the hypothalamic-pituitary-gonadal axis remains unclear. In the present study, we tested the hypothesis that the PPARG agonist rosiglitazone has a direct effect on the pituitary. In mouse LbetaT2 immortalized gonadotrophs, rosiglitazone treatment inhibited GnRH stimulation of the stress kinases p38MAPK and MAPKs/JNKs, but did not alter activation of ERKs, both in the presence and absence of activin. Furthermore, p38MAPK signaling was critical for both Lhb and Fshb promoter activity, and rosiglitazone suppressed the GnRH-mediated induction of Lhb and Fshb mRNA. Depletion of PPARG using a lentivirally encoded short hairpin RNA abolishes the effect of rosiglitazone to suppress activation of JNKs and induction of the transcription factors EGR1 and FOS as well as the gonadotropin genes Lhb and Fshb. Lastly, we show conditional knockout of Pparg in pituitary gonadotrophs caused an increase in luteinizing hormone levels in female mice, a decrease in follicle-stimulating hormone in male mice, and a fertility defect characterized by reduced litter size. Taken together, our data support a direct role for PPARG in modulating pituitary function in vitro and in vivo.
Keywords: female fertility, follicle-stimulating hormone, gonadotropin, gonadotropin-releasing hormone, luteinizing hormone, NR4A1 (Nur77), PPARG, PCOS, pituitary, pituitary hormones, rosiglitazone
PPARG represses LH and FSH expression in vitro, and a null mutation causes elevated LH in females as well as decreased litter sizes.
INTRODUCTION
Thiazolidinediones (TZDs) are synthetic ligands for the peroxisome proliferators-activated receptor γ (PPARG) and are used clinically to treat insulin resistance in type 2 diabetes [1–4] and polycystic ovarian syndrome (PCOS). The beneficial clinical effects of TZDs include increased insulin sensitivity, decreased blood pressure, correction of dyslipidemia, and improvement in inflammation [5, 6]. Although TZDs generally are thought to improve insulin sensitivity through effects on skeletal muscle and adipose tissue, TZDs are now also known to have a number of biological effects in other systems. For example, PPARG is expressed in macrophage cells of the immune system, and TZDs exert anti-inflammatory effects by suppressing macrophage gene expression. Indeed, recent studies suggest that a major portion of the beneficial clinical effects of TZDs may result from their anti-inflammatory properties.
Polycystic ovarian syndrome is a common endocrine disorder affecting 5–10% of women of reproductive age and is the major cause of anovulation and infertility [7]. In PCOS, inappropriate pituitary gonadotropin secretion leads to increased circulating luteinizing hormone (LH) and normal or decreased follicle-stimulating hormone (FSH) levels [8, 9]. Basal LH secretion is elevated, and pituitary LH responsiveness to gonadotropin-releasing hormone (GnRH) is exaggerated. However, it is not known whether this reflects a hypothalamic or a pituitary defect. Genetic studies have also linked polymorphisms in PPARG to PCOS, which provides additional support for a connection between PPARG activation and hypothalamic-pituitary-gonadal (HPG) axis function [10, 11]. Insulin-sensitizer therapy with TZDs in PCOS corrects the underlying insulin resistance and metabolic defects, decreases serum LH and androgen levels, and increases ovulation rate [12, 13]. Although the insulin-sensitizing effects of these drugs are well documented [12, 14], neither the mechanism underlying this amelioration nor the site of action on the HPG axis is understood. Clinical studies by Mehta et al. [15] showed that pioglitazone therapy reduces peak serum LH responses to multidose GnRH, which indicates that absolute pituitary responsiveness, but not sensitivity, is altered in vivo. This could be a direct effect of TZDs on the pituitary or an indirect effect resulting from changes in circulating androgens that might in turn alter feedback to the hypothalamus.
In view of the multiple potential actions of TZDs, we were interested in examining the mechanism by which PPARG agonists regulate reproductive hormone responses. We hypothesized that TZDs might have direct effects on the pituitary to alter gonadotropin synthesis and secretion. The LβT2 immortalized gonadotroph cells express the Lhb and Fshb subunit genes and respond to GnRH and activin. They also express PPARG and respond to rosiglitazone and so are a useful model to study the interaction of PPARG and GnRH signaling [16]. In the present study, we show that treatment of LβT2 cells with rosiglitazone suppresses the GnRH activation of the stress kinases p38 mitogen-activated protein kinases (MAPKs) and JNKs and the expression of the gonadotropin Lhb and Fshb subunit genes. We also show that pituitary-specific deletion of PPARG leads to elevated LH levels in female mice and a fertility defect characterized by reduced litter size, supporting the idea that PPARG can modulate pituitary function and alter the HPG axis.
MATERIALS AND METHODS
Materials
The GnRH was from Dr. A.F. Parlow (National Hormone and Pituitary Program, Harbor UCLA Medical Center, Torrance, CA). The inhibitors PD98059, SP600125, and PD169316 were purchased from Calbiochem. Activin A was purchased from R&D Systems. Antibodies to phosphorylated and total p38MAPKs, JNKs, ERKs, ATF2, PPARG, phosphorylated c-jun (JUN), CREB, and SRF were from Cell Signaling Technology. β-Tubulin and horseradish peroxidase (HRP)-linked secondary antibodies were purchased from Santa Cruz Biotechnology. All other reagents were purchased from either Sigma or Fisher Scientific. Vehicle for all experiments was dimethyl sulfoxide (DMSO), unless stated otherwise.
Cell Culture and Agonist Treatment
The LβT2 cells were maintained in monolayer cultures in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and antibiotics in a humidified 10% CO2 atmosphere at 37°C. Cells were treated with 10 μM rosiglitazone for 24, 48, or 72 h. Twenty-four hours before GnRH stimulation, the cells were starved in media containing 0.1% bovine serum albumin (BSA) and simultaneously treated with activin A (25 ng/ml) in the starvation media for 24 h wherever indicated. GnRH was added at the indicated concentrations for 30 min. Inhibitors of MEKs (PD98059: 5, 10, and 20 μM), JNKs (SP600125: 2.5, 5, and 10 μM), and p38MAPKs (PD169316: 1.2, 2.5, and 5 μM) were added 30 min before GnRH stimulation.
Western Blot Analysis
After treatment, cells were washed once with chilled PBS and solubilized in radioimmunoprecipitation lysis buffer containing a Protease inhibitor cocktail (Roche), 2% SDS, and 4% β-mercaptoethanol. Lysates were then subjected to SDS-PAGE and transferred onto a polyvinylidene fluoride membrane (Immobilon-P; Millipore). Following blocking with 5% milk in Tris-buffered saline containing 0.1% Tween 20, membranes were probed with primary antibodies at the dilutions recommended by the manufacturer and subsequently with HRP-linked secondary antibodies for chemiluminescent detection (Pierce). Following phospho-protein analysis, blots were stripped in 62.5 mM Tris-HCl (pH 6.7), 100 mM β-mercaptoethanol, and 2% SDS in a shaking water bath at 50°C for 30 min and reprobed for respective total proteins.
Plasmids and Transfections
The LβT2 cells were treated with rosiglitazone (10 μM) or DMSO as vehicle for a total of 72 h. After 24 h of rosiglitazone treatment, the cells were transiently transfected with 500 ng of 1.8-kb rat Lhb-luc reporter gene [17] or 1-kb mouse Fshb-luc reporter gene [18]. As an internal control for transfection efficiency, cells were cotransfected with 50 ng of the TK-LacZ plasmid containing β-galactosidase driven by the herpesvirus thymidine kinase promoter. After overnight incubation, the cells were washed and starved in DMEM medium containing 0.1% BSA and treated with hormones, inhibitors, and agonists as indicated. At the end of 8 h, the cells were washed once with PBS and then lysed. After lysing, the cells were assayed for luciferase activity using a buffer containing 100 mM Tris-HCl (pH 7.8), 15 mM MgSO4, 10 mM ATP, and 65 μM luciferin. β-Galactosidase activity was measured by chemiluminescence (Galacto-Light; Tropix). Both luciferase and β-galactosidase activity were measured using a Veritas microplate luminometer (Turner Design).
Lentivirus Production and Infection of LβT2 Cells
The lentivirus expressing a short hairpin (sh) RNA against Pparg or the control gene luciferase has been described previously [19]. Briefly, the infection of cells with shLuciferase (shLuc) and shPPARG lentivirus was carried out by addition of lentivirus along with polybrene to the LβT2 cell culture. Following overnight incubation, the medium was changed to fresh serum containing medium. After 3–4 days, the confluent cells were trypsinized and centrifuged at 4000 rpm for 5 min. The cell pellet was first washed in PBS and then in PBS containing 5% BSA. Approximately 8 × 106 cells were used for sorting by fluorescence-activated cell sorting. The green fluorescent protein positive cells were collected and cultured until they were confluent.
Real-Time PCR
Total RNA was extracted from the cells using TRIzol (Invitrogen) following the manufacturer's instructions. First-strand cDNA was synthesized using a High Capacity cDNA Synthesis kit (Roche). Samples were run in 20-μl triplicate reactions on an MJ Research Chromo4 instrument using SYBR Green Master Mix (Bio-Rad) and the sequence-specific primers for Lhb (forward, GTGCCGGCTACTGTCCTAGCATGGTC; reverse, CAGCTGAGGGCTACAGGAAAGGAGAC), Fshb (forward, CCACTTGGTGTGCGGGCTACTG; reverse, GTGTAGAGGGAGTCTGAGTGGCGGGC), Egr1 (forward, CCCTATGAGCACCTGACCAC; reverse, TCGTTTGGCTGGGATAACTC), Fos (forward, AACCTGGTGCTGGATTGTAT; reverse, CGAAAGACCTCAGGGTAGAA), Nr4a1 (forward, CTGCACAGCTTGGGTGTTGATGTTCC; reverse, TCCTGGAGCCCGTGTCGATCAGTG), Pparg (forward, CTGTCGGTT TCAGAAGTGCCT; reverse, CCCAAACCTGATGGCATTGTGAGACA), and M36B (forward, ACCCTGAAGTGCTCGACATCACAG; reverse, GCAGGGGCAGCAGCCGCAAATGC) under the following conditions: initial activation of DNA polymerase at 95°C for 3 min, followed by 45 cycles of denaturing at 94°C for 15 sec, annealing at 60°C for 30 sec, and extension at 72°C for 30 sec. Gene expression levels were calculated after normalization to the housekeeping gene, M36B, using the ΔΔCt method and expressed as relative mRNA levels compared with the control.
Generation of Pituitary Pparg Knockout Mice
Pituitary Pparg knockout (PKO) mice were created using the cre/loxP strategy by crossing pituitary-specific Lhb-cre mice [20] with Ppargflox/flox mice (official symbol, Ppargtm2Rev) [21]. Heterozygous mice (Ppargflox/+:Lhb-cre) from the F1 generation were back-crossed with Ppargflox/flox mice or with heterozygous cre-ve littermates to generate PKO mice. Genotypes were determined by PCR of tail DNA as previously described [22]. All mice were housed under controlled light (12L:12D) and temperature (22°C) conditions and had free access to food and water. Fertility was assessed by mating four pairs of PKO or control littermate mice over 6 mo. The number of litters and each litter size were recorded. All procedures were in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee of the University of California, San Diego.
Hormone Measurements
Animals used for hormone measurements were age matched at 10–12 wk. Blood was collected from PKO and cre-ve control mice from the tail vein. Estrous cycles were determined by vaginal lavage. Vaginal smears were taken daily between 0800 and 0900 h for 3 wk in normal saline. Based on the cytology, the smears were classified into various stages of the estrous cycle: A diestrous smear had almost exclusively leukocytes; a proestrous smear showed both leukocytes and nucleated epithelial cells, but predominantly nucleated epithelial cells; estrous smears had large, irregularly shaped, cornified, and clumped cells; and metestrous smears showed approximately equal numbers of leukocytes and epithelial cells. Blood was collected during diestrus. Plasma obtained after centrifugation using Adams MHCT II Microhematocrit centrifuge at 13 700 × g for 5 min was stored at −80°C until hormone analysis. Twenty-five microliters of plasma were used to measure LH and FSH levels by Luminex assay kit (Millipore Corporation) following the manufacturer's instructions.
Statistical Analysis
All the assays and SDS-PAGE data presented here are representative examples of at least three separate experiments. Differences between groups are shown as the mean ± SEM of at least three separate experiments and were analyzed for statistical significance using ANOVA followed by Tukey post-test (JMP IN Version 5.1; SAS Institute, Inc.). Sigmoidal dose-response curves were fit using the program Prism (Version 4.0; GraphPad Software) and tested both against the null hypothesis (i.e., no difference in the curves) and for significant differences in curve parameters. Hormone measurements were founds to be nonnormally distributed by Kolmogorov-Smirnov, D'Agostino-Pearson, and Shapiro-Wilk tests (P < 0.001), so data were analyzed by nonparametric Mann-Whitney and Wilcoxon signed-rank tests (Prism). A P value of 0.05 or less was considered to be statistically significant.
RESULTS
Rosiglitazone Suppresses GnRH- and Activin-Induced Activation of JNKs and p38MAPKs
We and others have reported that the three MAPK families—ERKs, p38MAPKs, and JNKs—are activated by GnRH [23]. To investigate the effects of TZDs on pituitary LβT2 gonadotroph cells, we pretreated cells with rosiglitazone for increasing times (24, 48, and 72 h) and measured the dose-dependent activation of the MAPKs by GnRH. At 10 μM rosiglitazone, we observed a dose-dependent increase in the phosphorylation levels of p38MAPKs, JNKs, and ERKs upon GnRH treatment, with maximum induction observed at 10–100 nM GnRH (Fig. 1A). Treatment with rosiglitazone for 24–72 h caused a significant decrease in phosphorylation of the stress kinases p38MAPKs and JNKs in response to GnRH (Fig. 1A). We saw a slight but significant increase in ERK activation at 48 h, but not at 24 or 72 h, of rosiglitazone treatment. These changes were verified by quantification of multiple blots (Fig. 1B). None of these treatments caused any visible effects on cell viability. Inhibition, albeit somewhat weaker, was also seen at 1 μM rosiglitazone; therefore, all further experiments used the maximal dose of 10 μM.
FIG. 1.

Effect of rosiglitazone (Rosi) on GnRH-induced activation of p38MAPKs, JNKs, and ERKs. LβT2 cells (1.5 × 106 cells/well) were treated with rosiglitazone (10 μM) or DMSO as vehicle for the indicated time periods. After 0, 24, or 48 h of pretreatment with rosiglitazone, the cells were incubated in serum-free DMEM containing 0.1% BSA for 24 h before treatment with varied concentrations of GnRH (0, 0.01, 0.1, 1, 10, or 100 nM) for 30 min. Whole-cell lysates were separated on SDS-PAGE and immunoblotted with an antibody for phospho-p38MAPKs (p-p38), phospho-JNKs (p-JNK), and phospho-ERKs (p-ERK; A). The blots were stripped and reblotted for p38MAPKs, JNKs, or ERKs, respectively, to determine the total protein loading. The blots are representative experiments, each repeated three times. Quantification of the GnRH dose response in the presence of rosiglitazone is also shown (B). Sigmoidal dose-response curves were modeled and tested for significance. Rosiglitazone caused a statistically significant decrease in the dose-response curve for p38MAPK and JNK activation by GnRH (P < 0.05) and a significant increase in the dose-response curve for ERK activation (P < 0.05).
Previously, we have shown that activin enhances the GnRH activation of the JNKs and p38MAPKs [23]. Therefore, we tested the effect of rosiglitazone on activin-enhanced activation of the stress kinases. After pretreatment for 48 h with 10 μM rosiglitazone, the cells were incubated in serum-free DMEM containing 25 ng/ml of activin and 10 μM rosiglitazone for another 24 h before treatment with GnRH (10 or 100 nM) for 30 min. Rosiglitazone reduced the effect of activin in enhancing the GnRH-mediated activation of JNKs and p38MAPKs but had no effect on ERKs (Fig. 2).
FIG. 2.

Effect of rosiglitazone (Rosi) on activin-enhanced GnRH activation of p38MAPKs and JNKs. LβT2 cells (1.5 × 106 cells/well) were treated with rosiglitazone (10 μM) or DMSO as vehicle for a total of 72 h. After 48 h of rosiglitazone pretreatment, the cells were incubated in serum-free DMEM containing activin A (25 ng/ml) for 24 h before treatment with GnRH (1 or 100 nM) for 30 min. Whole-cell lysates were separated on SDS-PAGE and immunoblotted with an antibody for phospho-p38MAPKs (p-p38), phospho-JNKs (p-JNK), and phospho-ERKs (p-ERK). The blots were stripped and reblotted for JNKs to demonstrate equal protein loading. The blots are representative experiments, each repeated three times. Rosiglitazone caused a statistically significant decrease in levels of phosphorylation of p38MAPKs and JNKs, but not of ERKs, induced by 1 and 100 nM of GnRH in the presence of activin compared to no rosiglitazone treatment (P < 0.05).
Rosiglitazone Inhibits Phosphorylation of Downstream Targets of p38MAPKs and JNKs
To further examine the inhibitory effects of rosiglitazone in the presence of activin and GnRH, we measured the phosphorylation of known downstream targets of the p38MAPKs. Treatment with rosiglitazone inhibited the phosphorylation of ATF2 and JUN in the presence of GnRH and activin (Fig. 3). In contrast, rosiglitazone had no effect on the phosphorylation of CREB, which is a downstream target of the phosphokinase A pathway. To verify that these transcription factors are indeed targets for the stress kinases, we used specific inhibitors of p38MAPKs, MEKs, and JNKs. Phosphorylation of ATF2 was blocked only by the p38 inhibitor (Fig. 4). ATF2 appears to be a relatively specific target, because the MEK and JNK inhibitors had no effect, or even slightly increased, the phosphorylation of ATF2. Inhibition of either JNKs or p38MAPKs reduced JUN phosphorylation, indicating that both kinases target this transcription factor. As a control, the MEK inhibitor reduced phosphorylation of SRF, a known target for ERKs. Interestingly, inhibition of p38MAPKs also reduced phosphorylation of SRF in response to GnRH. The inhibitors alone had no effect on basal phosphorylation of ATF2, JUN, or SRF (data not shown). These data indicate that rosiglitazone reduces GnRH activation of JNKs and p38MAPKs and downstream transcription factor targets.
FIG. 3.

Effect of rosiglitazone (Rosi) on GnRH-stimulated phosphorylation of ATF2, CREB, and JUN. LβT2 cells (1.5 × 106 cells/well) were treated with rosiglitazone (10 μM) or DMSO as vehicle for a total of 72 h. After 48 h of rosiglitazone pretreatment, the cells were incubated in serum-free DMEM containing activin A (25 ng/ml) for 24 h before treatment with GnRH (1 or 100 nM) for 30 min. Whole-cell lysates were separated on SDS-PAGE and immunoblotted with an antibody for phospho-ATF2 (p-ATF2), phospho-JUN (p-JUN), or phospho-CREB (p-CREB). The blots were stripped and reblotted for β-tubulin to demonstrate equal protein loading. The blots are representative experiments, each repeated three times. Rosiglitazone caused a statistically significant decrease in the level of ATF2 phosphorylation induced by 1 and 100 nM GnRH in the presence of activin and a significant decrease in the level of JUN phosphorylation in response to GnRH (100 nM) and activin (P < 0.05).
FIG. 4.

Effect of p38MAPK, JNK, and MEK inhibitors on the GnRH-induced phosphorylation of ATF2, SRF, and JUN. LβT2 cells (1.5 × 106 cells/well) were seeded in DMEM containing 10% serum and incubated overnight. This was followed by serum starving the cells through incubation in serum-free DMEM containing 0.1% BSA. After 24 h, the cells were treated with the inhibitors of MEKs (PD98059: 5, 10, and 20 μM), JNKs (SP600125: 2.5, 5, and 10 μM), or p38MAPKs (PD169316: 1.2, 2.5, and 5 μM) for 30 min, followed by treatment with 100 nM GnRH for 30 min. Whole-cell lysates were separated on SDS-PAGE and immunoblotted with an antibody for phospho-ATF2 (p-ATF2), phospho-JUN (p-JUN), or phospho-SRF (p-SRF). The blots were stripped and reblotted for ATF2, JUN, or β-tubulin to demonstrate equal protein loading. The blots are representative experiments, each repeated three times. PD169316 at a concentration of 2.5 and 5 μM caused a significant decrease in the GnRH-induced ATF2, JUN, and SRF phosphorylation (P < 0.05). SP600125 caused a significant decrease in GnRH-induced JUN phosphorylation, and PD98059 at 5, 10, and 20 μM significantly suppressed the GnRH-induced phosphorylation of SRF (P < 0.05).
p38MAPKs Are Critical for Induction of Lhb and Fshb Promoter Activity
Inhibition of the p38MAPK pathway impairs GnRH and activin induction of the Fshb promoter [24]. Therefore, we tested whether any of these kinases is responsible for the GnRH activation of Lhb promoter activity. Cells were transfected with a 1.8-kb rat Lhb promoter luciferase reporter [17]. Twenty-four hours after transfection, cells were serum-starved overnight, pretreated with inhibitors for MEKs (20 μM PD98059), JNKs (10 μM SP600125), or p38MAPKs (5 μM PD169316) for 30 min, then stimulated with 100 nM GnRH for 8 h. We observed a statistically significant decrease in GnRH-stimulated Lhb promoter activity in the presence of the p38MAPK inhibitor (Fig. 5A). We then tested whether inhibition of p38MAPKs altered sensitivity of the Lhb promoter to GnRH stimulation. Cells were transfected with the 1.8-kb rat Lhb promoter, serum-starved in the absence or presence of 25 ng/ml of activin for 16 h, treated with p38MAPK inhibitor or vehicle for 30 min, then stimulated with increasing doses of GnRH (0.01–100 nM) for 8 h. Inhibition of p38MAPK caused a decrease in responsiveness of the Lhb promoter but did not change the sensitivity to GnRH (Fig. 5, B and C). Similar experiments were performed with a 1-kb mouse Fshb promoter-luciferase reporter [18]. Pretreatment with the p38MAPK inhibitor, but not with the JNK or MEK inhibitors, caused a significant decrease in Fshb promoter activity (Fig. 5D) and decreased responsiveness of the promoter without changing sensitivity to GnRH (Fig. 5E). In the presence of activin, the basal promoter activity and the maximal response were increased (Fig. 5F), as expected. Interestingly, inhibition of p38MAPKs not only caused a decreased basal activity and maximal response to GnRH but also significantly altered the sensitivity to GnRH (median effective dose, 0.2 nM vs. 0.009 nM in the presence of PD169316; P < 0.05) (Fig. 5F).
FIG. 5.

GnRH signals to the Lhb and Fshb promoters via p38MAPKs. Induction of the Lhb and Fshb promoters requires p38MAPK, but not ERK or JNK, signaling. LβT2 cells (5 × 105 cells/well) were transiently transfected with 500 ng of the 1.8-kb Lhb luc reporter gene along with 50 ng of TK-LacZ plasmid containing β-galactosidase. After overnight starvation in serum-free DMEM, the cells were treated with the inhibitors of MEKs (PD98059: 20 μM), JNKs (SP600125: 10 μM), or p38MAPKs (PD169316: 5 μM) or with DMSO for 30 min followed by treatment with 100 nM GnRH for 30 min (A). Cells were lysed and assayed for Lhb promoter activity and β-galactosidase activity. The graph shows promoter activity as luciferase activity (relative light units) normalized to the β-galactosidase activity (relative light units) from the control plasmid. Cells were also transfected with the Lhb promoter as described above. After overnight starvation in serum-free media with or without 25 ng/ml of activin, cells were treated with 5 μM PD169316 or DMSO for 30 min, then with increasing doses of GnRH (0, 0.01, 0.1, 1, 10, or 100 nM). Graphs show GnRH dose-response curves for Lhb promoter activity in the absence (B) or presence (C) of activin pretreatment. Experiments were then repeated using the 1-kb mouse Fshb promoter luciferase reporter gene. Treatment with PD169316 inhibited FSHB induction by GnRH (D). Dose-response curves for Fshb promoter activity showed a decreased maximal response in the presence of the p38MAPK inhibitor (E). GnRH dose-response curves showed increased basal and maximal promoter activity in the presence of activin (F). PD169316 reduced basal promoter activity and maximal response but increased sensitivity. Asterisks indicate statistical significance (P < 0.05) compared to the corresponding sample without inhibitor. Dose-response curves were compared by testing the best-fit sigmoidal curves against the global null hypothesis curve. ED50, median effective dose.
Rosiglitazone Inhibits GnRH-Stimulated Lhb and Fshb mRNA Expression
Because GnRH-mediated activation of p38MAPKs was suppressed by rosiglitazone and p38MAPK inhibition impaired transcription of the Lhb (Fig. 6A) and Fshb (Fig. 6B) promoters, we examined the effect of rosiglitazone on Lhb and Fshb gene expression. Cells were treated with 10 μM rosiglitazone or vehicle for 48 h, then serum-starved in the presence of 25 ng/ml of activin plus rosiglitazone overnight and finally stimulated with 100 nM GnRH for 8 h. GnRH tended to increase, though not significantly, Lhb mRNA (Fig. 6A) but to up-regulate the expression of the Fshb mRNA by 10-fold (Fig. 6B). Rosiglitazone treatment significantly repressed Lhb expression down to basal levels and also repressed Fshb induction by 80%. Activin pretreatment enhanced the basal expression levels of the Lhb gene by 3-fold and levels of the Fshb gene by 80-fold, with no significant change upon rosiglitazone treatment. Furthermore, cotreatment with GnRH and activin caused a synergistic increase in both Lhb (5-fold) and Fshb (160-fold), as published previously [24]. Treatment with rosiglitazone eliminated this synergistic induction and decreased Lhb and Fshb mRNA levels to those induced by activin alone. Elimination of the synergistic effect on Fshb expression was consistent with published data showing that inhibition of p38MAPKs abolishes synergy on the Fshb promoter [24].
FIG. 6.
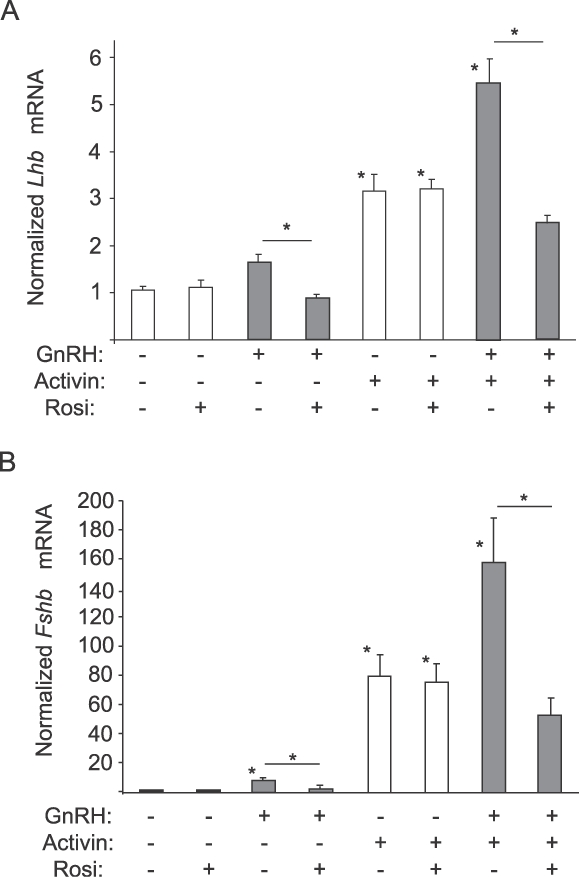
Rosiglitazone (Rosi) represses gonadotropin gene expression. LβT2 cells were treated with rosiglitazone (10 μM) or DMSO as vehicle for a total of 72 h. After 48 h of rosiglitazone treatment, the cells were starved in serum-free DMEM with or without activin A (25 ng/ml). After a further 24 h, the cells were treated with GnRH (100 nM) as indicated and incubated for 8 h. RNA was harvested and analyzed by quantitative PCR for Lhb expression (A) or Fshb expression (B). Data for Lhb or Fshb mRNA are normalized to the housekeeping gene M36B4 and presented as fold-induction compared to untreated samples. Asterisks indicate a statistically significant (P < 0.05) decrease in Lhb or Fshb mRNA levels compared to the untreated control or to the matched, non-rosiglitazone-treated sample.
PPARG Mediates Rosiglitazone Suppression of GnRH Induction of JNKs and Lhb and Fshb mRNA Expression
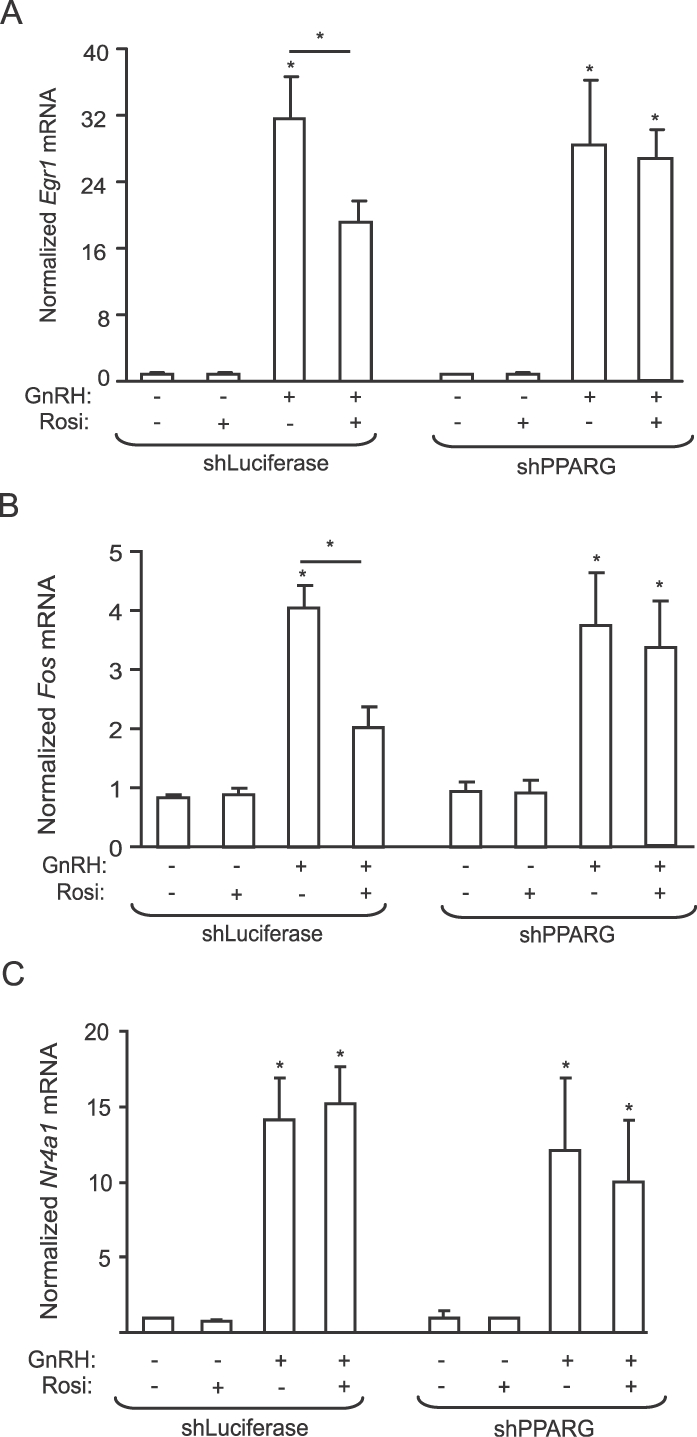
Although rosiglitazone is a well-accepted ligand for PPARG, PPARG-independent effects have also been documented in the pituitary [25] and other tissues [26, 27]. Therefore, to determine whether the observed suppressive effects of rosiglitazone on the stress kinases and gonadotropin mRNA expression are mediated via PPARG, we knocked down Pparg by infecting LβT2 cells with a lentivirus encoding an shRNA against Pparg or Luciferase as a control. A 70% decrease in PPARG protein and mRNA (Fig. 7A) was obtained. Pparg knockdown abolished the effect of rosiglitazone on GnRH-induced phosphorylation of JNKs and p38MAPKs (Fig. 7B). At the level of gonadotropin gene expression, rosiglitazone suppressed the GnRH induction of Lhb (Fig. 7C) and Fshb (Fig. 7D) mRNA expression in shLuc cells, but the suppression was abolished in shPPARG cells.
FIG. 7.

PPARG depletion abolishes the effect of rosiglitazone (Rosi) on GnRH-induced JNK phosphorylation and gonadotropin gene expression. Quantitative PCR analysis of Pparg mRNA expression and immunoblot for protein expression in shLuc and shPPARG cells were performed. The blot was stripped and reprobed for β-tubulin to demonstrate equal protein loading (A). To assess stress kinase activation, shLuc and shPPARG cells were treated with rosiglitazone (10 μM) or DMSO vehicle for 72 h. After 48 h of rosiglitazone treatment, the cells were starved for 24 h in serum-free DMEM with or without rosiglitazone, then treated with GnRH (100 nM) for 5 or 30 min. Whole-cell extracts were immunoblotted for phospho-p38MAPKs (p-p38) and phospho-JNKs (p-JNK), then stripped and reblotted for β-tubulin or JNKs, respectively (B). The blots are representative experiments, each repeated three times. JNK and p38MAPK phosphorylation was significantly decreased by rosiglitazone treatment only in the shLuc cells (P < 0.05). To assess gonadotropin subunit gene expression, shLuc and shPPARG cells were treated with rosiglitazone (10 μM) or DMSO vehicle for 72 h. After 48 h of rosiglitazone treatment, the cells were starved for 24 h in serum-free DMEM with or without rosiglitazone, then treated with GnRH (100 nM) for 8 h. RNA was analyzed by quantitative PCR for Lhb expression as before (C), and the same RNA was analyzed for Fshb expression as before (D). Data for Lhb, Fshb, or Pparg mRNA are presented as fold-induction compared to the control or untreated samples. Asterisks indicate statistical significance (P < 0.05) compared to the untreated controls or to the matched, non-rosiglitazone-treated samples.
PPARG Activation Suppresses GnRH Induction of Transcription Factors EGR1 and FOS But Not NR4A1
To define the mechanism leading to the PPARG-mediated suppression of gonadotropin gene expression, we analyzed the effects of rosiglitazone and GnRH on the expression of Egr1 and Fos (transcription factors for LHB and FSHB, respectively) in shLuc and shPPARG cells. GnRH induced Egr1 by 24-fold and Fos by 4-fold at the mRNA level. Rosiglitazone suppressed the GnRH induction of both Egr1 and Fos only in shLuc cells (Fig. 8A), not in shPPARG cells (Fig. 8B). A similar repression of EGR1 and FOS protein expression was observed. Rosiglitazone treatment caused a 20% decrease in both EGR1 and FOS protein (both P < 0.05) in shLuc cells but no decrease in shPPARG cells (data not shown). To verify the specificity of the PPARG effect, we analyzed the induction of NR4A1, an orphan nuclear receptor known to be induced by GnRH [27]. Nr4a1 mRNA expression was increased by 15-fold upon GnRH stimulation; however, the induction was unchanged by rosiglitazone in either shLuc or shPPARG cells (Fig. 8C). These data reinforce the conclusion that the effect of rosiglitazone is mediated by PPARG, and they suggest that suppression of Egr1 and Fos may underlie the suppression of Lhb and Fshb mRNA expression, respectively.
FIG. 8.
Rosiglitazone (Rosi) suppresses GnRH-induced Egr1 and Fos, but not of N4a1, mRNA expression. To assess Egr1 and Fos gene expression, shLuc and shPPARG cells were treated with rosiglitazone (10 μM) or DMSO vehicle for 72 h. After 48 h of rosiglitazone treatment, the cells were starved for 24 h in serum-free DMEM with or without rosiglitazone, then treated with GnRH (100 nM) for 8 h. RNA was analyzed by quantitative PCR for Egr1 (A), Fos (B), or Nr4a1 (Nur77; C) mRNA expression. Data for Egr1, Fos, or Nr4a1 mRNA are normalized to the housekeeping gene M36B4 and presented as fold-induction compared to the untreated samples. Asterisks indicate statistical significance (P < 0.05) compared to the untreated controls or to the matched, non-rosiglitazone-treated samples.
Pituitary-Specific Deletion of PPARG Elevates LH Levels In Vivo
To study the role of PPARG in vivo in the pituitary, we generated PKO mice following the cre-loxP strategy by crossing Pparg-floxed mice with Lhb-cre mice that express the cre recombinase in LH-positive pituitary gonadotroph cells with little or no expression in other organs [20]. The PKO mice had the genotype Ppargflox/flox:Lhb-cre, and the control group was made up of littermates with the genotype Ppargflox/flox but lacking the Lhb-cre allele. As expected, recombination of PPARG alleles occurred in the pituitaries of PKO mice and not in the control cre-ve littermates as determined by PCR (Fig. 9A). The PKO females had a 3-fold elevated plasma diestrous LH (286 ng/ml) compared to the control female mice (85 ng/ml) (Fig. 9B). This alteration was specific for the females; the LH levels in PKO male mice were unchanged compared to controls. The difference was not related to a hypomorphic Ppargflox/flox allele, because the LH levels in Ppargflox/flox mice (n = 17) were not significantly different from those in Ppargflox/+ mice (n = 18). No difference was observed in the plasma FSH levels in control and PKO female mice, but the male PKO mice had slightly, but significantly, lower FSH (Fig. 9B). The female PKO mice showed normal patterns of estrous cycles by vaginal lavage over a 2-wk period before hormone measurements. To analyze the PKO mice for reproductive phenotype, we bred four independent pairs of PKO or control mice over the course of 6 mo. The PKO mice were fertile and produced litters at the same rate as control mice. The total number of mice produced by the PKO breeding pairs was lower than that in the wild-type mice, and the mean litter size was significantly smaller (Fig. 9B).
FIG. 9.

Pituitary-specific deletion of Pparg causes reduction in the litter size and alters plasma gonadotropin profile in female mice. Genotyping of floxed Pparg allele and cre transgene in tail DNA shows that all mice contained the floxed Pparg allele but that only the PKO mice carried the Lhb-cre allele (A). PKO mice had the genotype Ppargflox/flox:Lhb-cre, and the control mice had the genotype Ppargflox/flox, but without the Lhb-cre. Analysis of DNA extracted from the pituitaries after mice were euthanized demonstrates the presence of the deleted knockout (KO) Pparg allele only in PKO mice. Reproductive hormones were measured during diestrus in females and randomly in males (B). Median hormone levels are given, because data are not normally distributed. Significance was assessed using nonparametric Mann-Whitney and Wilcoxon signed-rank tests. Fertility was assessed by breeding four pairs of PKO or control mice over 6 mo. Total number of litters and pups are shown along with the litter size (mean ± SEM). Significance was assessed using Student t-test. Asterisks indicate statistical significantly (P < 0.05) compared to the untreated controls.
DISCUSSION
In the present study, we show that rosiglitazone down-regulates GnRH-mediated phosphorylation of the JNKs and p38MAPKs in LβT2 cells. These kinases are activated by many cytokines and inflammatory signals, so this TZD effect is consistent with its known anti-inflammatory actions. This inhibition is also seen in the presence of activin, which we showed previously to enhance phosphorylation of the stress kinases by GnRH [23]. The elevated p38MAPK signaling in the presence of activin and GnRH likely causes a synergistic increase in the expression of Lhb and Fshb, which could contribute to the preovulatory LH and FSH surge when inhibin levels are low. Our results are consistent with a previous report [28] that both PPARA and PPARG agonists reduced the basal activity of the gonadotropin gene promoters and increased inhibin-α and follistatin levels, which would inhibit activin signaling. Therefore, it is possible that rosiglitazone suppresses the activin-enhanced stress kinase activation by inducing the antagonistic hormones follistatin and inhibin.
The GnRH receptor is a classical G protein-coupled receptor that couples to Gq/11, leading to activation of phosphokinase C and CaMK [29, 30], and to Gs, leading to activation of phosphokinase A [31, 32]. Both the Gq/11 and Gs pathways are upstream of ERK activation and the Gq/11 pathway is upstream of JNKs, but the signals leading to p38MAPKs are not known [32–34]. All these kinases phosphorylate specific nuclear targets to mediate transcriptional activation. GnRH up-regulates phosphorylation of both JNK2 and p38MAPKs in primary rat pituitary cultures [35] and in pituitary gonadotroph cell lines [32, 33]. Our results suggest that p38MAPKs are the main mediator for GnRH-stimulated Lhb and Fshb promoter activity, which disagrees with a report that JNKs are the critical regulator of the Lhb gene in primary rat pituitary cultures [35]. It is possible that GnRH uses different stress kinases to induce the Lhb gene in mouse and rat pituitary cells, because many of the transcription factor targets are redundant. Another study utilized a dominant-negative JUN expression vector to demonstrate that JNK signaling is critical in LβT2 cells [34], but in the present study, we show that JUN is a target for both JNKs and p38MAPKs. Therefore, the data in the previous study could be consistent with p38MAPK signaling to Lhb.
Conflicting reports also suggest the ERKs play a role in the transcriptional activation of Lhb in response to GnRH [36]. Blockade of ERK and p38MAPK signaling has an additive effect to reduce transcription of the Lhb promoter, both the minimal proximal promoter and one containing the upstream response element [37]. Blocking ERK, but not p38MAPK, signaling reduced EGR1 expression, yet blocking p38MAPK signaling eliminated EGR1 DNA binding but not expression, so the two signals may regulate different aspects of Lhb induction [37]. The role of ERKs has been supported by a pituitary double knockout of MAPK3/1 (ERK1/2) that led to decreased expression of Egr1 and Lhb [38], but that study is complicated by the observed lack of specificity of the GSU-cre mouse, which also expresses cre recombinase in the ovary and testis [39]. Our observed suppression of both JNK and p38MAPK activation and Lhb and Fshb gene expression by rosiglitazone is supported by the finding that rosiglitazone impairs induction of EGR1 and FOS. Female Egr1 knockout mouse are infertile because of low LH levels [40], and Egr1 regulates the Lhb promoter in vitro. Similar studies have shown the importance of FOS for induction of Fshb and that female Fos knockout mice are infertile, although it has not been shown whether this is the result of decreased FSH [24, 41]. The suppressive effect of rosiglitazone on Egr1 expression is confirmed by a report that rosiglitazone attenuates postoperative ileus in the mouse colon through PPARG-dependent down-regulation of EGR1 [26]. Whereas the effect of rosiglitazone on Egr1 and Fos expression is modest compared to the suppression of Lhb and Fshb, PPARG and p38MAPKs may also be involved in posttranscriptional regulation of DNA binding [37], which could further contribute to the repression of gonadotropin expression.
In agreement with the in vitro data, we find that PPARG regulates fertility in vivo, because female PKO mice have elevated plasma LH and reduced litter size compared to the control cre-negative littermates. PPARG may provide a tonic repression of inflammatory signaling in female gonadotrophs that is lost in the PKO mice, resulting in elevated LH. Although our in vitro data also point to repression of Fshb expression, the female PKO mice have normal FSH, but male PKO mice have reduced FSH, as expected. Activin is a physiologically more important stimulus of FSH levels than GnRH in vivo, and rosiglitazone does not repress activin-induced Fshb. Therefore, the normal FSH levels in female PKO mice may be a reflection of normal activin signaling, but we do not yet have an explanation for the reduced FSH in male PKO mice. Activin expression is responsive to inflammatory stimuli, so it is possible that activin tone is reduced in the male PKO mice [42]. Would the elevated LH alone cause the subfertility in the female mice? Transgenic mice with a 10- to 12-fold overexpression of a stabilized form of LH causes chronic anovulation with enlarged ovaries, ovarian cysts, or ovarian tumors, so the 3-fold overexpression seen in the PKO mice might be sufficient to reduce fertility. A similar phenotype of increased LH levels and reduced litter size is reported for a subset of subfertile female mice lacking estrogen receptor alpha (ESR1) in pituitary gonadotrophs [43]. Estrogen has anti-inflammatory effects similar to those of PPARG agonists and prevents metabolic complications of diet-induced obesity. It is conceivable that ESR1 and PPARG have similar effects in the gonadotroph to suppress inflammatory signaling that would otherwise elevate LH levels. This overlapping role is seen in other tissues as well: Muscle-specific knockout of Pparg causes insulin resistance and glucose intolerance, and knock out of Esr1 causes a similar phenotype, with muscle insulin resistance and increased tissue inflammation [21, 44]. The ability of ESR1 to repress inflammation is only seen in estrogenized females. Males and postmenopausal females are not protected because of low estrogen levels [45]. The PPARG effects also appear to be sex-specific: LH is not elevated in males compared to control littermates. It is possible that PPARG expression is low in male mice, that they do not produce endogenous PPARG ligands, or that other pathways, such as androgen receptor repression of Lhb, may compensate for lack of PPARG signaling in male PKO mice.
If GnRH uses the same signaling pathways as inflammation, then one would predict that states of inflammation would alter the reproductive axis. Indeed, this has been seen in both humans and animals. Chronic filarial infection lowers LH levels in women [46], and obese PCOS is associated with elevated inflammatory markers [47, 48]. Men with untreated systemic lupus erythematosus, rheumatoid arthritis, type 2 diabetes, metabolic syndrome, or chronic obstructive pulmonary disease—all conditions that are associated with increased inflammation—also have alterations in the HPG axis [49–52]. In animals, endotoxin (lipopolysaccharide) administration decreases serum LH in female and male rats [53, 54] and decreases GnRH pulsatility and pituitary responsiveness in sheep [53, 55]. Some of these effects can be replicated by cytokine treatment alone. For example, interleukin 1β suppresses LH in female rats [56, 57] and suppresses kisspeptin expression but increases gamma-aminobutyric acid in the hypothalamus [58, 59], so the effect can be central as well as on the pituitary.
In conclusion, the ability of PPARG to repress Lhb and the increased LH in PKO female mice provide an interesting model of reproductive dysfunction. A more complete analysis of the reproductive and inflammatory phenotype of the PKO mice is underway to fully understand how lack of PPARG signaling in the gonadotroph influences the HPG axis.
Acknowledgments
We thank Dr. Djurdjica Coss, Dr. WuQiang Fan, and members of the Webster Lab for helpful discussions and assistance.
Footnotes
Supported by the NICHD/NIH through a cooperative agreement (U54 HD012303) as part of the Specialized Cooperative Centers Program in Reproduction Research (N.J.G.W., J.M.O., P.L.M.). N.J.G.W., J.M.O., and P.L.M. are faculty members of the UCSD Biomedical Sciences Graduate Program. J.M.O. is supported by NIH grants DK033651 and DK074868, N.J.G.W. by NIH grant HD047400, and P.L.M. by NIH grant HD020377.
These authors contributed equally to this work.
REFERENCES
- Olefsky JM. Treatment of insulin resistance with peroxisome proliferator-activated receptor gamma agonists. J Clin Invest 2000; 106: 467 472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn CR, Chen L, Cohen SE. Unraveling the mechanism of action of thiazolidinediones. J Clin Invest 2000; 106: 1305 1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai A, Bang H, Green C. Metformin and glitazones: do they really help PCOS patients? J Fam Pract 2007; 56: 444 453 [PubMed] [Google Scholar]
- Zhou R, Bruns CM, Bird IM, Kemnitz JW, Goodfriend TL, Dumesic DA, Abbott DH. Pioglitazone improves insulin action and normalizes menstrual cycles in a majority of prenatally androgenized female rhesus monkeys. Reprod Toxicol 2007; 23: 438 448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhindsa G, Bhatia R, Dhindsa M, Bhatia V. Insulin resistance, insulin sensitization and inflammation in polycystic ovarian syndrome. J Postgrad Med 2004; 50: 140 144 [PubMed] [Google Scholar]
- Rautio K, Tapanainen JS, Ruokonen A, Morin-Papunen LC. Rosiglitazone treatment alleviates inflammation and improves liver function in overweight women with polycystic ovary syndrome: a randomized placebo-controlled study. Fertil Steril 2007; 87: 202 206 [DOI] [PubMed] [Google Scholar]
- Lujan ME, Chizen DR, Pierson RA. Diagnostic criteria for polycystic ovary syndrome: pitfalls and controversies. J Obstet Gynaecol Can 2008; 30: 671 679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dereli D, Dereli T, Bayraktar F, Ozgen AG, Yilmaz C. Endocrine and metabolic effects of rosiglitazone in nonobese women with polycystic ovary disease. Endocr J 2005; 52: 299 308 [DOI] [PubMed] [Google Scholar]
- Khan KA, Stas S, Kurukulasuriya LR. Polycystic ovarian syndrome. J Cardiometab Syndr 2006; 1: 125 130 [quiz 131–132]. [DOI] [PubMed] [Google Scholar]
- Korhonen S, Heinonen S, Hiltunen M, Helisalmi S, Hippelainen M, Koivunen R, Tapanainen JS, Laakso M. Polymorphism in the peroxisome proliferator-activated receptor-gamma gene in women with polycystic ovary syndrome. Hum Reprod 2003; 18: 540 543 [DOI] [PubMed] [Google Scholar]
- Orio F, Jr, Matarese G, Di Biase S, Palomba S, Labella D, Sanna V, Savastano S, Zullo F, Colao A, Lombardi G. Exon 6 and 2 peroxisome proliferator-activated receptor-gamma polymorphisms in polycystic ovary syndrome. J Clin Endocrinol Metab 2003; 88: 5887 5892 [DOI] [PubMed] [Google Scholar]
- Azziz R, Ehrmann D, Legro RS, Whitcomb RW, Hanley R, Fereshetian AG, O'Keefe M, Ghazzi MN. Troglitazone improves ovulation and hirsutism in the polycystic ovary syndrome: a multicenter, double-blind, placebo-controlled trial. J Clin Endocrinol Metab 2001; 86: 1626 1632 [DOI] [PubMed] [Google Scholar]
- Coffler MS, Patel K, Dahan MH, Yoo RY, Malcom PJ, Chang RJ. Enhanced granulosa cell responsiveness to follicle-stimulating hormone during insulin infusion in women with polycystic ovary syndrome treated with pioglitazone. J Clin Endocrinol Metab 2003; 88: 5624 5631 [DOI] [PubMed] [Google Scholar]
- Froment P, Fabre S, Dupont J, Pisselet C, Chesneau D, Staels B, Monget P. Expression and functional role of peroxisome proliferator-activated receptor-gamma in ovarian folliculogenesis in the sheep. Biol Reprod 2003; 69: 1665 1674 [DOI] [PubMed] [Google Scholar]
- Mehta RV, Patel KS, Coffler MS, Dahan MH, Yoo RY, Archer JS, Malcom PJ, Chang RJ. Luteinizing hormone secretion is not influenced by insulin infusion in women with polycystic ovary syndrome despite improved insulin sensitivity during pioglitazone treatment. J Clin Endocrinol Metab 2005; 90: 2136 2141 [DOI] [PubMed] [Google Scholar]
- Heaney AP, Fernando M, Melmed S. PPAR-gamma receptor ligands: novel therapy for pituitary adenomas. J Clin Invest 2003; 111: 1381 1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen KA, Santos SJ, Kreidel MK, Diaz AL, Rey R, Lawson MA. Acute regulation of translation initiation by gonadotropin-releasing hormone in the gonadotrope cell line LbetaT2. Mol Endocrinol 2004; 18: 1301 1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackray VG, McGillivray SM, Mellon PL. Androgens, progestins, and glucocorticoids induce follicle-stimulating hormone beta-subunit gene expression at the level of the gonadotrope. Mol Endocrinol 2006; 20: 2062 2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W, Nguyen MT, Yoshizaki T, Favelyukis S, Patsouris D, Imamura T, Verma IM, Olefsky JM. Suppression of PPAR-gamma attenuates insulin-stimulated glucose uptake by affecting both GLUT1 and GLUT4 in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab 2007; 293: 219 227 [DOI] [PubMed] [Google Scholar]
- Charles MA, Mortensen AH, Potok MA, Camper SA. Pitx2 deletion in pituitary gonadotropes is compatible with gonadal development, puberty, and fertility. Genesis 2008; 46: 506 513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevener AL, Olefsky JM, Reichart D, Nguyen MT, Bandyopadyhay G, Leung HY, Watt MJ, Benner C, Febbraio MA, Nguyen AK, Folian B, Subramaniam S,, et al. Macrophage PPARγ is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest 2007; 117: 1658 1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell 1999; 4: 585 595 [DOI] [PubMed] [Google Scholar]
- Zhang H, Bailey JS, Coss D, Lin B, Tsutsumi R, Lawson MA, Mellon PL, Webster NJ. Activin modulates the transcriptional response of LbetaT2 cells to gonadotropin-releasing hormone and alters cellular proliferation. Mol Endocrinol 2006; 20: 2909 2930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coss D, Hand CM, Yaphockun KK, Ely HA, Mellon PL. p38 Mitogen-activated protein kinase is critical for synergistic induction of the FSH(beta) gene by gonadotropin-releasing hormone and activin through augmentation of c-Fos induction and Smad phosphorylation. Mol Endocrinol 2007; 21: 3071 3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery MN, Leontiou C, Bonner SE, Merulli C, Nanzer AM, Musat M, Galloway M, Powell M, Nikookam K, Korbonits M, Grossman AB. PPAR-gamma expression in pituitary tumors and the functional activity of the glitazones: evidence that any antiproliferative effect of the glitazones is independent of the PPAR-gamma receptor. Clin Endocrinol (Oxf) 2006; 65: 389 395 [DOI] [PubMed] [Google Scholar]
- De Backer O, Elinck E, Priem E, Leybaert L, Lefebvre RA. Peroxisome proliferator-activated receptor gamma activation alleviates postoperative ileus in mice by inhibition of Egr-1 expression and its downstream target genes. J Pharmacol Exp Ther 2009; 331: 496 503 [DOI] [PubMed] [Google Scholar]
- Hamid T, Malik MT, Millar RP, Kakar SS. Protein kinase A serves as a primary pathway in activation of Nur77 expression by gonadotropin-releasing hormone in the LbetaT2 mouse pituitary gonadotroph tumor cell line. Int J Oncol 2008; 33: 1055 1064 [PubMed] [Google Scholar]
- Takeda M, Otsuka F, Otani H, Inagaki K, Miyoshi T, Suzuki J, Mimura Y, Ogura T, Makino H. Effects of peroxisome proliferator-activated receptor activation on gonadotropin transcription and cell mitosis induced by bone morphogenetic proteins in mouse gonadotrope LbetaT2 cells. J Endocrinol 2007; 194: 87 99 [DOI] [PubMed] [Google Scholar]
- Cheng KW, Leung PC. The expression, regulation and signal transduction pathways of the mammalian gonadotropin-releasing hormone receptor. Can J Physiol Pharmacol 2000; 78: 1029 1052 [PubMed] [Google Scholar]
- Winters SJ, Ghooray D, Fujii Y, Moore JP, Jr, Nevitt JR, Kakar SS. Transcriptional regulation of follistatin expression by GnRH in mouse gonadotroph cell lines: evidence for a role for cAMP signaling. Mol Cell Endocrinol 2007; 271: 45 54 [DOI] [PubMed] [Google Scholar]
- Bachir LK, Laverriere JN, Counis R. Isolation and characterization of a rat nitric oxide synthase type I gene promoter that confers expression and regulation in pituitary gonadotrope cells. Endocrinology 2001; 142: 4631 4642 [DOI] [PubMed] [Google Scholar]
- Liu F, Austin DA, Mellon PL, Olefsky JM, Webster NJ. GnRH activates ERK1/2 leading to the induction of c-fos and LHbeta protein expression in LbetaT2 cells. Mol Endocrinol 2002; 16: 419 434 [DOI] [PubMed] [Google Scholar]
- Roberson MS, Zhang T, Li HL, Mulvaney JM. Activation of the p38 mitogen-activated protein kinase pathway by gonadotropin-releasing hormone. Endocrinology 1999; 140: 1310 1318 [DOI] [PubMed] [Google Scholar]
- Yokoi T, Ohmichi M, Tasaka K, Kimura A, Kanda Y, Hayakawa J, Tahara M, Hisamoto K, Kurachi H, Murata Y. Activation of the luteinizing hormone beta promoter by gonadotropin-releasing hormone requires c-Jun NH2-terminal protein kinase. J Biol Chem 2000; 275: 21639 21647 [DOI] [PubMed] [Google Scholar]
- Haisenleder DJ, Burger LL, Walsh HE, Stevens J, Aylor KW, Shupnik MA, Marshall JC. Pulsatile gonadotropin-releasing hormone stimulation of gonadotropin subunit transcription in rat pituitaries: evidence for the involvement of Jun N-terminal kinase but not p38. Endocrinology 2008; 149: 139 145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris D, Bonfil D, Chuderland D, Kraus S, Seger R, Naor Z. Activation of MAPK cascades by GnRH: ERK and Jun N-terminal kinase are involved in basal and GnRH-stimulated activity of the glycoprotein hormone LHbeta-subunit promoter. Endocrinology 2002; 143: 1018 1025 [DOI] [PubMed] [Google Scholar]
- Maudsley S, Naor Z, Bonfil D, Davidson L, Karali D, Pawson AJ, Larder R, Pope C, Nelson N, Millar RP, Brown P. Proline-rich tyrosine kinase 2 mediates gonadotropin-releasing hormone signaling to a specific extracellularly regulated kinase-sensitive transcriptional locus in the luteinizing hormone beta-subunit gene. Mol Endocrinol 2007; 21: 1216 1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss SP, Miller A, Navratil AM, Xie J, McDonough SP, Fisher PJ, Landreth GE, Roberson MS. ERK signaling in the pituitary is required for female but not male fertility. Mol Endocrinol 2009; 23: 1092 1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlenhaut NH, Jakob S, Anlag K, Eisenberger T, Sekido R, Kress J, Treier AC, Klugmann C, Klasen C, Holter NI, Riethmacher D, Schütz G. Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell 2009; 139: 1130 1142 [DOI] [PubMed] [Google Scholar]
- Topilko P, Schneider-Maunoury S, Levi G, Trembleau A, Gourdji D, Driancourt MA, Rao CV, Charnay P. Multiple pituitary and ovarian defects in Krox-24 (NGFI-A, Egr-1)-targeted mice. Mol Endocrinol 1998; 12: 107 122 [DOI] [PubMed] [Google Scholar]
- Johnson RS, Spiegelman BM, Papaioannou V. Pleiotropic effects of a null mutation in the c-fos proto-oncogene. Cell 1992; 71: 577 586 [DOI] [PubMed] [Google Scholar]
- Phillips DJ, de Kretser DM, Hedger MP. Activin and related proteins in inflammation: not just interested bystanders. Cytokine Growth Factor Rev 2009; 20: 153 164 [DOI] [PubMed] [Google Scholar]
- Singh SP, Wolfe A, Ng Y, DiVall SA, Buggs C, Levine JE, Wondisford FE, Radovick S. Impaired estrogen feedback and infertility in female mice with pituitary-specific deletion of estrogen receptor alpha (ESR1). Biol Reprod 2009; 81: 488 496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas V, Nguyen MTA, Henstridge DC, Nguyen AK, Beaven SW, Watt MJ, Hevener AL. Impaired oxidative metabolism and inflammation are associated with insulin resistance in ERα-deficient mice. Am J Physiol Endocrinol Metab 2010; 298: E304 E319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers NH, Perfield JW, II, Strissel KJ, Obin MS, Greenberg AS. Reduced energy expenditure and increased inflammation are early events in the development of ovariectomy-induced obesity. Endocrinology 2009; 150: 2161 2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavoungou D, Poaty-Mavoungou V, Ongali B, Akoume MY, Maka G, Mavoungou E. Hypothalamic-pituitary-gonadal axis and immune response imbalance during chronic filarial infections. Trop Med Int Health 2005; 10: 1180 1186 [DOI] [PubMed] [Google Scholar]
- González F, Rote NS, Minium J, Weaver AL, Kirwan JP. Elevated circulating levels of macrophage migration inhibitory factor in polycystic ovary syndrome. Cytokine 2010; 51: 240 244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samy N, Hashim M, Sayed M, Said M. Clinical significance of inflammatory markers in polycystic ovary syndrome: their relationship to insulin resistance and body mass index. Dis Markers 2009; 26: 163 170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köller MD, Templ E, Riedl M, Clodi M, Wagner O, Smolen JS, Luger A. Pituitary function in patients with newly diagnosed untreated systemic lupus erythematosus. Ann Rheum Dis 2004; 63: 1677 1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tengstrand B, Carlström K, Hafström I. Gonadal hormones in men with rheumatoid arthritis—from onset through 2 years. J Rheumatol 2009; 36: 887 892 [DOI] [PubMed] [Google Scholar]
- Dandona P, Dhindsa S, Chaudhuri A, Bhatia V, Topiwala S, Mohanty P. Hypogonadotrophic hypogonadism in type 2 diabetes, obesity and the metabolic syndrome. Curr Mol Med 2008; 8: 816 828 [DOI] [PubMed] [Google Scholar]
- Karadag F, Ozcan H, Karul AB, Yilmaz M, Cildag O. Sex hormone alterations and systemic inflammation in chronic obstructive pulmonary disease. Int J Clin Pract 2009; 63: 275 281 [DOI] [PubMed] [Google Scholar]
- Herman AP, Tomaszewska-Zaremba D. Effect of endotoxin on the expression of GnRH and GnRHR genes in the hypothalamus and anterior pituitary gland of anestrous ewes. Anim Reprod Sci 2010; 120: 105 111 [DOI] [PubMed] [Google Scholar]
- O'Bryan MK, Schlatt S, Phillips DJ, de Kretser DM, Hedger MP. Bacterial lipopolysaccharide-induced inflammation compromises testicular function at multiple levels in vivo. Endocrinology 2000; 141: 238 246 [DOI] [PubMed] [Google Scholar]
- Daniel JA, Whitlock BK, Wagner CG, Sartin JL. Regulation of the growth hormone and luteinizing hormone response to endotoxin in sheep. Domest Anim Endocrinol 2002; 23: 361 370 [DOI] [PubMed] [Google Scholar]
- Sirivelu MP, Shin AC, Perez GI, MohanKumar PS, MohanKumar SM. Effect of l-dopa on interleukin-1beta-induced suppression of luteinizing hormone secretion in intact female rats. Hum Reprod 2009; 24: 718 725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra PS, Edwards TG, Xu B, Jain M, Kalra SP. The antigonadotropic effects of cytokines: the role of neuropeptides. Domest Anim Endocrinol 1998; 15: 321 332 [DOI] [PubMed] [Google Scholar]
- Sirivelu MP, Burnett R, Shin AC, Kim C, MohanKumar PS, MohanKumar SM. Interaction between GABA and norepinephrine in interleukin-1beta-induced suppression of the luteinizing hormone surge. Brain Res 2009; 1248: 107 114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasa T, Matsuzaki T, Murakami M, Shimizu F, Kuwahara A, Yasui T, Irahara M. Decreased expression of kisspeptin mediates acute immune/inflammatory stress-induced suppression of gonadotropin secretion in female rat. J Endocrinol Invest 2008; 31: 656 659 [DOI] [PubMed] [Google Scholar]