

Abstract
Surfactant protein-A (SP-A) is an important antimicrobial protein that opsonizes and permeabilizes membranes of microbial pathogens in mammalian lungs. Previously, we have shown that Pseudomonas aeruginosa flagellum-deficient mutants are preferentially cleared in the lungs of wild-type mice by SP-A-mediated membrane permeabilization, and not by opsonization. In this study, we report a flagellum-mediated mechanism of P. aeruginosa resistance to SP-A. We discovered that flagellum-deficient (ΔfliC) bacteria are unable to produce adequate amounts of exoproteases to degrade SP-A in vitro and in vivo, leading to its preferential clearance in the lungs of SP-A+/+ mice. In addition, ΔfliC bacteria failed to degrade another important lung antimicrobial protein lysozyme. Detailed analyses showed that ΔfliC bacteria are unable to upregulate the transcription of lasI and rhlI genes, impairing the production of homoserine lactones necessary for quorum-sensing, an important virulence process that regulates the production of multiple exoproteases. Thus, reduced ability of ΔfliC bacteria to quorum-sense attenuates production of exoproteases and limits degradation of SP-A, thereby conferring susceptibility to this major pulmonary host defense protein.
Keywords: Surfactant Protein-A, membrane permeabilization, Pseudomonas aeruginosa, flagellum, exoproteases
Introduction
Pseudomonas aeruginosa is one of the most common causes of nosocomial infections in humans and lung infection in patients with cystic fibrosis (CF) (Lyczak et al., 2002). It is also a primary cause of sepsis and death in immunocompromised individuals (e.g., burns, cancer chemotherapy, HIV). Antibiotic-resistant P. aeruginosa is an emerging clinical problem that can lead to denial of lung transplantation in children and adults with pulmonary failure, and to death in premature infants (Conway et al., 2003). Thus, there is an urgent need to explore alternative strategies for better management of P. aeruginosa infections.
Through its carbohydrate recognition domain, the calcium-dependent pulmonary collectin surfactant protein-A (SP-A) binds and opsonizes a myriad of microbes, enhancing their clearance (Hawgood and Shiffer, 1991; Crouch and Wright, 2001, Wright 2005). Severely depleted levels of SP-A have been associated with several respiratory diseases including bacterial pneumonia, adult respiratory distress syndrome (Baughman et al., 1993; Gunther et al., 1996; Levine et al., 1996) and CF (Griese et al., 1997; Postle et al., 1999; Noah et al., 2003). SP-A−/− mice are more susceptible to lung infection with P. aeruginosa and other pathogens (Crouch and Wright, 2001). Furthermore, replacement of collectins in animals deficient in SP-A and surfactant protein-D (SP-D) corrected defects in clearance of microbes from the lungs of rodents, suggesting a possible role for these proteins in human therapies (Crouch and Wright, 2001).
Recently, we and others have reported that SP-A also directly kills microbes in a macrophage-independent manner, by increasing the permeability of microbial membranes (Wu et al., 2003; McCormack et al., 2003; Zhang et al., 2005, Zhang et al., 2007). However, the mechanism by which SP-A disrupts microbial cell membranes and its relative importance in lung defense are poorly defined. Apart from lipopolysaccharide (LPS), which is required for resistance to SP-A-mediated membrane permeabilization (Schaeffer et al., 2004; Zhang et al., 2005; Kuzmenko et al., 2006; Zhang et al., 2007), the mechanism(s) by which microbes protect themselves from SP-A is unknown. By comparative signature-tagged mutagenesis screens of a P. aeruginosa mutant library in the lungs of SP-A+/+ versus SP-A−/− mice, we identified a P. aeruginosa mutant that is incapable of making flagellum (flgE), is preferentially cleared in the SP-A+/+ mouse lungs (Zhang et al., 2007). Strikingly, the flgE mutant bacteria were more susceptible to SP-A-mediated membrane permeabilization, an event that was independent of macrophage-mediated killing. Further analysis revealed that the flagellum-deficient mutants flgE, fliC, and fliD are attenuated in their ability to synthesize adequate amounts of LPS, resulting in a compromised outer membrane, thus rendering them more susceptible to SP-A-mediated membrane permeabilization than wild-type bacteria (Zhang et al., 2007).
During routine bacterial culturing, we have noticed that flagellar hook (flgE) and flagellin (ΔfliC) deficient P. aeruginosa mutant strains produce less pyocyanin, a redox-active toxic secondary metabolite (Lau et al., 2004). Because the production of pyocyanin and other virulence factors including exoproteases are regulated by the bacterial intercellular communication process called quorum-sensing (QS), and because the flagellum is a large cell surface appendage emanating from the bacterial membrane, we surmise that the loss of flagellum may cause pleiotropic defects that render bacteria susceptible to SP-A-mediated killing. In this study, we demonstrate that ΔfliC P. aeruginosa bacteria are unable to produce adequate amounts of homoserine lactones required to positively upregulate the production of exoproteases. This results in reduced degradation of SP-A during lung infection which in turn contributed to enhanced clearance of the ΔfliC bacteria in the lungs of SP-A+/+ mice.
Results
The ΔfliC mutant is cleared more efficiently following lung infection in SP-A+/+ but not SP-A−/− mice
Previously, we have reported that the flagellar hook-deficient mutant flgE is susceptible to clearance in the lungs of SP-A+/+ mice (Zhang et al., 2007). In this study, we examined the ΔfliC mutant in single infection studies. In the absence of infection, histopathological features of SP-A−/− mouse lungs were not significantly different when compare to the lungs of SP-A+/+ mice (data not shown). Eighteen hr after intranasal inoculation with the wild-type strain PAO1, SP-A+/+ mice showed signs of infection and respiratory distress but were not moribund. In contrast, PAO1-infected SP-A−/− mice were moribund and had to be euthanized (data not shown). The number of viable wild-type bacteria in SP-A−/− were 0.9 log higher than in SP-A+/+ mice (Fig. 1A). Eighteen hr after infection with the ΔfliC mutant bacteria, the lungs of SP-A+/+ mice showed little sign of disease. In contrast, SP-A−/− infected with ΔfliC mutant bacteria developed more serious signs of infection with respiratory distress. The viable counts of ΔfliC mutant were 1.8 log lower than PAO1 in SP-A+/+ mice. However, the number of ΔfliC bacteria was 1.75 log higher in SP-A−/− mice than in SP-A+/+ mice, and was statistically indistinguishable when compared to the number of PAO1 bacteria in the SP-A+/+ mice (Fig. 1A). These results suggest that ΔfliC bacteria are more virulent in the lungs of SPA−/− mice than in the lungs of SP-A+/+ mice. The attenuation of virulence of ΔfliC bacteria was not due to reduced growth rate as wild-type PAO1, ΔfliC and PAOC-fliC bacteria have virtually identical growth kinetics (Fig. 1B). Histopathological analysis suggests that PAO1 caused broncho-pneumonia with pulmonary infiltrates whereas the ΔfliC mutant caused only alveolitis in the lungs of SP-A+/+ mice (Fig. 1C; Zhang et al., 2005). Furthermore, PAO1 caused more extensive consolidation with more areas of lobar pneumonia than those caused by the ΔfliC bacteria in SP-A−/− lungs. As presented in Fig. 1C, ΔfliC bacteria caused mild alveolitis in SP-A+/+ mice with little pulmonary changes. In contrast, SP-A−/− mice developed lobar pneumonia with areas of densely consolidated alveoli (Fig. 1C). These results indicate that the flagellum plays an important protective role against anti-P. aeruginosa activity mediated by SP-A.
Fig. 1. The ΔfliC mutant is attenuated for virulence in SP-A+/+ mice.
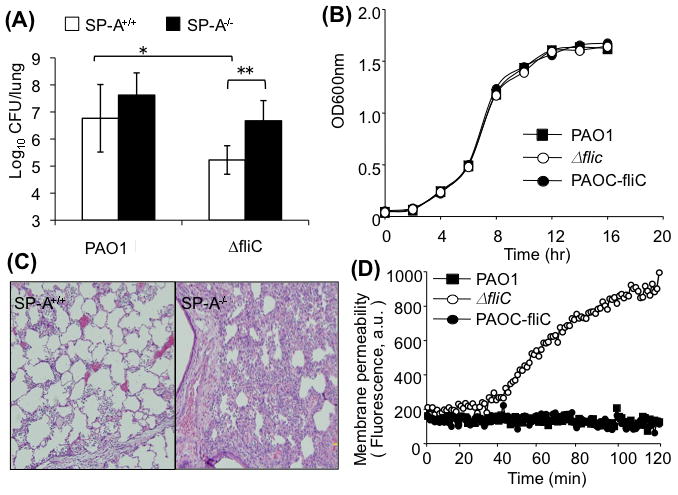
A. Respiratory tract infections with wild-type PAO1 or ΔfliC mutant bacteria were performed by intranasal inoculation of anesthetized SP-A+/+ or SP-A−/− mice. CFU in lung homogenates were enumerated 18 hr after inoculation. Data are the mean CFU ± SE (n = 5 per group). * p < 0.05 when comparing lungs of SP-A+/+ mice infected with PAO1 versus ΔfliC; ** p < 0.01 when compared between SP-A+/+ and SP-A−/− mice infected with ΔfliC bacteria.
B. Attenuation of ΔfliC bacteria in mouse lungs was not due to a slower growth rate. Bacterial growth was assessed by comparing the absorbance at OD600. The data from one of the three independent experiments are shown.
C. Representative H&E-stained lung sections from SP-A+/+ and SP-A−/− mice 18-hr post intranasal instillation of ΔfliC bacteria. Original magnification: 10x.
D. The flagellum-deficient ΔfliC bacteria are more susceptible to SP-A-mediated membrane permeabilization. Bacterial cells from the wild-type PAO1, mutant strain ΔfliC, or genetically complemented PAOC-fliC were exposed to 50 μg/ml human SP-A in the presence of the phosphatase substrate ELF97. Fluorescence was measured for 120 min using a fluorimeter and expressed as arbitrary units (a.u.). Data are means of five experiments.
The ΔfliC mutant is susceptible to SP-A-mediated membrane permeabilization
Previous studies have demonstrated that flagella act as a major ligand for nonopsonic phagocytosis of P. aeruginosa, and are required to trigger internalization (Mahenthiralingam et al., 1994; Mahenthiralingam and Speert, 1995). Our recently published studies confirm that, while flagella enhance internalization of P. aeruginosa into macrophages and neutrophils, SP-A-mediated macrophage and neutrophil phagocytosis is not responsible for the preferential clearance of flagellum-deficient flgE mutant bacteria from SP-A+/+ mice (Zhang et al., 2007). In addition, the susceptibility to SP-A-mediated membrane permeabilization is dependent on the presence of intact flagellum and flagellum-regulated LPS biosynthesis, rather than flagellum-dependent motility (Zhang et al., 2007). Here, we show that ΔfliC bacteria exhibit 4.9 fold greater susceptibility to SP-A-mediated membrane permeabilization than wild-type PAO1 bacteria after 90 min exposure to SP-A (Fig. 1D). The levels of susceptibility of ΔfliC mutant bacteria are comparable to the flgE mutant (Zhang et al., 2007), and to the E. coli K12 (Wu et al., 2003). Importantly, resistance to SP-A-mediated membrane permeabilization was fully restored to wild-type level in complemented PAOC-fliC bacteria (Fig. 1D). These results suggest that an intact flagellum or flagellar function is essential for resistance to SP-A-mediated membrane permeabilization, and are consistent with previous findings that the flagellum is required for acute lung infection by P. aeruginosa (Feldman et al., 1998).
Flagellum-deficient mutants are deficient in their ability to produce the quorum-sensing regulated exotoxin pyocyanin and SP-A-degrading exoproteases
Because the flagellum is the single largest surface appendage on the P. aeruginosa, the loss of flagellum may result in pleiotropic defects that render such bacteria susceptible to SP-A. During routine bacterial culturing, we have noticed that flagellum-deficient P. aeruginosa mutant strains flgE and ΔfliC (Table 1) produce less pyocyanin. Pyocyanin is a redox-active tricyclic secondary metabolite previously shown to be important for P. aeruginosa virulence (Lau et al., 2004, Caldwell et al., 2009). The biosynthesis of multiple secreted virulence factors including pyocyanin and exoproteases in P. aeruginosa is regulated by an intercellular communication process called quorum-sensing (QS) (Lau et al., 2004, Juhas et al., 2005). We compared the ability of wild-type PAO1, complemented strain PAOC-fliC and ΔfliC mutant to produce the QS-regulated exotoxin pyocyanin. The ΔfliC and flgE mutant bacteria produced 41% and 48% less pyocyanin than PAO1, respectively (Fig. 2A–2B). In contrast, pyocyanin production was fully restored in PAOC-fliC (Fig. 2A).
Table 1.
Bacterial Strains and Plasmids Used
| Bacterial Strains | Relevant characteristics | Reference |
|---|---|---|
| P. aeruginosa | ||
| PAO1 | Wild-type | Dasgupta et al., 2003 |
| ΔfliC | PAO1 fliC::Gmr | Fleiszig et al., 2001 |
| PAOC-fliC | ΔfliC complemented on the chromosome with the PAO1 fliC gene at the attB site | Arora et al., 2005 |
| PAO1 | Wild-type | Holloway et al., 1979 |
| FlgE | PAO1 miniTn5Km2flgE, flagellar mutant | Zhang et al., 2007 |
| ΔlasB (PDO240) | Elastase-deficient mutant derived from PAO1 | McIver et al., 1995 |
| ΔlasI ΔrhlI | PAI-1 and PAI-2 deficient double knockout mutant derived from PAO1 | Thaden et al., 2010 |
| E. coli | ||
| DH5-α | F-f80 ΔlacZΔM15 endA1 recA1 hsdR17 (r−km+k) supe44 thi-1 l-gyr A96 relA1 Δ(lacZYA-argF) U169 | Miller et al., 1988 |
| Plasmid | ||
| pUTminiTn5Km2 | R6K-based suicide delivery plasmid, Kmr | Potvin et al., 2003 |
| pSW205 | ori (colE1) ori (P. aeruginosa) promoterless lacZ Apr | Gambello et al., 1993 |
| pTS400 | lasB'-lacZ translational fusion on pSW205 | Passador et al., 1993 |
| pECP62.5 | lasB'-lacZ from pTS400 on pJPP8, tacp-lasR | Pearson et al., 1997 |
| pECP60 | rhlA'-lacZ translational fusion on pSW205 | Pesci et al., 1997 |
| pECP61.5 | rhlA'-lacZ from pECP60 on pJPP8, tacp-rhlR | Pearson et al., 1997 |
Fig. 2. Flagellum-deficient P. aeruginosa mutants produce less pyocyanin and SP-A-degrading exoproteases.
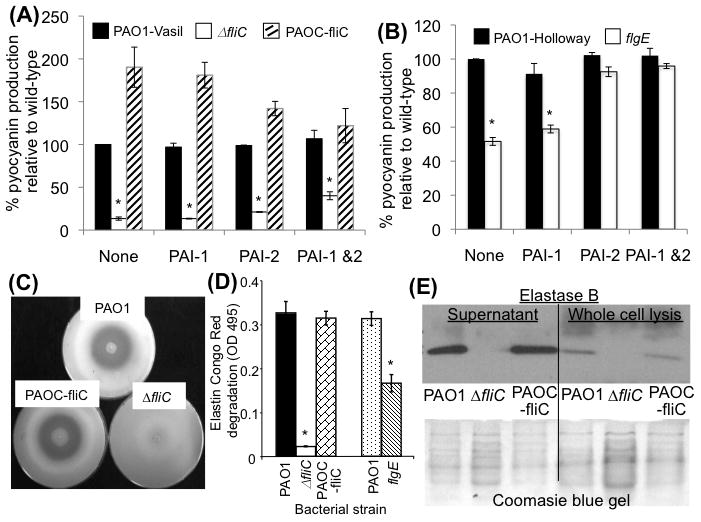
A-B. Flagellum-deficient ΔfliC and flgE mutant bacteria are attenuated in pyocyanin production. PCN production is partially restored in ΔfliC by a combination of PAI-1 and PAI-2 (A), and fully restored in flgE mutant by either PAI-1 or PAI-2. Pyocyanin concentration was measured by OD690. The experiments were performed in triplicates and repeated three times. The means ± SD from one experiments are shown. *p < 0.01 when comparing the pyocyanin levels of PAO1 or PAOC-fliC against ΔfliC.
C. Flagellum-deficient ΔfliC mutant bacteria are attenuated in the production total exoproteases. Skim milk-embedded agarose plates were inoculated with a suspension of wild-type PAO1, genetically complemented PAOC-fliC or ΔfliC bacteria. The reduced zone of clearance indicates that the ΔfliC mutant is severely attenuated in production of exoproteases. The images from one of the three independent experiments are shown.
D. Flagellum-deficient ΔfliC mutant bacteria are attenuated in their elastase capability. Elastase activity was assessed with Elastin Congo Red assays. Both ΔfliC & ΔflgE mutants are significantly attenuated in elastase activity when compared to their respective parental PAO1 strains. The experiments were performed in triplicates and repeated three times. The means ± SD from one experiments are shown. *p < 0.01 when comparing the elastase activities of PAO1 or PAOC-fliC against ΔfliC.
E. Western analysis of elastase B production by wild-type PAO1, ΔfliC and PAOC-fliC from culture supernatant and from whole cell lysis. Western blot from one of the three independent experiments are shown.
Previous studies have shown that P. aeruginosa secretes elastase and protease IV, enzymes that degrade SP-A and SP-D in vitro (Mariencheck et al., 2003; Alcorn and Wright, 2004; Beatty et al., 2005; Malloy et al., 2005). Because the production of exoproteases also is regulated by QS, we examined whether the ability of ΔfliC mutant to produce exoproteases was compromised. As shown in Fig. 2C, wild-type PAO1 and the complemented PAOC-fliC bacteria produced similar amounts of exoproteases that degrade proteins in skim milk. In contrast, the ΔfliC bacteria failed to clear skim milk. Elastin Congo Red analysis showed that ΔfliC bacteria were severely reduced in their elastase activities (Fig. 2D) when compared to the PAO1 bacteria or the complemented PAOC-fliC bacteria. Importantly, flgE mutant bacteria were also attenuated in their ability to degrade elastin, although the levels of reduction were not as dramatic as in the ΔfliC bacteria (Fig. 2D). Western blot analysis on proteins in whole cell extracts and culture supernatant confirmed that ΔfliC mutant bacteria produced and secreted less elastase B than PAO1 and PAOC-fliC when grown in tryptic soy broth (Fig. 2E).
The flagellum-deficient ΔfliC mutant is impaired in its ability to degrade SP-A
We assessed the implication of impaired production of exoproteases by ΔfliC mutant on its ability to degrade SP-A. SP-A (50 μg) was incubated with 1 × 108 PAO1, ΔfliC or complemented PAOC-fliC bacteria for the indicated time intervals. After 6 hr of incubation, degradation of SP-A by PAO1 and by PAOC-fliC bacteria was clearly visible. By 12 hr post incubation, SP-A was almost completely degraded PAO1 and PAOC-fliC cells (Fig. 3A). In contrast, SP-A incubated with ΔfliC bacteria remained intact, with little indication of degradation 18 hr post-coincubation. As control for lack of SP-A degradation, SP-A was co-incubated with an elastase-deficient ΔlasB mutant PDO240. Very little SP-A degradation was detected (Fig. 3B). These results further confirm that inability of ΔfliC to degrade SP-A is due to its deficiency in exoprotease activity.
Fig. 3. SP-A-degrading ability is reduced in ΔfliC and ΔlasB mutant bacteria in vitro.
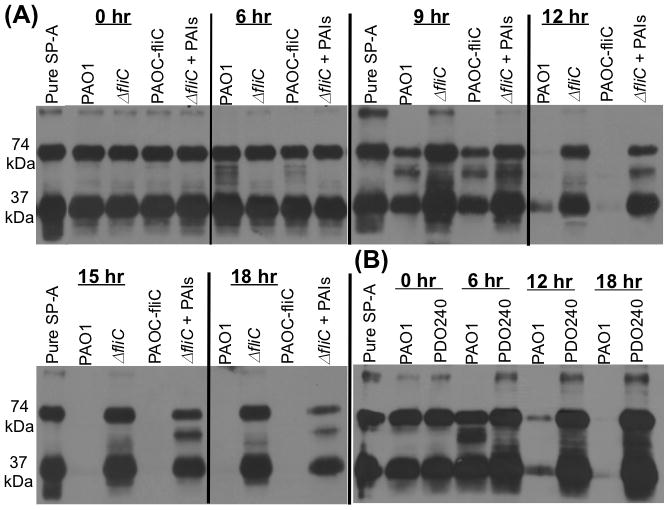
A. SP-A (25 μg) was incubated with 1 × 108 PAO1, ΔfliC or PAOC-fliC bacteria for the indicated time intervals. A subset of ΔfliC bacteria-SP-A mixture was supplemented with N-acyl homoserine lactones PAI-1 and PAI-2 (PAIs). SP-A degradations were assessed qualitatively by Western analyses of 20 μl of SP-A/bacterial suspension. Western blots were probed with anti-SP-A antibody. The results of one out three independent experiments are shown.
B. SP-A (25 μg) was incubated with 1 × 108 PAO1 or ΔlasB mutantPDO240 bacteria for the indicated time intervals. SP-A degradation was assessed as described in A. The western blot from one of the three independent experiments is shown.
Decreased production of exoproteases attenuates the ability of ΔfliC bacteria to degrade SP-A during infection of SP-A+/+ mouse lungs
Although in vitro studies have shown that P. aeruginosa can secrete elastase and protease IV to degrade SP-A and SP-D (Mariencheck et al., 2003; Alcorn and Wright, 2004; Beatty et al., 2005; Malloy et al., 2005), the biological importance of P. aeruginosa exoproteases in the removal of SP-A and the resulting resistance to SP-A-mediated membrane permeabilization during infection of SP-A+/+ lungs is unknown. To examine whether exoprotease deficiency may have contributed to enhanced clearance of ΔfliC from SP-A+/+ mouse lungs, we compared in vivo SP-A degradation by wild-type PAO1, the ΔfliC mutant, the complemented strain PAOC-fliC. An elastase deficient ΔlasB mutant PDO240, was included as control that lacks the ability to degrade SP-A. Bronchoalveolar lavage (BAL) samples from mice infected with wild-type PAO1 or genetically complemented PAOC-fliC had significantly decreased SP-A (Fig. 4A) in this western blot analysis. In contrast, we detected intact SP-A and partially degraded SP-A from BAL of mice infected with ΔfliC and ΔlasB bacteria (Fig. 4A). Coomassie Blue staining indicated that equal loading of BAL proteins were used for western blot analysis (Fig. 4B). These results indicate that P. aeruginosa is capable of evading host defenses by secreting exoproteases to degrade SP-A, and that the flagellum-deficient ΔfliC mutant is unable to degrade SP-A resulting from an inability to synthesize adequate exoproteases. This culminates in the enhanced clearance of the ΔfliC mutant from the SP-A+/+ mouse lungs.
Fig. 4. Flagellum-deficient ΔfliC mutant degrade less SP-A during lung infection.
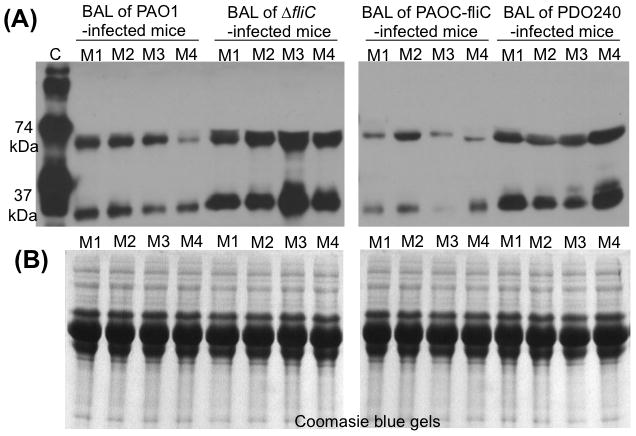
A. Intact SP-A protein was reduced in the BAL fluid from PAO1- or PAOC-fliC-infected SP-A+/+ mice (n = 4), suggesting that SP-A was degraded in mouse lungs. In contrast, more abundant SP-A was clearly visible in the BAL fluids from ΔfliC (n=4). BAL of mice infected with ΔlasB mutant PDO240 (n = 4) was used as negative control for degradation of SP-A. C = Purified human SP-A. M1 - M4 = BAL of four mice infected with P. aeruginosa. Western blot analyses were performed using a monoclonal antibody against SP-A.
B. Coomassie blue-stained gels indicate equal amounts of BAL proteins from mouse lungs were loaded into each lane in A.
BAL samples of mouse lungs infected with ΔfliC mutant bacteria but not wild-type PAO1 or complimented PAOC-fliC bacteria are capable of permeabilizing bacterial membranes
A previous study suggested that SP-A is a principal microbial permeabilizing factor in the alveolar lining fluid (Kuzmenko et al., 2005). Given this information, we next compared the membrane permeabilizing ability of BAL samples (from Fig. 4) from SPA+/+ mouse lungs following infection with PAO1, PAOC-fliC or ΔfliC bacteria. BAL samples from PAO1- (Fig. 5A) or PAOC-fliC-infected (Fig. 5B) SP-A+/+ mice were unable to permeabilize PAO1, PAOC-fliC, ΔfliC or E. coli DH5-α bacteria. In contrast, BAL fluids from ΔfliC-infected mice possessed 3.8- to 4-fold higher capacity to permeabilize both ΔfliC and E. coli DH5-α bacteria (Fig. 5C) relative to BAL samples from PAO1- or PAOC-fliC-infected mice (compare Fig. 5C against Figs. 5A and 5B). Furthermore, BAL samples from ΔfliC-infected mice were able to permeabilize both PAO1 and PAOC-fliC bacteria about 2.3-fold higher than BAL samples from PAO1 or PAOC-fliC-infected mice. These results suggest that elaboration of exoproteases is an important protective mechanism against the antimicrobial activities of SP-A during lung infection by P. aeruginosa and therefore a significant mechanism of evading host defenses.
Fig. 5. Mouse BAL from ΔfliC-infected animals contains intact SP-A that permeabilizes bacterial membranes. Pooled BAL fluids (Fig. 4) from SP-A+/+ mice infected with PAO1 or PAOC-fliC or ΔfliC bacteria (50 μg/ml total proteins) were used for membrane permeabilization assays.
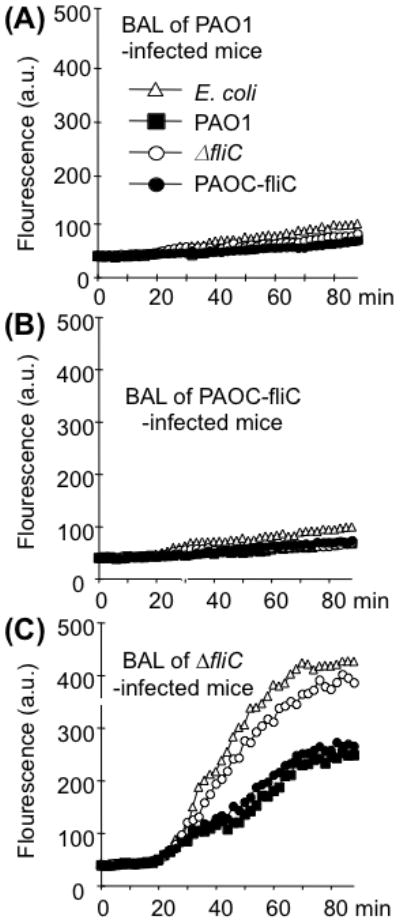
A-B. BAL samples from PAO1 and PAOC-fliC infected mice fail to permeabilize bacterial membranes. Data are expressed as the mean of four experiments.
C. BAL samples from ΔfliC-infected mice are able to permeabilize bacterial membranes of PAO1 and PAOC-fliC bacteria at low level, and E. coli DH5-α and ΔfliC bacteria at higher levels. Data are expressed as the mean of four experiments.
Protease-deficient ΔfliC mutant is defective in its ability to degrade lysozyme in vivo
We have previously shown that wild-type P. aeruginosa strain PAO1 is highly resistant to human SP-A-mediated membrane permeabilization in vitro (Zhang et al., 2005; Zhang et al., 2007). However, membrane permeabilization studies shown that BAL samples from ΔfliC mutant were able, albeit weakly, to permeabilize membranes of PAO1 and PAOC-fliC (Fig. 5). One possible explanation for this observation is that additional pulmonary innate immunity proteins upregulated during infection by ΔfliC were acting synergistically to permeabilize the membranes of PAO1 and PAOC-fliC bacteria. Given this unanticipated twist, we examined BAL samples for the presence of lysozyme, a known lung innate immune protein that is capable of permeabilizing bacterial membranes. As shown in Fig. 6A, BAL samples from mice infected with PAO1 or PAOC-fliC had trace amounts of lysozyme. In contrast, BAL samples from mice infected with ΔfliC or ΔlasB mutant still contained intact lysozyme. Thus, infection by P. aeruginosa likely induced the expression of lysozyme, which was subsequently degraded by exoproteases produced by PAO1 or PAOC-fliC. In contrast, due to inability of the ΔfliC and ΔlasB mutants to produce adequate exoproteases, SP-A and lysozyme remained intact, and might have acted synergistically to enhance the permeabilization of PAO1 and PAOC-fliC cells. To confirm lysozyme degradation, we incubated 20 ng/ml lysozyme with 1 × 108 PAO1, ΔfliC, PAOC-fliC or ΔlasB mutant PDO240 bacteria. After 18 hr incubation, lysozyme exposed to ΔfliC or PDO240 mutant remained intact (Fig. 6B–6C). In contrast, PA01 or PAOC-fliC bacteria were able to degrade lysozyme (Fig. 6B–6C). PAO1Antimicrobial synergy between various combinations of lung innate proteins has been demonstrated in vitro (Singh et al., 2000; Yan and Hancock, 2001). To assess this possibility, we performed permeabilization assays using wild-type PAO1 bacteria with SP-A and lysozyme, singly or in combination. As shown in Fig. 6D, control PAO1 bacteria (without exposure to SP-A or lysozyme) or PAO1 exposed to SP-A showed little or no membrane permeabilization. By itself, lysozyme was more efficient than SP-A in permeabilizing the membrane of PAO1. However, when PAO1 bacteria were exposed to a combination of SP-A and lysozyme, there was a synergistic membrane permeabilization effect.
Fig. 6. Exoprotease-deficient ΔfliC mutant is defective in its ability to degrade lysozyme in vivo and in vitro.
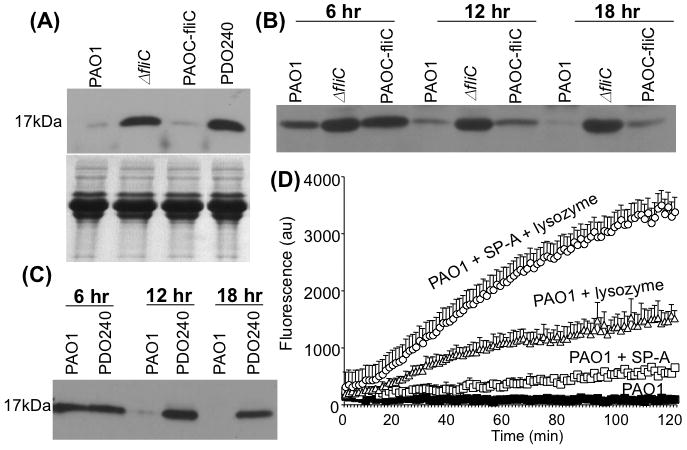
A. Western blot analysis of proteins within BAL samples derived from infected mice (from Fig. 4) indicates that PAO1 and PAOC-fliC were able to degrade lysozyme. In contrast, the ΔfliC and the ΔlasB− PDO240 mutants were not able to degrade lysozyme in vivo. The western blot from one of the three independent experiments is shown.
B-C. Lysozyme (5μg) was incubated with 1 × 108 PAO1, ΔfliC, or PAOC-fliC (B) or PAO1 versus PDO240 (C) for the indicated time intervals. Lysozyme degradations were assessed qualitatively by Western blotting analyses using 10 μl of lysozyme/bacterial suspension. Immunoblot was probed with anti-lysozyme antibody. The western blot from one of the three independent experiments is shown.
D. SP-A and lysozyme act synergistically to permeabilize the wild-type PAO1 membrane. PAO1 bacteria were incubated in the presence of SP-A and lysozyme singly or in combination. Experiments were performed in triplicates and repeated three times. The mean ± SD from one typical experiment is shown.
Flagellum deficiency abolishes the ability of P. aeruginosa to produce quorum-sensing autoinducers PAI-1 and PAI-2
It is well established that the production of exoproteases in P. aeruginosa is regulated by the QS signaling cascade (reviewed in Juhas et al., 2005, Lau et al., 2005). Because the single polar flagellum is required to mediate chemotactic signaling in response to environmental changes (reviewed in Bren and Eisenbach, 2000), we hypothesize that the exoprotease deficiency in ΔfliC mutant bacteria is due to their attenuated ability to produce or sense quorum sense QS signaling homoserine lactones, PAI-1 and PAI-2, which are required to activate the synthesis of exoproteases. Flagella provide a positive feedback mechanism to maintain the continuous biosynthesis of QS molecules. We compared the production of QS autoinducers PAI-1 and PAI-2 between wild-type PAO1, PAOC-fliC or ΔfliC bacteria. E. coli strains harboring lasB'-lacZ translational fusions in the presence (lasB'-lacZ, tacp-lasR) or absence (lasB'-lacZ, no tacp-lasR) of LasR transcription factor, and E. coli strains harboring rhlA'-lacZ translational fusions in the presence (rhlA'-lacZ, tacp-rhlR) or absence (rhlA'-lacZ, no tacp-rhlR) of RhlR transcription factor were exposed to PAI-1 or PAI-2 extracts derived from wild-type PAO1, PAOC-fliC or ΔfliC bacteria, respectively. As shown in Fig. 7A, PAI-1/PAI-2 extracts from wild-type PAO1 or complemented PAOC-fliC strains activated the expression of lasB-lacZ in E. coli at levels 45-fold and 23-fold higher than extracts from ΔfliC bacteria, respectively. Similarly, PAI-1/PAI-2 extracts from wild-type PAO1 and complemented PAOC-fliC strains activated the expression of rhlA-lacZ in E. coli at levels 15-fold and 10-fold higher than extracts from ΔfliC bacteria, respectively (Fig. 7B). These results suggest that the ΔfliC bacteria are either defective in the expression of QS autoinducers PAI-1 and PAI-2 or unable to sense the presence of QS molecules PAI-1 and PAI-2. To examine these two possibilities, we compared the transcript levels of lasI and rhlI genes, which encode PAI-1 and PAI-2, respectively, by quantitative real time PCR (qRT-PCR) between the wild-type PAO1 and the ΔfliC mutant. A ΔlasI ΔrhlI mutant strain (Table 1) was used as negative control for the transcription of these two genes. As expected, neither lasI nor rhlI transcript was detected in the ΔlasI ΔrhlI mutant strain (data not shown). The expression of lasI and rhlI genes in the wild-type PAO1 bacteria was maintained at high levels, and was between 3-5 logs above the expression of lasI and rhlI genes in the ΔfliC mutant throughout both log and stationary phases of growth. Interestingly, the only time point when the expression of lasI and rhlI genes in the ΔfliC mutant increased was during the transition from early log to late log phase, but the increase was transient and returned to basal levels throughout early to late staionary phases of growth. These results suggest that the expression of lasI and rhlI in the ΔfliC mutant were severely attenuated when compared to the wild-type PAO1.
Fig. 7. The ΔfliC mutant bacteria are reduced in their ability to produce quorum-sensing homoserine lactones.
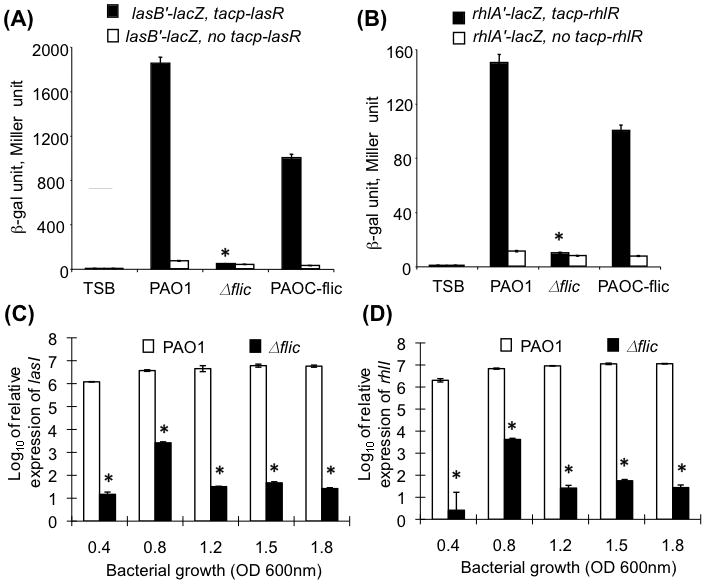
A-B. PAI-1 and PAI-2 extracts from the wild-type PAO1 and complemented strain PAOC-fliC were able to induce the expression of lasB-lacZ (A) and rhlA-lacZ (B) in E. coli whereas extracts from ΔfliC mutant bacteria failed to induce the expression of both lasB-lacZ (A) and rhlA-lacZ (B) in E. coli. Experiments were performed in triplicates and repeated three times. The means ± SD of one typical experiment are shown. *p < 0.01 when compared the β-gal activities of PAO1 or PAOC-fliC against ΔfliC.
C-D. qRT-PCR analysis of lasI and rhlI expression in flagellum-deficient ΔfliC mutant. The transcript levels of lasI (C) and rhlI (D) in ΔfliC bacteria were significantly reduced when compared to wild-type PAO1 through all phases of growth. Experiments were performed in triplicates and repeated three times. The means ± SD of one typical experiment are shown. *p < 0.001 when compared the transcript levels of lasI/rhlI of PAO1 against ΔfliC.
Provision of exogenous autoinducer PAI-1, PAI-2 and PQS restored the ability of ΔfliC to produce exoprotease
If the defective exoprotease production in the ΔfliC bacteria is solely due to decreased transcription of lasI and rhlI genes, but not due to its inability to respond to PAI-1 and PAI-2, then provision of these QS molecules should restored the production of exoproteases to the ΔfliC bacteria. The production of exoproteases is dependent of homoserine lactone molecules PAI-1, PAI-2, the quinolones PQS (Diggle et al., 2006), as well as divalent cations Ca2+ and Zn2+ (Morihara, 1964; Olson and Ohman, 1992; Sarkisova et al., 2005). We examined the exoprotease activities of bacteria in the Ca2+ and Zn2+-sufficient tryptic soy broth (TSB) or TSB treated with Chelex 100 to deplete cations (TSBD). Bacteria were grown to stationary phase (to allow for maximal QS) in TSB culture media or in TSBD. As shown in Fig. 8A, in the Ca2+- and Zn2+-deficient TSBD medium, provision of PAI-1, PAI-2 or PQS, a third signaling component of signaling homoserine lactones, alone or in combination could not induce the exoprotease activity in PAO1, PAOC-fliC and ΔfliC. Exoprotease activity was only detected in Ca2+- and Zn2+-sufficient TSB cultures of PAO1 and PAOC-fliC bacteria, and not in culture supernatants of Δ ΔfliC bacteria. These results suggest that Ca2+ and Zn2+ are indispensable for the activities of exoproteases; and that provision of QS molecules PAI-1, PAI-2 and PQS alone is not sufficient to induce the production of exoproteases.
Fig. 8. Production of exoproteases in ΔfliC was restored by the provision of quorum–sensing molecules PAI-1 and PAI-2, and PQS.
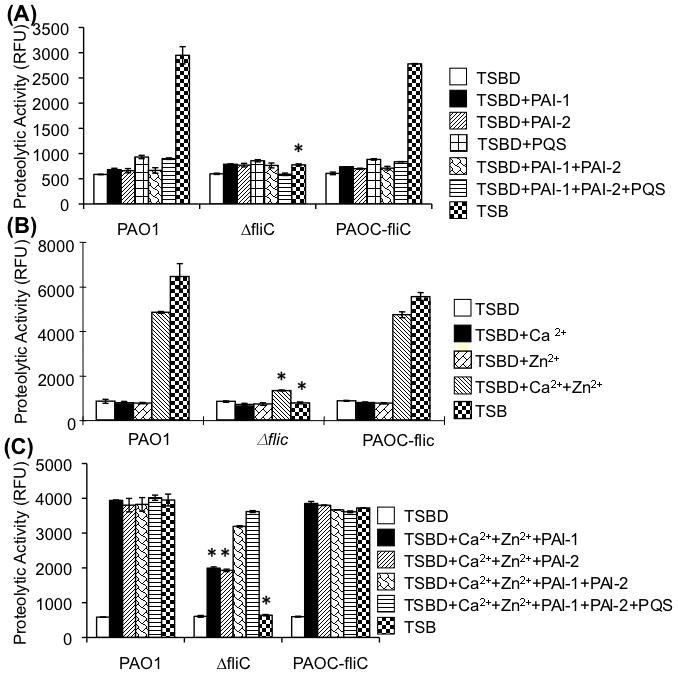
A. Wild type PAO1, ΔfliC mutant, or complemented strain PAOC-fliC were cultured in tryptic soy broth (TSB) or TSB treated with Chelex 100 to deplete all the divalent cations, Ca2+ and Zn2+ (TSBD). TSBD was supplemented singly or in combinations with PAI-1, PAI-2 or PQS. In the absence of PAI-1, PAI-2 and PQS, no exoprotease activity was detected in TSBD from any of the bacterial strains. However, both stationary phase PAO1 and PAOC-fliC were able to produce proteases in Ca2+/Zn2+-sufficient TSB. In contrast, the ΔfliC mutant was unable to produce proteases when grown in TSB. Experiments were performed in triplicates and repeated three times. The means ± SD of one typical experiment are shown. *p < 0.01 when compared the exoprotease activities of PAO1 or PAOC-fliC against ΔfliC.
B. Bacteria were cultured in Ca2+/Zn2+-sufficient TSB or in TSBD. Wild-type PAO1 and complemented strain PAOC-fliC were able to respond to Ca2+ and Zn2+ with increased proteolytic activities. In contrast, ΔfliC mutant failed to respond to both cations and was unable to produce exoproteases. Experiments were performed in triplicates and repeated three times. The means ± SD of one typical experiment are shown. *p < 0.01 when compared the exoprotease activities of PAO1 or PAOC-fliC against ΔfliC.
C. Exogenously supplied quorum sensing molecules restored the exoprotease activity in ΔfliC. Provision of either PAi-1 or PAI-2 fully restored exoprotease activity in PAO1 or PAOC-fliC, and only partially restored the exprotease activity in ΔfliC. The combination of exogenously supplied PAI-1, PAI-2 and PQS was able to restore the production of exoproteases by ΔfliC in TSBD supplemented with Ca2+/Zn2+. Experiments were performed in triplicates and repeated three times. The means ± SD of one typical experiment are shown. *p < 0.01 when compared the exoprotease activities of PAO1 or PAOC-fliC against ΔfliC.
We then assayed for the activities of exoproteases by bacteria cultured in the presence or absence of cations Ca2+ and/or Zn2+ alone, in the absence of QS molecules. Provision of Ca2+ or Zn2+ alone was insufficient to activate the production of exoproteases in TSBD medium (Fig. 8B) suggesting that exoprotease activities required the presence of both cations. As expected, wild-type PAO1 bacteria or complemented PAOC-fliC bacteria were able to produce abundant exoprotease activities in cation-sufficient TSB or TSBD supplemented with both Ca2+ and Zn2+ (Fig. 8B). In contrast, ΔfliC mutant bacteria were unable to produce exoprotease activities when cultured in TSB or in TSBD supplemented with both Ca2+ and Zn2+ (Fig. 8B). These results suggest that PAO1 and PAOC-fliC but not ΔfliC were able to produce their own QS molecules and activate the production of exoproteases in the presence of sufficient Ca2+ and Zn2+. We next examined whether provision of both cations and QS molecules could restore the exoprotease activities in ΔfliC mutant bacteria. As shown in Fig. 8C, in the presence of both Ca2+ and Zn2+, provision of PAI-1 or PAI-2 alone or both PAI-1 and PAI-2 or PAI-1/PAI-2/PQS in TSBD was sufficient to induce the production of exoproteases in wild-type PAO1 and the complemented strain PAOC-fliC. In contrast, in the presence of both Ca2+ and Zn2+, provision of PAI-1 or PAI-2 alone could only restore ~40% of exoprotease activities in ΔfliC mutant. Provision of PAI-1/PAI-2 or PAI-1/PAI-2/PQS induced 80.5% and 92.5% of exoprotease activity respectively, to the ΔfliC mutant. These results suggest that PAI-1 and PAI-2 are the main regulators of exoprotease activities in P. aeruginosa. The contribution of PQS to the exoprotease activities may be less significant. However, we cannot rule out that our protease assays may be at the maximum level of detection in the presence of excess PAI-1/PAI-2 and cations, masking the effects of PQS.
We have also examined whether provision of PAI-1 and PAI-2 could restore the production of pyocyanin in both ΔfliC and flgE mutant bacteria. As shown in Fig. 2A–B, provision of PAI-2 but not PAI-1 could minimally restore the production of pyocyanin in ΔfliC mutant bacteria. Provision of PAI-2 also partially restored the production of pyocyanin in flgE mutant, albeit to a higher level than in ΔfliC bacteria. Collectively, these results suggest that PAI-1/PAI-2/PQS synergistically regulate the production of exoproteases whereas PAI-2 alone positively regulates the production of pyocyanin.
Discussion
In this study, we examined the molecular mechanisms underlying the increased susceptibility of a flagellum-deficient ΔfliC mutant of P. aeruginosa to clearance by SP-A. We showed that the ΔfliC mutant was more readily cleared from SP-A+/+ mouse lungs, but was virulent in SP-A−/− mouse lungs. Because we have previously shown that SP-A-mediated opsonization does not play a role in the preferential clearance of ΔfliC bacteria (Zhang et al., 2007), the attenuation of these flagellum-deficient bacteria in mouse lungs is likely caused by increased susceptibility to SP-A-mediated membrane permeabilization. For the first time, we demonstrate that the ΔfliC bacteria are deficient in the protective activities of exoproteases that are required to degrade SP-A in vitro, and during infection of mouse lungs. The ΔfliC bacteria are also unable to degrade another pulmonary innate immunity protein lysozyme. We show that P. aeruginosa flagella appear to function as a positive feedback loop that is required to maintain transcription of QS molecules PAI-1 and PAI-2, although the mechanism for this effect on gene regulation is unknown. The loss of flagella in ΔfliC bacteria severely reduces transcripts of lasI and rhlI genes. The defect in exoprotease activity is not caused by the inability of ΔfliC bacteria to sense the environment as provision of excess PAI-1/PAI-2/PQS together with Ca2+/Zn2+ could completely restore the production of exoproteases.
Previously, Feldman et al (1998) reported that the flagellum of P. aeruginosa plays a role in virulence in a mouse model of acute pneumonia. Nonmotile ΔfliC mutant bacteria were found to cause local inflammation at the site of inoculation, but failed to disseminate throughout the mouse lung or systemically via the bloodstream. Various mechanisms of abrogation of virulence have been proposed, including an inability to attach to host mucin (Adamo et al., 2004; Prince, 2006) and failure to initiate biofilm formation (O’Toole et al., 1998; Wagner and Iglewski, 2008). Another explanation is that flagellum-mediated chemotaxis (Dasgupta et al., 2003) may be required for P. aeruginosa to localize to alveolar micro-compartments devoid of SP-A. However, our previously published in vitro data refute this hypothesis. Genetic and biochemical analyses have revealed that the stator-deficient ΔmotAB ΔmotCD mutant, which has an intact flagellum structure but is nonmotile (Toutain et al., 2005), is as resistant to SP-A-mediated membrane permeabilization as its isogenic wild-type parental strain (Zhang et al., 2007). Our experimental data provide additional clues to enhanced clearance of flagellum-deficient P. aeruginosa mutants from the lung. Firstly, the lack of flagella reduces the ability of P. aeruginosa to synthesize adequate LPS to stabilize the outer membrane in order to withstand the “assault” of SP-A and the detergent 0.25% SDS (Zhang et al., 2007). Secondly, as shown in the current study, flagellum-deficient P. aeruginosa mutants are reduced in their ability to upregulate the transcription of lasI and rhlI QS signaling molecules that are essential for the production of exoproteases required to degrade innate host defense proteins, including SP-A and lysozyme, during lung infections.
The production of many P. aeruginosa exoproteases is tightly regulated by QS, an inter-cellular signaling process (Juhas et al., 2005) as well as by the availability of cations Zn2+ and Ca2+. In fact, the expression and processing of the predominant QS-regulated P. aeruginosa protease, elastase, is dependent on the presence of Zn2+ and Ca2+ (Morihara, 1964; Olson and Ohman, 1992; Sarkisova et al., 2005). As a metalloprotease, elastase requires Zn2+ for its catalytic activity, which is stabilized by Ca2+ (Morihara, 1964; Thayer et al., 1991). These analyses were subsequently confirmed by Sarkisova et al., (2005), who showed that Zn2+ and Ca2+ drastically induced the production of exoproteases in P. aeruginosa biofilms that formed under in vitro conditions. We have shown that the ΔfliC bacteria are severely attenuated in the transcription and production of QS autoinducers PAI-1 and PAI-2, and QS-regulated elastase and pyocyanin. The loss of exoproteases, pyocyanin and QS molecules is not due to inability of ΔfliC bacteria to undergo chemotaxis sensing of environments because exogenously supplied QS molecules in combination with Ca2+ and Zn2+ completely restore their exoprotease activities.
The cause-and-effect relationship between the lack of exoprotease and pyocyanin production and exoprotease activities in the ΔfliC bacteria is not immediately obvious. One could argue that reduced exoprotease and pyocyanin production in the ΔfliC bacteria is caused by a secondary mutation within the QS genetic machinery. However, several reasons argue against this notion: Firstly, production of exoproteases and pyocyanin was fully rescued in the genetically complemented strain, PAOC-fliC. Secondly, another flagella mutant, the flgE mutant that is derived from a different wild-type PAO1 strain, is also attenuated in the production of QS-regulated exoproteases and pyocyanin. Thirdly, as we have previously reported, flagellum-deficient mutants flgE, ΔfliC and ΔfliD exhibit heightened susceptibility to SP-A-mediated membrane permeabilization and 0.25% SDS. This is caused by the reduced ability of these mutants to upregulate the biosynthesis of LPS (Zhang et al., 2007). QS was originally thought not to regulate LPS biosynthesis (De Kievit et al., 2001). However, more recent sophisticated microarray analyses suggest that QS negatively regulates the expression of some LPS genes at low levels (Wagner et al., 2004). Thus, if the ΔfliC mutant carries a secondary mutation within the QS genetic machinery that resulted in loss of exoprotease production, it should also have resulted in increased expression of LPS. However, our previously published results demonstrate that various flagellum-deficient mutants are attenuated in their ability to synthesize LPS (Zhang et al., 2007). Instead, we propose that the ΔfliC bacteria are unable to provide a positive feedback regulation that is required to maintain a high levels of lasI/rhlI transcription, attenuating its ability to synthesize adequate PAI- and PAI-2 to induce the production of exoproteases. Interestingly, the expression of both lasI and rhlI genes was visibly increased by 2 and 3 logs respectively in ΔfliC bacteria when they entered the late log phase (OD600 0.8), but dropped back to basal levels when these bacteria transitioned into early stationary phase of growth. These results suggest that the positive feedback loop maintaining the high transcription of lasI and rhlI genes have been disrupted in the ΔfliC bacteria, impairing their ability to produce exoproteases and pyocyanin.
The important role of proteases in host-pathogen interactions is well documented. For example, neutrophil elastase has been reported to degrade SP-A and SP-D (Pison et al., 1989; Liau et al., 1996; Hirche et al., 2004; Rubio et al., 2004; Cooley et al., 2008) and implicated in the pathogenesis of chronic airway diseases, including CF (Voynow et al., 2008). Multiple in vitro studies have shown that P. aeruginosa secretes exoproteases to degrade host innate immune proteins, including SP-A and SP-D (Lema et al., 2000; Mariencheck et al., 2003; Alcorn and Wright, 2004; Beatty et al., 2005; Malloy et al., 2005), cytokines (reviewed by Galloway, 1991), several complement components, including the opsonin C3 and the chemotactic peptide C5 (Morihara 1964; Schultz et al., 1974; Schad et al., 1987) and immunoglobulin G (Schultz et al., 1974). Previously, Kuzmenko et al (2005) have suggested that SP-A is a principal microbial permeabilizing factor in the alveolar lining fluid. Thus, our in vivo studies, which demonstrate for the first time that P. aeruginosa secretes exoproteases that degrade SP-A and thereby evades clearance from the lung, may have potentially important clinical and therapeutic implications. We show that degradation of SP-A abolishes its ability to permeabilize P. aeruginosa membranes. We predict that it will also incapacitate the ability of degraded or partially degraded SP-A to serve as an effective opsonin. Of additional important is the fact that exoproteases secreted by P. aeruginosa degrade lysozyme, which is known to have antimicrobial activities (Markart et al., 2004). In contrast, ΔfliC mutant bacteria, which are impaired in the production of exoproteases, are unable to degrade SP-A and lysozyme. As a result, the BAL fluid from ΔfliC-infected SP-A+/+ mice is still capable of permeabilizing the bacterial membranes.
Finally, based on the results presented herein and on our previously published data (Zhang et al., 2007), we propose a model to illustrate the offensive and defensive mechanisms elaborated by P. aeruginosa flagella to confer resistance against SP-A (Fig. 9). We term defensive mechanisms for those factors that are involved in protection of P. aeruginosa without resulting in inactivation of SP-A. In contrast, offensive measures describe those factors secreted by P. aeruginosa that resulted in inactivation or degradation of SP-A and other major host defense proteins. An intact flagellum structure is required for positive feedback transcriptional regulation of genes involve in QS, including lasI and rhlI. The loss of flagella interrupts the continual transcription and accumulation of QS molecules, and reduced production of offensive exoproteases necessary to degrade SP-A in vitro and during infection of mouse lungs (Fig. 9A). The loss of flagella also results in the reduced amounts of defensive LPS required to stabilize the outer membrane (Zhang et al., 2007) (Fig. 9B). Thus, the dual roles of the flagellum in influencing the expression of LPS and exoprotease production combine to confer resistance to SP-A-mediated membrane permeabilization. Therapeutic strategies aimed at blocking flagellar biosynthesis, or aerosolizing SP-A or SP-A fragments into infected airways where SP-A levels are severely depleted, may be reasonable therapeutic options as adjunctive treatment strategies.
Fig. 9.
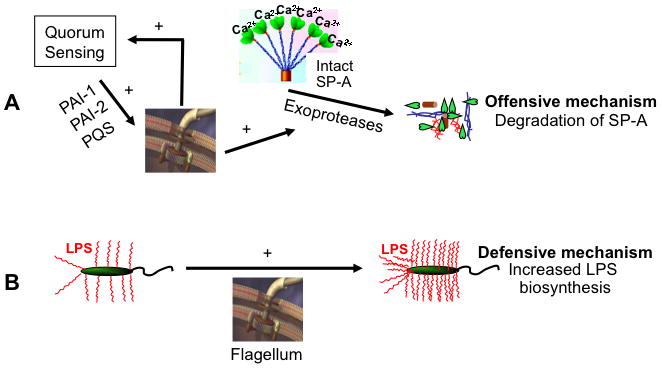
Model of P. aeruginosa flagellum-mediated resistance to membrane permeabilization by SP-A. Two mechanisms are proposed: (A) Flagella positively feedback regulate and maintain the production of PAI-1, PAI-2 and PQS, which in turn upregulate the production of exoproteases to degrade SP-A, and (B) Flagella positively regulate the production of LPS to resist membrane permeabilization.
Experimental procedures
Reagents
All chemicals, except where noted, were obtained from SigmaChemical Co. (St. Louis, MO).
Bacterial strains, plasmids, media and growth conditions
All bacterial strains, plasmid vectors, and their derivatives are described in Table 1. The parental wild-type P. aeruginosa PAO1 (Dasgupta et al., 2003; originally from Michael Vasil (Table 1), the ΔfliC (Fleiszig et al., 2001) mutant, and the complemented fliC strain PAOC-fliC (Arora et al., 2005), are the same as previously described. The pUTminiTn5Km2-generated flgE mutant was previously described and derived from a PAO1 strain originally from Holloway (Zhang et al., 2007). The ΔlasB mutant PDO240 (McIver et al., 1995) was a gift from Dr. Dennis Ohman. The lacZ reporter E. coli strains are described in Table 1. Bacterial strains were grown in LB (Luria-Bertani Broth), TSB (Tryptic Soy Broth) or TSBD (TSB treated with Chelex 100) for 16 hr at 37°C, and then suspended inLB with 20% glycerol and frozen in aliquotsat −80°C. Before each assay, aliquots of the bacteria were cultured from frozen stocks in appropriate media with or without antibiotics to stationary growth phase (OD600 nm 3.0) as indicated. The optical density at 600 nm was determined spectrophotometrically and correlated with numbers of viable bacteria by colony-formingunits (cfu) after plating serial dilutions on agar plates. When required, antibiotics were used at the following concentrations: for P. aeruginosa, carbenicillin (300 μg/ml), gentamycin (30 μg/ml), and kanamycin (100 μg/ml); for E. coli, carbenicillin (100 μg/ml), tetracyclin (20 μg/ml), and kanamycin (50 μg/ml).
Purification of human SP-A
Human SP-A was purified from the lung washings of patients with alveolar proteinosis as previously published (Suwabe et al., 1996) and stored in 5 mM Tris, 150mM NaCl, pH 7.4, at −20°C. The preparations were deemed free of EDTA by a modified spectrophotometric assay, using β-phenanthrolene–disulfonic acid as the indicator ( Kratochvil and White, 1965).
Protein assays
Routine protein concentrations were determined by the bicinchoninic acid protein assay kit (BCA; Pierce Chemical Co., Rockford, IL, USA), using bovine serum albumin (BSA) as a standard. Protein samples were resolved on 8–16% SDS-PAGE gel and stained with Coomassieblue or silver nitrate.
Animal husbandry
Swiss Black SP-A−/− mice (a gift of J. Whitsett/T. Korfhagen) were derived from embryonic stem cells after disruption of the mouse SP-A gene by homologous recombination and were maintained by breeding with Swiss Black mice, as previously reported (Korfhagen et al., 1996). The SP-A null allele was backrossed into the C3H/HeNgenetic background through nine generations as described Wu et al., (2003). C3H/HeN control (SP-A+/+) mice were purchased from Charles River Laboratory (Boston, MA). All comparisons made with the SP-A−/− mice were with age- and strain-matched C3H/HEN controls. All animals were housed in positively ventilatedmicroisolator cages with automatic recirculating water located in a room with laminar, high efficiency particulate-filteredair. The animals received autoclaved food, water, and bedding. Mice were handled in accordance with approved protocols through the Institutional Animal Care and Use Committee at the Universityof Illinois at Urbana-Champaign.
Mouse infection
Single infections of SP-A+/+ and SP-A−/− mice (five per group) were performed with 1 × 107 wild-type PAO1, ΔfliC, PAOC-fliC or PDO240 bacteria by the intranasal route as we have previously published (Zhang et al., 2005; Zhang et al., 2007). After 18 hr, lungs were harvested for determination of bacterial load, or BAL for protein and membrane permeabilization analyses. Attenuation was defined as the log10 difference in CFU of ΔfliC bacteria recovered from the lung tissues of SP-A+/+ versus SP-A−/− mice 18 hr after inoculation.
BAL
BAL was performed on P. aeruginosa-infected mice as we have previously described (Zhang et al., 2005). The trachea was exposed and intubated with a 1.7-mm outer diameter polyethylene catheter. BAL was performed by instilling PBS in 1 ml aliquots. Two ml of PBS were instilled per mouse. The BAL samples were pooled for membrane permeabilization assays and for western blot analysis.
Histological analysis
For histological analysis of mouse lungs infected with P. aeruginosa, lungs were inflation-perfused with 10% phosphate-buffered formalin at a constant pressure (25 cm H2O). The inflated lungs were post-fixed for 24 h and then embedded in paraffin wax. Five-micron sections were stained with haematoxylin and eosin (H&E) and scored for pathological changes by a pathologist that was masked as to the genotype and treatment of the mice.
Membrane permeabilization assays
The effect of the SP-A or lysozyme on P. aeruginosa and E. coli cell wall integrity was assessed by determining permeability to a phosphatase substrate, Enzyme-Labeled Fluorescence 97 (ELF-97, Molecular Probes), as we have previously described (Zhang et al., 2005, Zhang et al., 2007). SP-A (50 μg/ml) and/or lysozyme (5 μg/ml) was incubated with 1 × 108 logarithmic or stationary phase bacterial cells/ml in 100 μl of 5 mM Tris and 150 mM NaCl for 15 min at 37 °C, and 100 μM ELF97 phosphatase substrate was added. Fluorescence was measured at excitation and emission wavelengths of 355 and 535 nm, respectively, for a period of 120 min.
Exoprotease assays
Total exoprotease assays were performed using M9 minimal agar containing 5% skim milk spotted with P. aeruginosa strains PAO1 (wild-type), flagellar-deficient ΔfliC, and the genetically complemented PAOC-fliC bacteria. Exoprotease activities were determined by the SensolyteTM Red Protease Assay Kit (AnaSpec, Inc, San Jose, CA, Cat # 71140) using stationary phase culture supernatant of PAO1, ΔfliC or PAOC-fliC grown in TSB or TSBD with or without homoserine lactones PAI-1 (1.33 μM) and/or PAI- 2 (2.34 μM) and/or quinolone PQS (50 μM). Elastase activities were measured by Elastin Congo Red assay as previously published (Rust et al., 1994).
In vitro SP-A and lysozyme degradation assays
P. aeruginosa strains PAO1, ΔfliC, and PAOC-fliC bacteria were cultured in TSB overnight to late stationary phase. SP-A (25 μg) or lysozyme (5 μg) was added to 1 × 108 bacterial cells resuspended in 250 μl of fresh TSB supplemented with 2 mM CaCl2. A subset of bacteria-SP-A mixture was supplemented with the homoserine lactones PAI-1 (1.33 μM) and/or PAI-2 (2.34 μM). At indicated time intervals, a 20 μl aliquot of each bacterial-SPA mixture was mixed with loading buffer for SDS-PAGE and Western blot analysis. Degradation of chicken lysozyme (Sigma, St. Louis, MA) was performed as described for SP-A using a polyclonal antibody against lysozyme (Markart et al., 2004).
Western blot
Western blot analyses were performed using standard protocols as described (Sambrook et al., 1989). Briefly, protein samples (purified SP-A or mouse BAL fluids) were resolved by SDS-PAGE and electro-blotted onto Immobilon P polyvinylidene difluoride membranes (Millipore, Bedford, MA). The membranes were then incubated for 60 min at room temperature in blocking solution (PBS containing 3% bovine serum albumin), followed by a 4-hr incubation with monoclonal antibody against mouse SP-A (Santa Cruz Biotecnology Inc, Santa Cruz, CA) or a polyclonal anti-mouse lysozyme (Markart et al., 2004), respectively. The membranes were hybridized with horseradish peroxidase-conjugated goat anti-mouse (SP-A) or anti-rabbit (lysozyme) IgG secondary antibody. The immune complexes were visualized using the ECL Western Blotting Detection System (Amershan Biosciences, Piscataway, NJ) and Kodak BIOMAX (Kodak, Rochester, NY) X-ray films.
PAI-1 and PAI-2 extraction and lacZ reporter assays
The P. aeruginosa autoinducers PAI-1 and PAI-2 were extracted with ethyl acetate from 5 ml stationary phase cultures of P. aeruginosa strains PAO1, PAOC-fliC and ΔfliC grown in TSB as described (Pearson et al., 1995). The extracts were added to E. coli DH5-α carrying the lacZ translational fusion constructs pTS400, pECP62.5, pECP60, and pECP61.5 (Table 1) to assay for the induction of β-galactosidase.
Quantitative real-time PCR (qRT-PCR) of lasI and rhlI
Wild-type PAO1 and isogenic ΔfliC and ΔlasI ΔrhlI mutants were cultured in LB until indicated density. Total RNA was extracted by using the Qiagen RNeasy Mini Kit. cDNA were prepared from same amount of total RNA as the templates of Taqman Real-time PCR. The probes and primers for the P. aeruginosa lasI and rhlI genes were designed by the Applied Biosystems (Carlsbad, CA). qRT-PCR was carried out on the ABI 9700 system. The P. aeruginosa house keeping gene rpsL was used as endogenous control. All the experiments are repeated at least three times.
Statistical analyses
Statistical analysis was performed using the Student’s t-test and one-way analyses of variance (ANOVA). A significant difference was considered to be p < 0.05.
Acknowledgments
We thank Jayme Jeffries and Emma Pearson for critical reading of the manuscript. We thank Professor Reuben Ramphal (University of Florida College of Medicine) for his generous gifts of the wild-type PAO1, ΔfliC, and the complemented PAO-fliC strain; Professors Jeff Whitsett and Tom Korfhagen (Cincinnati Children Hospital) for the gift of the Swiss Black SP-A−/− mice, Professor Frank McCormack (University of Cincinnati College of Medicine) for the C3H SP-A−/− mice; Professor Dennis Ohman (Virginia Commonwealth University) for the gift of rabbit anti-elastase B antibody; Professor Steve Lory (Harvard Medical School) for the gift of ΔlasI ΔrhlI strain; and Dr. Francois Lepine (INRS-Institut Armand-Frappier, Canada) for the gift of quinolone PQS. This work is supported in part by NIH grants AI057915 and HL090699 to GWL. This investigation was conducted in a facility constructed with the support from Research Facilities Improvement Program Grant Number C06 RR 16515-01 from the National Center for Research Resources, National Institutes of Health.
References
- Adamo R, Sokol S, Soong G, Gomez MI, Prince A. Pseudomonas aeruginosa flagella activate airway epithelial cells through asialoGM1 and toll-like receptor 2 as well as toll-like receptor 5. Am J Respir Cell Mol Biol. 2004;30:627–634. doi: 10.1165/rcmb.2003-0260OC. [DOI] [PubMed] [Google Scholar]
- Alcorn JF, Wright JR. Degradation of pulmonary surfactant protein D by Pseudomonas aeruginosa elastase abrogates innate immune function. J Biol Chem. 2004;279:30871–30879. doi: 10.1074/jbc.M400796200. [DOI] [PubMed] [Google Scholar]
- Arora SK, Neely AN, Blair B, Lory S, Ramphal R. Role of motility and flagellin glycosylation in the pathogenesis of Pseudomonas aeruginosa burn wound infections. Infect Immun. 2005;73:4395–4398. doi: 10.1128/IAI.73.7.4395-4398.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman RP, Sternberg RI, Hull W, Buchsbaum JA, Whitsett J. Decreased surfactant protein A in patients with bacterial pneumonia. Am Rev Respir Dis. 1993;147:653–657. doi: 10.1164/ajrccm/147.3.653. [DOI] [PubMed] [Google Scholar]
- Beatty AL, Malloy JL, Wright JR. Pseudomonas aeruginosa degrades pulmonary surfactant and increases conversion in vitro. Am J Respir Cell Mol Biol. 2005;32:128–134. doi: 10.1165/rcmb.2004-0276OC. [DOI] [PubMed] [Google Scholar]
- Bren A, Eisenbach M. How signals are heard during bacterial chemotaxis: protein-protein interactions in sensory signal propagation. J Bacteriol. 2000;182:6865–6873. doi: 10.1128/jb.182.24.6865-6873.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell CC, Chen Y, Goetzmann HS, Hao Y, Borchers MT, Hassett DJ, Young LR, Mavrodi D, Thomashow L, Lau GW. Pseudomonas aeruginosa exotoxin pyocyanin causes cystic fibrosis airway pathogenesis. Am J Pathol. 2009;175:2473–2488. doi: 10.2353/ajpath.2009.090166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway SP, Brownlee KG, Denton M, Peckham DG. Antibiotic treatment of multidrug-resistant organisms in cystic fibrosis. Am J Respir Med. 2003;2:321–332. doi: 10.1007/BF03256660. [DOI] [PubMed] [Google Scholar]
- Cooley J, McDonald B, Accurso FJ, Crouchm EC, Remold-O’Donnell E. Patterns of neutrophil serine protease-dependent cleavage of surfactant protein D in inflammatory lung disease. J Leukoc Biol. 2008;83:946–955. doi: 10.1189/jlb.1007684. [DOI] [PubMed] [Google Scholar]
- Crouch E, Wright JR. Surfactant proteins A and D and pulmonary host defense. Annu Rev Physiol. 2001;63:521–554. doi: 10.1146/annurev.physiol.63.1.521. [DOI] [PubMed] [Google Scholar]
- Dasgupta N, Wolfgang MC, Goodman AL, Arora SK, Jyot J, Lory S, Ramphal R. A four-tiered transcriptional regulatory circuit controls flagellar biogenesis in Pseudomonas aeruginosa. Mol Microbiol. 2003;50:809–824. doi: 10.1046/j.1365-2958.2003.03740.x. [DOI] [PubMed] [Google Scholar]
- De Kievit TR, Gillis R, Marx S, Brown C, Iglewski BH. Quorum-sensing genes in Pseudomonas aeruginosa biofilms: their role and expression patterns. Appl Environ Microbiol. 2001;67:1865–73. doi: 10.1128/AEM.67.4.1865-1873.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggle SP, Cornelis P, Williams P, Cámara M. 4-quinolone signalling in Pseudomonas aeruginosa: old molecules, new perspectives. Int J Med Microbiol. 2006;296:83–91. doi: 10.1016/j.ijmm.2006.01.038. [DOI] [PubMed] [Google Scholar]
- Feldman M, Bryan R, Rajan S, Scheffler L, Brunnert S, et al. Role of flagella in pathogenesis of Pseudomonas aeruginosa pulmonary infection. Infect Immun. 1998;66:43–51. doi: 10.1128/iai.66.1.43-51.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleiszig SM, Arora SK, Van R, Ramphal R. FlhA, a component of the flagellum assembly apparatus of Pseudomonas aeruginosa, plays a role in internalization by corneal epithelial cells. Infect Immun. 2001;69:4931–4937. doi: 10.1128/IAI.69.8.4931-4937.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway DR. Pseudomonas aeruginosa elastase and elastolysis revisited: recent developments. Mol Microbiol. 1991;5:2315–2321. doi: 10.1111/j.1365-2958.1991.tb02076.x. [DOI] [PubMed] [Google Scholar]
- Griese M, Birrer P, Demirsoy A. Pulmonary surfactant in cystic fibrosis. Eur Respir J. 1997;10:1983–1988. doi: 10.1183/09031936.97.10091983. [DOI] [PubMed] [Google Scholar]
- Gunther A, Siebert C, Schmidt R, Ziegler S, Grimminger F, et al. Surfactant alterations in severe pneumonia, acute respiratory distress syndrome, and cardiogenic lung edema. Am J Respir Crit Care Med. 1996;153:176–184. doi: 10.1164/ajrccm.153.1.8542113. [DOI] [PubMed] [Google Scholar]
- Hawgood S, Shiffer K. Structures and properties of the surfactant-associated proteins. Annu Rev Physiol. 1991;53:375–394. doi: 10.1146/annurev.ph.53.030191.002111. [DOI] [PubMed] [Google Scholar]
- Hirche TO, Crouch EC, Espinola M, Brokelman TJ, Mecham RP, DeSilva N, Cooley J, Remold-O’Donnell E, Belaaouaj A. Neutrophil serine proteinases inactivate surfactant protein D by cleaving within a conserved subregion of the carbohydrate recognition domain. J Biol Chem. 2004;279:27688–27698. doi: 10.1074/jbc.M402936200. [DOI] [PubMed] [Google Scholar]
- Juhas M, Eberl L, Tümmler B. Quorum sensing: the power of cooperation in the world of Pseudomonas. Environ Microbiol. 2005;7:459–471. doi: 10.1111/j.1462-2920.2005.00769.x. [DOI] [PubMed] [Google Scholar]
- Korfhagen TR, Bruno MD, Ross GF, Huelsman KM, Ikegami M, et al. Altered surfactant function and structure in SP-A gene targeted mice. Proc Natl Acad Sci USA. 1996;93:9594–9599. doi: 10.1073/pnas.93.18.9594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratochvil B, White MC. Spectrophotometric determination of microgram quantities of (ethylenedinitrilo)tetraacetic acid with bis(2,4,6-tripyridyl-S-triazine)iron(II) Anal Chem. 1965;37:111–113. doi: 10.1021/ac60220a028. [DOI] [PubMed] [Google Scholar]
- Kuzmenko AI, Wu H, Wan S, McCormack FX. Surfactant protein A is a principal and oxidation-sensitive microbial permeabilizing factor in the alveolar lining fluid. J Biol Chem. 2005;280:25913–25919. doi: 10.1074/jbc.M411344200. [DOI] [PubMed] [Google Scholar]
- Kuzmenko AI, Wu H, McCormack FX. Pulmonary collectins selectively permeabilize model bacterial membranes containing rough lipopolysaccharide. Biochemistry. 2006;45:2679–2685. doi: 10.1021/bi0522652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau GW, Ran H, Kong F, Hassett DJ, Mavrodi D. Pseudomonas aeruginosa pyocyanin is critical for lung infection in mice. Infect Immun. 2004;72:4275–8. doi: 10.1128/IAI.72.7.4275-4278.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau GW, Hassett DJ, Ran H, Kong F. The role of pyocyanin in Pseudomonas aeruginosa infection. Trends Mol Med. 2004;10:599–606. doi: 10.1016/j.molmed.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Lau GW, Hassett DJ, Britigan BE. Modulation of lung epithelial functions by Pseudomonas aeruginosa. Trends Microbiol. 2005;13:389–397. doi: 10.1016/j.tim.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Lema G, Dryja D, Vargas I, Enhorning G. Pseudomonas aeruginosa from patients with cystic fibrosis affects function of pulmonary surfactant. Pediatr Res. 2000;47:121–126. doi: 10.1203/00006450-200001000-00021. [DOI] [PubMed] [Google Scholar]
- LeVine AM, Lotze A, Stanley S, Stroud C, O’Donnell R, et al. Surfactant content in children with inflammatory lung disease. Crit Care Med. 1996;24:1062–1067. doi: 10.1097/00003246-199606000-00029. [DOI] [PubMed] [Google Scholar]
- Liau DF, Yin NX, Huang J, Ryan SF. Effects of human polymorphonuclear leukocyte elastase upon surfactant proteins in vitro. Biochim Biophys Acta. 1996;1302:117–128. doi: 10.1016/0005-2760(96)00042-2. [DOI] [PubMed] [Google Scholar]
- Lyczak JB, Cannon CL, Pier GB. Lung infections associated with cystic fibrosis. Clin Microbiol Rev. 2002;15:194–222. doi: 10.1128/CMR.15.2.194-222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahenthiralingam E, Campbell ME, Speert DP. Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect Immun. 1994;62:596–605. doi: 10.1128/iai.62.2.596-605.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahenthiralingam E, Speert DP. Nonopsonic phagocytosis of Pseudomonas aeruginosa by macrophages and polymorphonuclear leukocytes requires the presence of the bacterial flagellum. Infect Immun. 1995;63:4519–4523. doi: 10.1128/iai.63.11.4519-4523.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malloy JL, Veldhuizen RA, Thibodeaux BA, O’Callaghan RJ, Wright JR. Pseudomonas aeruginosa protease IV degrades surfactant proteins and inhibits surfactant host defense and biophysical functions. Am J Physiol Lung Cell Mol Physiol. 2005;288:L409–418. doi: 10.1152/ajplung.00322.2004. [DOI] [PubMed] [Google Scholar]
- Mariencheck WI, Alcorn JF, Palmer SM, Wright JR. Pseudomonas aeruginosa elastase degrades surfactant proteins A and D. Am J Respir Cell Mol Biol. 2003;28:528–537. doi: 10.1165/rcmb.2002-0141OC. [DOI] [PubMed] [Google Scholar]
- Markart P, Faust N, Graf T, Na CL, Weaver TE, Akinbi HT. Comparison of the microbicidal and muramidase activities of mouse lysozyme M and P. Biochem J. 2004;380:385–392. doi: 10.1042/BJ20031810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack FX, Gibbons R, Ward SR, Kuzmenko A, Wu H, Deepe GS., Jr Macrophage-independent fungicidal action of the pulmonary collectins. J Biol Chem. 2003;278:36250–36256. doi: 10.1074/jbc.M303086200. [DOI] [PubMed] [Google Scholar]
- McIver KS, Kessler E, Olson JC, Ohman DE. The elastase propeptide functions as an intramolecular chaperone required for elastase activity and secretion in Pseudomonas aeruginosa. Mol Microbiol. 1995;18:887–889. doi: 10.1111/j.1365-2958.1995.18050877.x. [DOI] [PubMed] [Google Scholar]
- Morihara K. Production of elastase and proteinase by Pseudomonas aeruginosa. J Bacteriol. 1964;88:745. doi: 10.1128/jb.88.3.745-757.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noah TL, Murphy PC, Alink JJ, Leigh MW, Hull WM, et al. Bronchoalveolar lavage fluid surfactant protein-A and surfactant protein-D are inversely related to inflammation in early cystic fibrosis. Am J Respir Crit Care Med. 2003;168:685–691. doi: 10.1164/rccm.200301-005OC. [DOI] [PubMed] [Google Scholar]
- Olson JC, Ohman DE. Efficient production and processing of elastase and LasA by Pseudomonas aeruginosa require zinc and calcium ions. J Bacteriol. 1992;174:4140–4147. doi: 10.1128/jb.174.12.4140-4147.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole GA, Kolter R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol. 1998;30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- Pearson JP, Passador L, Iglewski BH, Greenberg EP. A second N- acylhomoserine lactone signal produced by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 1995;92:1490–1494. doi: 10.1073/pnas.92.5.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pison U, Tam EK, Caughey GH, Hawgood S. Proteolytic inactivation of dog lung surfactant-associated proteins by neutrophil elastase. Biochim Biophys Acta. 1989;992:251–257. doi: 10.1016/0304-4165(89)90082-2. [DOI] [PubMed] [Google Scholar]
- Postle AD, Mander A, Reid KB, Wang JY, Wright SM, et al. Deficient hydrophilic lung surfactant proteins A and D with normal surfactant phospholipid molecular species in cystic fibrosis. Am J Respir Cell Mol Biol. 1999;20:90–98. doi: 10.1165/ajrcmb.20.1.3253. [DOI] [PubMed] [Google Scholar]
- Prince A. Flagellar activation of epithelial signaling. Am J Respir Cell Mol Biol. 2006;34:548–551. doi: 10.1165/rcmb.2006-0022SF. [DOI] [PubMed] [Google Scholar]
- Rubio F, Cooley J, Accurso FJ, Remold-O’Donnell E. Linkage of neutrophil serine proteases and decreased surfactant protein-A (SP-A) levels in inflammatory lung disease. Thorax. 2004;59:318–323. doi: 10.1136/thx.2003.014902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust L, Messing CR, Iglewski BH. Elastase assays. Methods Enzymol. 1994;235:554–562. doi: 10.1016/0076-6879(94)35170-8. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: a Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sarkisova S, Patrauchan MA, Berglund D, Nivens DE, Franklin MJ. Calcium-induced virulence factors associated with the extracellular matrix of mucoid Pseudomonas aeruginosa biofilms. J Bacteriol. 2005;187:4327–4337. doi: 10.1128/JB.187.13.4327-4337.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schad PA, Bever RA, Nicas TI, Leduc F, Hanne LF, Iglewski BH. Cloning and characterization of elastase genes from Pseudomonas aeruginosa. J Bacteriol. 1987;169:2691–2696. doi: 10.1128/jb.169.6.2691-2696.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer LM, McCormack FX, Wu H, Weiss AA. Bordetella pertussis lipopolysaccharide resists the bactericidal effects of pulmonary surfactant protein A. J Immunol. 2004;173:1959–1965. doi: 10.4049/jimmunol.173.3.1959. [DOI] [PubMed] [Google Scholar]
- Schultz DR, Miller KD. Elastase of Pseudomonas aeruginosa: inactivation of complement components and complement-derived chemotactic and phagocytic factors. Infect Immun. 1974;10:128–135. doi: 10.1128/iai.10.1.128-135.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh PK, Tack BF, McCray PB, Jr, Welsh MJ. Synergistic and additive killing by antimicrobial factors found in human airway surface liquid. Am J Physiol Lung Cell Mol Physiol. 2000;279:L799–L805. doi: 10.1152/ajplung.2000.279.5.L799. [DOI] [PubMed] [Google Scholar]
- Suwabe A, Mason RJ, Voelker DR. Calcium dependent association of surfactant protein A with pulmonary surfactant: application to simple surfactant protein A purification. Arch Biochem Biophys. 1996;327:285–291. doi: 10.1006/abbi.1996.0123. [DOI] [PubMed] [Google Scholar]
- Thaden JT, Lory S, Gardner TS. Quorum-sensing regulation of a copper toxicity system in Pseudomonas aeruginosa. J Bacteriol. 2010;192:2557–68. doi: 10.1128/JB.01528-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer MM, Flaherty KM, McKay DB. Three-dimensional structure of the elastase of Pseudomonas aeruginosa at 1.5-A resolution. J Biol Chem. 1991;266:2864–2871. doi: 10.2210/pdb1ezm/pdb. [DOI] [PubMed] [Google Scholar]
- Toutain CM, Zegans ME, O’Toole GA. Evidence for two flagellar stators and their role in the motility of Pseudomonas aeruginosa. J Bacteriol. 2005;187:771–777. doi: 10.1128/JB.187.2.771-777.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voynow JA, Fischer BM, Zheng S. Proteases and cystic fibrosis. Int J Biochem Cell Biol. 2008;40:1238–1245. doi: 10.1016/j.biocel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner VE, Iglewski BH. P. aeruginosa Biofilms in CF Infection. Clin Rev Allergy Immunol. 2008;35:124–134. doi: 10.1007/s12016-008-8079-9. [DOI] [PubMed] [Google Scholar]
- Wagner VE, Gillis RJ, Iglewski BH. Transcriptome analysis of quorum-sensing regulation and virulence factor expression in Pseudomonas aeruginosa. Vaccine. 2004;22(Suppl 1):S15–20. doi: 10.1016/j.vaccine.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol. 2005;5:58–68. doi: 10.1038/nri1528. [DOI] [PubMed] [Google Scholar]
- Wu H, Kuzmenko A, Wan S, Schaffer L, Weiss A, et al. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J Clin Invest. 2003;111:1589–1602. doi: 10.1172/JCI16889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Hancock RE. Synergistic interactions between mammalian antimicrobial defense peptides. Antimicrob Agents Chemother. 2001;45:1558–1560. doi: 10.1128/AAC.45.5.1558-1560.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Chen Y, Potvin E, Sanschagrin F, Levesque RC, McCormack FX, Lau GW. Comparative signature-tagged mutagenesis identifies Pseudomonas factors conferring resistance to the pulmonary collectin SP-A. PLoS Pathog. 2005;1:259–268. doi: 10.1371/journal.ppat.0010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, McCormack FX, Levesque RC, O’Toole GA, Lau GW. The flagellum of Pseudomonas aeruginosa is required for resistance to clearance by surfactant protein A. PLoS ONE. 2007;2:e564. doi: 10.1371/journal.pone.0000564. [DOI] [PMC free article] [PubMed] [Google Scholar]