

Abstract
In this review, we will discuss several well-accepted signaling pathways toward calcium-mediated mechanisms of cystic expansion. The second messenger calcium ion has contributed to a vast diversity of signal transduction pathways. We will dissect calcium signaling as a possible mechanism that contributes to renal cyst formation. Because cytosolic calcium also regulates an array of signaling pathways, we will first discuss cilia-induced calcium fluxes, followed by Wnt signaling that has attributed to much-discussed planar cell polarity. We will then look at the relationship between cytosolic calcium and cAMP as one of the most important aspects of cyst progression. The signaling of cAMP on MAPK and mTOR will also be discussed. We infer that while cilia-induced calcium fluxes may be the initial signaling messenger for various cellular pathways, no single signaling mediator or pathway is implicated exclusively in the progression of the cystic expansion.
Introduction
Polycystic kidney disease (PKD) is characterized by formation of fluid-filled cysts. For the past decade, many ideas and much hard work have been put forth to understand the disease, although the mechanisms of cyst formation and expansion remain speculations. Based on transmittance, PKD can be simply classified into acquired and hereditary forms. The acquired form of PKD can be found in patients who have had acute renal failure with subsequent dialysis. The majority of PKD cases, however, are transmitted hereditarily from the parents. The two most common hereditary forms of PKD are autosomal dominant PKD (ADPKD) and autosomal recessive PKD (ARPKD). The genes mutated in ADPKD include PKD1 and PKD2, whereas ARPKD is caused by mutation in PKHD1 gene (OMIMs: #601313, 613095, 263200). The prevalence of ADPKD and ARPKD is 1 in 1,000 and 1 in 20,000 live births, respectively.
The products encoded by these PKD genes are called cysto-proteins (Figure 1), which include polycystin-1 (PKD1), polycystin-2 (PKD1), and fibrocystin (PKHD1). Though the mechanism of cyst formation is still a mystery, abnormal function of these proteins results in cyst formation. In particular, cysto-proteins interact with one another (Figure 1). Thus, aberrant functions in any of these cysto-proteins may result in similar pathogenic phenotypes in the kidney, liver, pancreas and possibly other organs. In addition, these three proteins are localized within the same subcellular domain in the cell. Because these proteins are localized and have distinct functions in the primary cilia, ciliary hypothesis has been developed to explain a unifying pathogenic concept for PKD.
Figure 1. Cysto-protein complex.

Polycystin-1 and polycystin-2 form a complex with each other at their COOH termini. Functioning as an adaptor protein, Kif bridges the interaction of between fibrocystin and polycystin complex. Figure is reproduced from Kolb, et al with permission [39].
In this review, we will discuss calcium signaling as a possible mechanism that contributes to the functions of these cysto-proteins. Dysfunction of any of these proteins may thus interrupt calcium signaling pathways, which may promote abnormal downstream signal transductions of various signaling molecules participating in renal cyst formation.
Calcium signaling by primary cilia
When the PKD genes were discovered and cloned [1-3], their functions in cation transport immediately became of considerable interest in understanding the molecular functions of the cysto-proteins. In particular, sequence analysis of polycystin-2 showed putative homologies with other known calcium channels [4]. In addition to their physical and functional interactions with other calcium-regulated proteins [5-7], interactions of cysto-proteins with other calcium channels also provided further insights into the modulation of intracellular calcium signaling [8-12]. Thus, polycystin-2 has long been predicted to regulate cytosolic calcium [13, 14], including modulating intraorganellar calcium release [15] and extracellular calcium influx [16].
To function as a calcium channel, polycystin-2 depends on its interaction with polycystin-1 [16, 17]. Likewise, proper function of fibrocystin depends on the indirect interaction with the polycystin complex [18-20]. In addition, activation of polycystin-2 has been found to depend on its interaction with mammalian diaphanous-related forming 1 (mDia1). mDai1 is able to regulate polycystin-2 depending on the membrane potential or voltage levels in the cells. At resting potentials, mDai1 in an autoinhibited state binds to polycystin-2 thereby inhibiting its channel activity. However, at positive potentials, GTP-bound mDia1 releases and thereby allows activation of polycystin-2 [21].
Localization of cysto-proteins to primary cilia further confirms the roles of polycystins and fibrocystin in intracellular calcium signaling. In addition, it further elaborates the molecular functions of cysto-proteins as regulators for intracellular calcium signaling. Most important are the mechanosensory functions of cysto-proteins that have been independently described in the mouse and human kidney epithelia [20, 22-27], vascular endothelia [28-30], osteochondrocytes [31, 32], cholangiocytes [33, 34] and developing nodes [35-37]. It is now generally accepted that localization of these cysto-proteins to the primary cilia is important and necessary to initiate the first signaling cascade of intracellular calcium [38-40]. This signaling pathway may further provide other complex downstream signaling pathways.
In general, primary cilia are mechanosensory compartments that house many sensory proteins, including the cysto-proteins. Shear stress that produces enough drag force on the cell surface is able to bend the primary cilium (Figure 2). Subsequently, activation of the polycystins and other interacting proteins in this complex may result in cytosolic calcium increase. This paradigm was established based mainly on in vitro studies where cultured kidney cells were challenged with a fluid-flow shear stress [41, 42]. The ex vivo experiments using isolated gastrulation stage node, perfused tubules and arteries have further confirmed the mechanosensory function of the primary cilium [28, 36, 43]. In the vascular artery [28], the influx of extracellular calcium initiates the biochemical cascades that lead to production of nitric oxide vasodilator through endothelial nitric oxide (eNOS). This activation of eNOS depends on the contribution or activity of calmodulin, phosphoinositide kinase-3, protein kinase B and calcium-dependent protein kinase (Figure 3).
Figure 2. Primary cilia in signal transduction system.

Cilia sense fluid-shear stress on the apical membrane of the cells. Fluid flow that produces enough drag-force on the top of the cells will bend sensory cilia. Bending of cilia will activate the cysto-proteins, resulting in extracellular calcium influx. Figure is reproduced from AbouAlaiwi, et al with permission [38].
Figure 3. Mechanosensory cilia, cytosolic calcium and nitric oxide production.
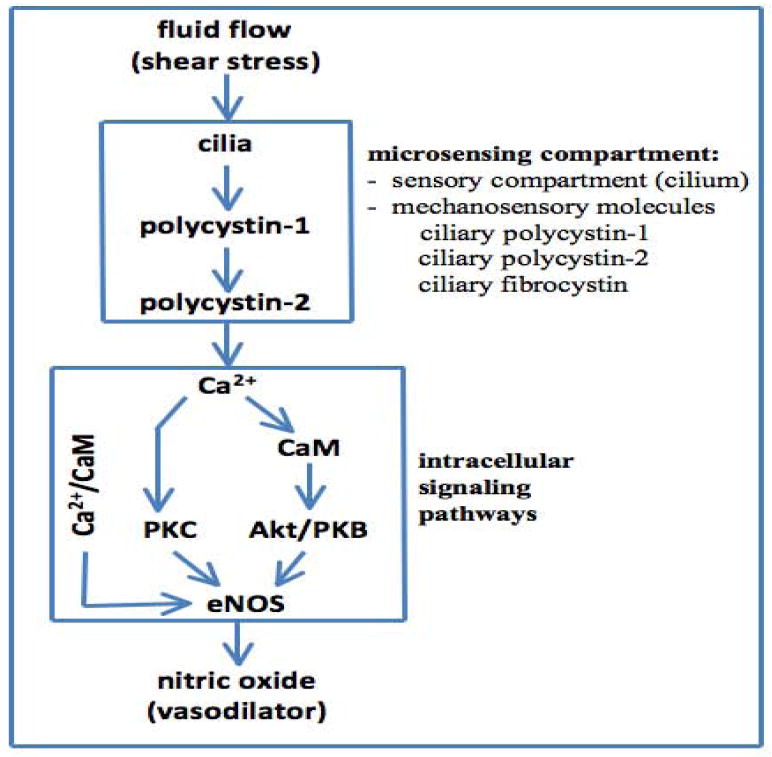
Bending or activation of the cilia involves mechanosensory polycystin-1 and polycystin-2 complex, which results in biochemical synthesis of NO. The signaling pathway requires extracellular calcium influx (Ca2+), followed by activation of various calcium-dependent proteins including calmodulin (CaM) and protein kinase C (PKC). Together with PKB, CaM and PKC are important downstream molecular components to activate endothelial nitric oxide synthase (eNOS).
In the next sections, we will discuss the downstream pathways that depend, directly or indirectly, on the initial calcium signaling. Because of the complexity in calcium signaling, we will discuss only those pathways that have possible relevance to renal cyst expansion.
Signaling by Wnt
Wnt signaling pathways are involved in many aspects of cell development, such as cell polarity determination, cell adhesion, growth, motility, and many others. The Wnt pathway involves a daunting number of secreted Wnt ligands and Frizzled receptors that regulate a large number of Wnt signaling molecules. In the simplest terms, Wnt signaling can activate three distinct pathways (Figure 4): 1) β-catenin dependent canonical pathway, 2) β-catenin independent non-canonical or PCP (planar cell polarity) pathway, and 3) Wnt-calcium pathway, which can influence both the canonical and non-canonical pathways[44].
Figure 4. Mechanosensory cilia, cytosolic calcium and Wnt signaling.

Bending or activation of the cilia results in maintaining cytosolic calcium (Ca2+), followed by activation of inversin. Calcium and inversin function as the molecular switches for Wnt signaling pathways. The Wnt canonical pathway involves β-catenin. Both β-catenin and E-cadherin regulate cell differentiation and proliferation. The Wnt non-canonical pathway involves small GTPase rho and JNK, both of which play an important role in planar cell polarity.
Many regulatory proteins involved in Wnt signaling are localized in the primary cilium and base of the cilium, also known as a basal body. It is thus speculated that flow-induced cytosolic calcium influx switches off the canonical Wnt pathway and activates the non-canonical Wnt/calcium signaling pathway (Figure 4). This molecular switch is regulated by inversin, which is a ciliary protein that can turn different Wnt signaling pathways on and off [45]. Of note is that abnormalities in inversin function result in polycystic kidney phenotype.
The zebrafish cystic kidney gene seahorse has also been found to be involved in a variety of cilia-mediated processes such as body curvature, kidney cyst formation, left-right asymmetry, and others including PCP signaling and inhibition of the canonical Wnt signaling [46]. Seahorse seems to be essential for a functional non-canonical Wnt signaling. It associates with Dishevelled (Dsh), the divergence point for the canonical Wnt and PCP pathways. The Seahorse gene encodes a highly conserved five leucine-rich repeats (LRR) and a leucine-rich repeat cap, from Drosphila to humans. One of the leucine-rich repeat proteins, LRRC50, has been found to be conserved in both zebrafish and humans. Lrrc50 in expressed in all ciliated tissues in zebrafish, resulting in cilary dysfunction in lrrc50 mutants. In humans and dog kidney cells, LRRC50 has been shown to localize at the mitotic spindle and cilium, implying it to be a ciliary protein in vertebrates [47].
It is generally accepted that Wnt signaling pathway is not regulated properly in polycystic kidney disease (Figure 4). In the canonical Wnt pathway, β-catenin in the nucleus mediates many gene induction events, and any deregulation of this pathway can result in uninhibited proliferation of cells [48, 49]. It is speculated that flow-induced cytosolic calcium influx is required to turn off the canonical Wnt pathway and activate the non-canonical Wnt/calcium signaling pathway. As such, over-activation of canonical Wnt pathway, by over-production of an activated form of β-catenin for an example, would result in polycystic kidney phenotype [50]. This view is consistent with the profiling gene expression study [51]. A consistently high level of Wnt signaling is observed in cystic tissues from ADPKD patients, but not in tissues which exhibited low level or no cyst formation from the same patients. Furthermore, abnormalities in polycystins enhance activity of Wnt signaling pathway [52-55].
E-cadherin is one of the interacting Wnt signaling molecules, and it plays an essential role in intercellular cell junction assembly. It is required for epithelial polarity and tubule formation. Disruption of E-cadherin could lead to abnormal levels of β-catenin and impeding renal epithelial polarization [54, 56]. Furthermore, the protein complex of polycystins, E-cadherin, and β-catenin is interrupted in cyst-lining epithelial cells from ADPKD patients [57]. In addition, the levels of β-catenin in the developing hearts and kidneys of Pkd1-/- mouse embryos compared to wild type embryos is decreased [53]. Overall, the data suggest that interruption in Wnt signaling pathway would result in less differentiated epithelial cells, yielding to proliferation and acceleration of cyst expansion [58].
Planar cell polarity, which involves non-canonical Wnt signaling, has recently been the most discussed topic toward understanding of renal cysts. Planar cell polarity is principally involved in the development of tissue architectures along a parallel axis, other than the apical-basolateral axis of a renal tubule (Figure 5). Oriented cell division is thought to be necessary for the elongation of the developing nephron. Abnormalities in the planar cell polarity, as reflected by the mitotic spindle, have been observed in various PKD mouse models [52, 59-63]. In these studies, cell division or mitotic spindle orientation within the tubular axis were measured. However, it is currently unclear whether such an alignment can be considered a process toward planar cell polarity [64]. In Drosophila, for example, mitotic spindle alignment is achieved only after centrosomes have been properly aligned [65]. Thus, there is a cell-cycle checkpoint for centrosomal positioning. The question remains whether such a checkpoint is disrupted in PKD.
Figure 5. Planar cell polarity and cystic kidney disease.
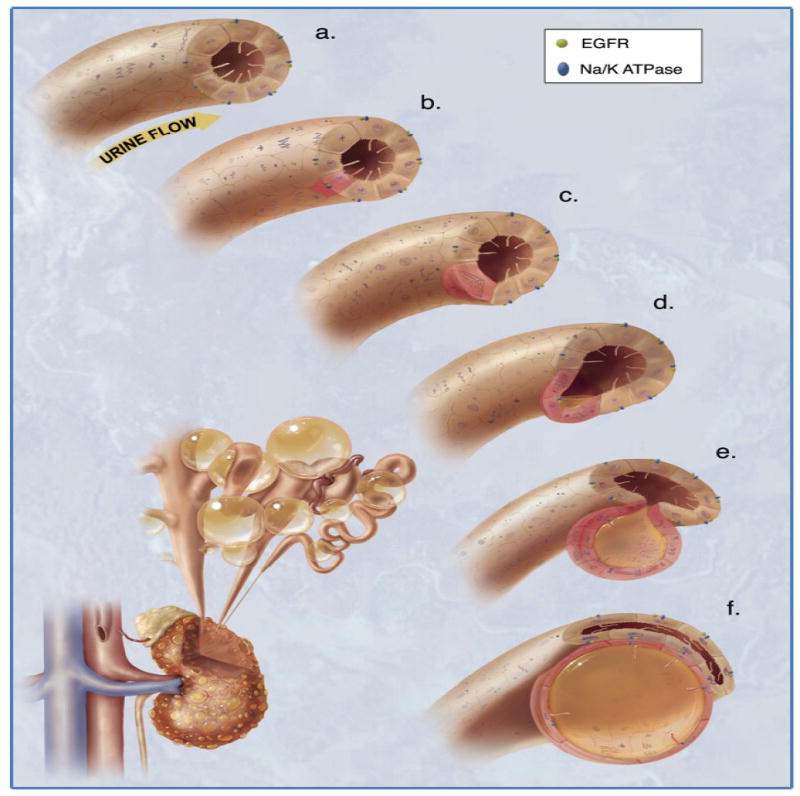
The illustration depicts the mechanism of mitotic spindle formation as an index of planar cell polarity in cyst expansion. a. Each cilium senses changes in urine flow. This message provides critical calcium signals to the cell. b. Abnormalities in any of the cysto-proteins results in abnormal ciliary function. c. The functional abnormality in ciliary sensing results in disorientation of the mitotic spindle, and hence the cell will lose planar cell polarity. d. Direction of cell division becomes randomized. This will result in increasing tubular diameter, rather than tubular elongation. e. Budding of a cyst from the renal tubule occurs. The abnormal localizations of epidermal growth factor receptor (EGFR) and Na+/K+ ATPase pump are typical characteristics of polycystic kidneys. f. The cyst is eventually enlarged and expanded further. Multiple cysts within a kidney are illustrated on the bottom left corner. Figure is reproduced from Kolb, et al with permission [39].
Within the non-canonical Wnt signaling, there is also a huge body of evidence indicating that JNK signaling system is involved in PKD [66-70]. The strongest evidence of non-canonical Wnt signaling, however, came from a study involving Rho small G protein. In the mouse model, mutation in the key regulator of Rho small G protein results in cystic kidney phenotype [71]. Taken together, interruption in Wnt signaling pathway would result in cyst formation due to planar cell polarity defects, and mitotic spindle orientation might be a contributing factor in the process.
Signaling by cAMP / MAPK
Cyclic adenosine monophosphate, or cAMP, has been identified as one of the most important players in cyst progression in both ADPKD and ARPKD (Figure 6). Two major processes that contribute to the expansion of the renal cysts in polycystic kidney disease are cell proliferation and fluid secretion. cAMP is intimately involved in accelerating both of these processes [72-75]. Tissues from the kidneys, liver, and vascular smooth muscles of PKD animal models exhibit increased levels of cAMP [76-78].
Figure 6. Cytosolic calcium and cAMP signaling.

Under normal conditions, the carboxy terminal tails of polycystin-1 and polycystin-2 interact, which enables calcium entry into the cell. This further stimulates the release of calcium from the calcium stores inside the cell. In the presence of high, steady state levels of calcium, calcium-stimulated phosphodiesterases are capable of degrading cAMP into AMP, thereby controlling the cAMP levels in the cell. The low levels of cAMP are unable to stimulate MAPK pathway. In polycystic kidneys, however, the resulting abnormality in cysto-proteins causes a lower level of intracellular calcium. Hence, the activity of phosphodiesterases is not stimulated and cAMP level increases, leading to activation of B-Raf of MAPK pathway.
cAMP is known to be one of most important players in effecting hormonal activation of intracellular pathways and is intimately involved in cell proliferation in almost all cell lines. However, cAMP does not produce the same effects in all cell lines. Though cAMP has been used for several years as an anti-proliferative agent [79, 80], cAMP has also been known to stimulate cell proliferation by activating the mitogen activated protein kinase (MAPK) pathway (Figure 6). For example, cAMP is anti-proliferative in normal tissues, but it stimulates cell proliferation in cystic epithelial cells [73, 81, 82].
The change in cAMP related phenotype - inhibition of cell proliferation in normal cells and stimulation of cell proliferation in cystic cells - is associated with the amount of intracellular calcium in PKD tissues and cells [82, 83]. The imbalance in the cytosolic calcium due to the disruptions in the cysto-proteins promotes abnormal function of phosphodiesterase (Figure 6). Phosphodiesterase 1 isoforms (PDE1a, PDE1b and PDE1c) are present in especially high levels in the kidneys [84]. Most importantly, the activity of this phosphodiesterase is regulated by intracellular calcium and cAMP. In PKD cells with a low level of intracellular calcium, phosphodiesterase activity is down-regulated. This results in aberrant conversion of cAMP to AMP. The resulting increase in cytosolic cAMP further stimulates the MAPK pathways, whichpromotes cyst expansion through higher cell proliferation.
In general, cystic epithelia in polycystic kidneys have exhibited high levels of both cAMP and MAPK activity compared to normal cells, resulting in cell proliferation in polycystic kidneys and an inhibition of cell proliferation in normal cells [85, 86]. In normal epithelial cells, cAMP agonists inhibit MAPK pathway by blocking activation of Raf-1 (Raf-C) through cAMP-dependent protein kinase (PKA). On the other hand, cAMP was found to stimulate the MAPK pathway in PKD cells, thereby stimulating cell proliferation. This difference has also been observed to result from an increased affinity of cAMP for B-Raf rather than A-Raf and Raf-1 (C-Raf) [82, 86]. As mentioned earlier, this change in cAMP-related signaling is also attributed to the level of intracellular calcium. The high levels of cAMP, combined with low levels of calcium fluxes in polycystic kidneys, could further result in a decrease of PI3K/Akt activity, thereby stimulating B-Raf activation and hence activation of the MAPK pathway of Ras/B-Raf/MEK/ERK [83, 87, 88].
Signaling by mTOR
The Ras/Raf/ERK pathway plays another important role in polycystic kidney disease by regulating the mammalian target of rapamycin (mTOR) pathway through molecular signaling of tuberin (Figure 7). Tuberin, which is also regulated by Akt, is a GTPase activating protein (GAP). Tuberin regulates the activity of Rheb, a small G-protein belonging to the Ras super family. GTP-bound Rheb is active, while GDP-bound Rheb is inactive. Hamartin and tuberin form a heterodimer which converts Rheb-GTP to Rheb-GDP, thereby inactivating Rheb. Rheb activates mTOR pathway. Hence, tuberin inactivates GTP-bound Rheb and inhibits the mTOR pathway [89-91].
Figure 7. Signaling pathway of mTOR.

In healthy kidney cells, the cysto-protein complex interacts with tuberin. Hamartin and tuberin form a complex and localize in the basal body. Tuberin is a GTPase activating protein (GAP) which controls the activity of Rheb. Tuberin inactivates Rheb, thereby inhibiting the mTOR pathway. In polycystic kidneys, however, tuberin is no longer protected in this complex and is phosphorylated by Akt, RSK (via ERK). As a result, tubulin is unable to form a heteradimer with hamartin. Hence, tuberin is no longer able inhibit Rheb, and the mTOR pathway is activated.
The respective protein products of TSC1 and TSC2, hamartin and tuberin, regulate formation of primary cilia [92, 93]. Most importantly, however, analysis of normal and diseased cells from ADPKD patients indicates that the cyst lining epithelial cells exhibit higher levels of mTOR signaling compared to the surrounding normal epithelium [94]. When mTOR pathway is inhibited with rapamycin, many murine models of ADPKD and ARPKD show a decrease in renal cyst expansion [94-100]. The consensus is that the cytoplasmic tail of polycystin-1 directly or indirectly interacts with both mTORC1 complex as well as tuberin, the protein product of Tsc2 which itself regulates mTORC1. Membrane localization of polycystin-1 and hamartin are therefore able to bind to tuberin, keeping it near the plasma membrane. Thus, membrane bound polycystin-1 is capable of controlling mTORC1 pathway and hence extensive cell proliferation. Disruption of polycystin-1 would imply the activation of the mTORC1 cascade yielding cell proliferation and subsequent cyst formation [101, 102].
In addition, the cysto-proteins are known to contribute to the activation of the Akt/PKB pathway [101, 103, 104]. Thus, the interaction of Akt and tuberin can further provide an additional regulation of mTOR pathway by the cysto-proteins. Although the role of calcium in mTOR signaling has still not been properly studied in polycystic kidney disease, there is a possibility that calcium-ERK-mTOR pathway could exist. In particular, calcium is known to be associated with cAMP/ERK activity (Figure 6), and ERK has been implicated in mTOR signaling (Figure 7). There is no doubt that further studies are needed to dissect the contributions of calcium, ERK and mTOR in cystogenesis.
Interestingly, clinical trials of mTOR inhibitors in ADPKD patients have not yielded a much anticipated results [105-107]. The two mTOR inhibitors tested in ADPKD patients were sirolimus and everolimus. Sirolimus was tested on 100 patients (18 to 40 years) exhibiting early stages of the disease, while everolimus was tried on 433 patients in advanced-stage II and III, with a renal baseline volume of 1500 mL. Sirolimus did not show any decrease in total kidney size in humans, though it showed promising results in mouse models. However, the animals were treated with a dose of 5 mg/kg of body weight [94], a dosage that is unsafe for humans, who were administered 2 mg sirolimus for 18 months. No difference in GFR was observed as these patients exhibited the initial stages of the disease [105]. Everolimus, on the other hand, slowed the increase in the total kidney volume without any effect on GFR. After a brief transient period, patients exhibited a rapid decline in GFR [106]. These studies imply that reducing the cyst size need not necessarily improve renal function in these patients.
Prospective
Cilia function and structure are important and necessary to maintain the architecture of kidney tissue. Abnormalities in the structure or function of cilia results in PKD. The mechanosensory cilia are crucial in maintaining intracellular calcium signaling. Many cell types, including renal epithelia, use cytosolic calcium as a second messenger to further regulate other cellular homeostasis through a very complex signal transduction system. This signaling system includes Wnt, cAMP - MAPK, Akt, mTOR, and other pathways that are not discussed in this review.
In understanding this complexity, an important lesson is that no single signaling mediator is implicated exclusively in the progression of the cystic expansion. Rather, all of these signaling pathways are intimately connected, thereby regulating the progression of the disease. Nonetheless, only by understanding such a complicated system do we have better insights to attain the most effective way to retard progression of polycystic kidney disease.
Acknowledgments
Due to restricted space, we apologize to those whose work is not described in this review. Authors are grateful for stimulating discussion about primary cilia given by research assistants, graduates, undergraduates and pharmacy students in our laboratory. Authors thank Drs. Robert Kolb, Stefan Somlo, Bradley Yoder, and Jing Zhou for valuable insights and use of their laboratory reagents. Authors also thank Charisse Montgomery for her editorial review of the manuscript as well as Maki Takahashi and Shao Lo for use of their figures and illustrations. Work from our laboratory that is cited in this review has been supported by grants from the NIH (DK080640) and the NIH Recovery Act Funds. We are thankful to The University of Toledo research programs, including the deArce Memorial Endowment Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. The International Polycystic Kidney Disease Consortium, Cell. 1995;81:289–298. doi: 10.1016/0092-8674(95)90339-9. [DOI] [PubMed] [Google Scholar]
- 2.Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, Kimberling WJ, Breuning MH, Deltas CC, Peters DJ, Somlo S. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein, Science. Vol. 272. New York, N.Y: 1996. pp. 1339–1342. [DOI] [PubMed] [Google Scholar]
- Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, Milliner DS, Torres VE, Harris PC. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nature genetics. 2002;30:259–269. doi: 10.1038/ng833. [DOI] [PubMed] [Google Scholar]
- Cai Y, Maeda Y, Cedzich A, Torres VE, Wu G, Hayashi T, Mochizuki T, Park JH, Witzgall R, Somlo S. Identification and characterization of polycystin-2, the PKD2 gene product. The Journal of biological chemistry. 1999;274:28557–28565. doi: 10.1074/jbc.274.40.28557. [DOI] [PubMed] [Google Scholar]
- Delmas P, Nomura H, Li X, Lakkis M, Luo Y, Segal Y, Fernandez-Fernandez JM, Harris P, Frischauf AM, Brown DA, Zhou J. Constitutive activation of G-proteins by polycystin-1 is antagonized by polycystin-2. The Journal of biological chemistry. 2002;277:11276–11283. doi: 10.1074/jbc.M110483200. [DOI] [PubMed] [Google Scholar]
- Li Y, Santoso NG, Yu S, Woodward OM, Qian F, Guggino WB. Polycystin-1 interacts with inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling with implications for polycystic kidney disease. The Journal of biological chemistry. 2009;284:36431–36441. doi: 10.1074/jbc.M109.068916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnell SC, Magenheimer BS, Maser RL, Rankin CA, Smine A, Okamoto T, Calvet JP. The polycystic kidney disease-1 protein, polycystin-1, binds and activates heterotrimeric G-proteins in vitro. Biochemical and biophysical research communications. 1998;251:625–631. doi: 10.1006/bbrc.1998.9514. [DOI] [PubMed] [Google Scholar]
- Bai CX, Giamarchi A, Rodat-Despoix L, Padilla F, Downs T, Tsiokas L, Delmas P. Formation of a new receptor-operated channel by heteromeric assembly of TRPP2 and TRPC1 subunits. EMBO reports. 2008;9:472–479. doi: 10.1038/embor.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottgen M, Buchholz B, Garcia-Gonzalez MA, Kotsis F, Fu X, Doerken M, Boehlke C, Steffl D, Tauber R, Wegierski T, Nitschke R, Suzuki M, Kramer-Zucker A, Germino GG, Watnick T, Prenen J, Nilius B, Kuehn EW, Walz G. TRPP2 and TRPV4 form a polymodal sensory channel complex. The Journal of cell biology. 2008;182:437–447. doi: 10.1083/jcb.200805124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wright JM, Qian F, Germino GG, Guggino WB. Polycystin 2 interacts with type I inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling. The Journal of biological chemistry. 2005;280:41298–41306. doi: 10.1074/jbc.M510082200. [DOI] [PubMed] [Google Scholar]
- Ma R, Li WP, Rundle D, Kong J, Akbarali HI, Tsiokas L. PKD2 functions as an epidermal growth factor-activated plasma membrane channel. Molecular and cellular biology. 2005;25:8285–8298. doi: 10.1128/MCB.25.18.8285-8298.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiokas L, Arnould T, Zhu C, Kim E, Walz G, Sukhatme VP. Specific association of the gene product of PKD2 with the TRPC1 channel. Proceedings of the National Academy of Sciences of the United States of America; 1999. pp. 3934–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perrett S, Kim K, Ibarra C, Damiano AE, Zotta E, Batelli M, Harris PC, Reisin IL, Arnaout MA, Cantiello HF. Polycystin-2, the protein mutated in autosomal dominant polycystic kidney disease (ADPKD), is a Ca2+-permeable nonselective cation channel. Proceedings of the National Academy of Sciences of the United States of America; 2001. pp. 1182–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev PM, Guo L, Chen XZ, Segal Y, Peng JB, Basora N, Babakhanlou H, Cruger G, Kanazirska M, Ye C, Brown EM, Hediger MA, Zhou J. Polycystin-2 is a novel cation channel implicated in defective intracellular Ca(2+) homeostasis in polycystic kidney disease. Biochemical and biophysical research communications. 2001;282:341–350. doi: 10.1006/bbrc.2001.4554. [DOI] [PubMed] [Google Scholar]
- Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, Ehrlich BE, Somlo S. Polycystin-2 is an intracellular calcium release channel. Nature cell biology. 2002;4:191–197. doi: 10.1038/ncb754. [DOI] [PubMed] [Google Scholar]
- Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, Sukhatme VP, Guggino WB, Germino GG. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature. 2000;408:990–994. doi: 10.1038/35050128. [DOI] [PubMed] [Google Scholar]
- Tsiokas L, Kim E, Arnould T, Sukhatme VP, Walz G. Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proceedings of the National Academy of Sciences of the United States of America; 1997. pp. 6965–6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Fu Y, Hui K, Moeckel G, Mai W, Li C, Liang D, Zhao P, Ma J, Chen XZ, George AL, Jr, Coffey RJ, Feng ZP, Wu G. Fibrocystin/polyductin modulates renal tubular formation by regulating polycystin-2 expression and function. J Am Soc Nephrol. 2008;19:455–468. doi: 10.1681/ASN.2007070770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Li C, Liang D, Chen XZ, Coffy RJ, Ma J, Zhao P, Wu G. Polycystin -2 expression is regulated by a PC2-binding domain in the intracellular portion of fibrocystin. The Journal of biological chemistry. 2008;283:31559–31566. doi: 10.1074/jbc.M805452200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Zhang J, Nauli SM, Li X, Starremans PG, Luo Y, Roberts KA, Zhou J. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Molecular and cellular biology. 2007;27:3241–3252. doi: 10.1128/MCB.00072-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai CX, Kim S, Li WP, Streets AJ, Ong AC, Tsiokas L. Activation of TRPP2 through mDia1-dependent voltage gating. The EMBO journal. 2008;27:1345–1356. doi: 10.1038/emboj.2008.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvet V, Tian X, Husson H, Grimm DH, Wang T, Hiesberger T, Igarashi P, Bennett AM, Ibraghimov-Beskrovnaya O, Somlo S, Caplan MJ. Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. The Journal of clinical investigation. 2004;114:1433–1443. doi: 10.1172/JCI21753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low SH, Vasanth S, Larson CH, Mukherjee S, Sharma N, Kinter MT, Kane ME, Obara T, Weimbs T. Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Developmental cell. 2006;10:57–69. doi: 10.1016/j.devcel.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nature genetics. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- Nauli SM, Rossetti S, Kolb RJ, Alenghat FJ, Consugar MB, Harris PC, Ingber DE, Loghman-Adham M, Zhou J. Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J Am Soc Nephrol. 2006;17:1015–1025. doi: 10.1681/ASN.2005080830. [DOI] [PubMed] [Google Scholar]
- Xu C, Rossetti S, Jiang L, Harris PC, Brown-Glaberman U, Wandinger-Ness A, Bacallao R, Alper SL. Human ADPKD primary cyst epithelial cells with a novel, single codon deletion in the PKD1 gene exhibit defective ciliary polycystin localization and loss of flow-induced Ca2+ signaling. American journal of physiology. 2007;292:F930–945. doi: 10.1152/ajprenal.00285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Shmukler BE, Nishimura K, Kaczmarek E, Rossetti S, Harris PC, Wandinger-Ness A, Bacallao RL, Alper SL. Attenuated, flow-induced ATP release contributes to absence of flow-sensitive, purinergic Cai2+ signaling in human ADPKD cyst epithelial cells. American journal of physiology. 2009;296:F1464–1476. doi: 10.1152/ajprenal.90542.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AbouAlaiwi WA, Takahashi M, Mell BR, Jones TJ, Ratnam S, Kolb RJ, Nauli SM. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circulation research. 2009;104:860–869. doi: 10.1161/CIRCRESAHA.108.192765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauli SM, Kawanabe Y, Kaminski JJ, Pearce WJ, Ingber DE, Zhou J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation. 2008;117:1161–1171. doi: 10.1161/CIRCULATIONAHA.107.710111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poelmann RE, Van der Heiden K, Gittenberger-de Groot A, Hierck BP. Deciphering the endothelial shear stress sensor. Circulation. 2008;117:1124–1126. doi: 10.1161/CIRCULATIONAHA.107.753889. [DOI] [PubMed] [Google Scholar]
- Hou B, Kolpakova-Hart E, Fukai N, Wu K, Olsen BR. The polycystic kidney disease 1 (Pkd1) gene is required for the responses of osteochondroprogenitor cells to midpalatal suture expansion in mice. Bone. 2009;44:1121–1133. doi: 10.1016/j.bone.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Zhang S, Mahlios J, Zhou G, Magenheimer BS, Guo D, Dallas SL, Maser R, Calvet JP, Bonewald L, Quarles LD. Cilia-like structures and polycystin-1 in osteoblasts/osteocytes and associated abnormalities in skeletogenesis and Runx2 expression. The Journal of biological chemistry. 2006;281:30884–30895. doi: 10.1074/jbc.M604772200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masyuk AI, Masyuk TV, Splinter PL, Huang BQ, Stroope AJ, LaRusso NF. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology. 2006;131:911–920. doi: 10.1053/j.gastro.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masyuk TV, Huang BQ, Ward CJ, Masyuk AI, Yuan D, Splinter PL, Punyashthiti R, Ritman EL, Torres VE, Harris PC, LaRusso NF. Defects in cholangiocyte fibrocystin expression and ciliary structure in the PCK rat. Gastroenterology. 2003;125:1303–1310. doi: 10.1016/j.gastro.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Bisgrove BW, Snarr BS, Emrazian A, Yost HJ. Polaris and Polycystin-2 in dorsal forerunner cells and Kupffer’s vesicle are required for specification of the zebrafish left-right axis. Developmental biology. 2005;287:274–288. doi: 10.1016/j.ydbio.2005.08.047. [DOI] [PubMed] [Google Scholar]
- McGrath J, Somlo S, Makova S, Tian X, Brueckner M. Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell. 2003;114:61–73. doi: 10.1016/s0092-8674(03)00511-7. [DOI] [PubMed] [Google Scholar]
- Vogel P, Read R, Hansen GM, Freay LC, Zambrowicz BP, Sands AT. Situs inversus in Dpcd/Poll-/-, Nme7-/-, and Pkd1l1 -/-mice. Veterinary pathology. 2010;47:120–131. doi: 10.1177/0300985809353553. [DOI] [PubMed] [Google Scholar]
- AbouAlaiwi WA, Lo ST, Nauli SM. Primary Cilia: Highly Sophisticated Biological Sensors. Sensors. 2009;9:7003–7020. doi: 10.3390/s90907003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb RJ, Nauli SM. Ciliary dysfunction in polycystic kidney disease: an emerging model with polarizing potential. Front Biosci. 2008;13:4451–4466. doi: 10.2741/3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauli SM, Zhou J. Polycystins and mechanosensation in renal and nodal cilia. Bioessays. 2004;26:844–856. doi: 10.1002/bies.20069. [DOI] [PubMed] [Google Scholar]
- Praetorius HA, Spring KR. Removal of the MDCK cell primary cilium abolishes flow sensing. The Journal of membrane biology. 2003;191:69–76. doi: 10.1007/s00232-002-1042-4. [DOI] [PubMed] [Google Scholar]
- Siroky BJ, Ferguson WB, Fuson AL, Xie Y, Fintha A, Komlosi P, Yoder BK, Schwiebert EM, Guay-Woodford LM, Bell PD. Loss of primary cilia results in deregulated and unabated apical calcium entry in ARPKD collecting duct cells. American journal of physiology. 2006;290:F1320–1328. doi: 10.1152/ajprenal.00463.2005. [DOI] [PubMed] [Google Scholar]
- Liu W, Murcia NS, Duan Y, Weinbaum S, Yoder BK, Schwiebert E, Satlin LM. Mechanoregulation of intracellular Ca2+ concentration is attenuated in collecting duct of monocilium-impaired orpk mice. American journal of physiology. 2005;289:F978–988. doi: 10.1152/ajprenal.00260.2004. [DOI] [PubMed] [Google Scholar]
- Habas R, Dawid IB. Dishevelled and Wnt signaling: is the nucleus the final frontier? Journal of biology. 2005;4:2. doi: 10.1186/jbiol22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, Schermer B, Benzing T, Cabello OA, Jenny A, Mlodzik M, Polok B, Driever W, Obara T, Walz G. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nature genetics. 2005;37:537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto N, Cao Y, Park A, Sun Z. Cystic kidney gene seahorse regulates cilia-mediated processes and Wnt pathways. Developmental cell. 2008;14:954–961. doi: 10.1016/j.devcel.2008.03.010. [DOI] [PubMed] [Google Scholar]
- van Rooijen E, Giles RH, Voest EE, van Rooijen C, Schulte-Merker S, van Eeden FJ. LRRC50, a conserved ciliary protein implicated in polycystic kidney disease. J Am Soc Nephrol. 2008;19:1128–1138. doi: 10.1681/ASN.2007080917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla V, Reiter JF. The primary cilium as the cell’s antenna: signaling at a sensory organelle Science. Vol. 313. New York, N.Y: 2006. pp. 629–633. [DOI] [PubMed] [Google Scholar]
- Veland IR, Awan A, Pedersen LB, Yoder BK, Christensen ST. Primary cilia and signaling pathways in mammalian development, health and disease. Nephron. 2009;111:39–53. doi: 10.1159/000208212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadi-Kheddouci S, Berrebi D, Romagnolo B, Cluzeaud F, Peuchmaur M, Kahn A, Vandewalle A, Perret C. Early development of polycystic kidney disease in transgenic mice expressing an activated mutant of the beta-catenin gene. Oncogene. 2001;20:5972–5981. doi: 10.1038/sj.onc.1204825. [DOI] [PubMed] [Google Scholar]
- Lal M, Song X, Pluznick JL, Di Giovanni V, Merrick DM, Rosenblum ND, Chauvet V, Gottardi CJ, Pei Y, Caplan MJ. Polycystin-1 C-terminal tail associates with beta-catenin and inhibits canonical Wnt signaling. Human molecular genetics. 2008;17:3105–3117. doi: 10.1093/hmg/ddn208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happe H, Leonhard WN, van der Wal A, van de Water B, Lantinga-van Leeuwen IS, Breuning MH, de Heer E, Peters DJ. Toxic tubular injury in kidneys from Pkd1-deletionmice accelerates cystogenesis accompanied by dysregulated planar cell polarity and canonical Wnt signaling pathways. Human molecular genetics. 2009;18:2532–2542. doi: 10.1093/hmg/ddp190. [DOI] [PubMed] [Google Scholar]
- Kim E, Arnould T, Sellin LK, Benzing T, Fan MJ, Gruning W, Sokol SY, Drummond I, Walz G. The polycystic kidney disease 1 gene product modulates Wnt signaling. The Journal of biological chemistry. 1999;274:4947–4953. doi: 10.1074/jbc.274.8.4947. [DOI] [PubMed] [Google Scholar]
- Kim I, Ding T, Fu Y, Li C, Cui L, Li A, Lian P, Liang D, Wang DW, Guo C, Ma J, Zhao P, Coffey RJ, Zhan Q, Wu G. Conditional mutation of Pkd2 causes cystogenesis and upregulates beta-catenin. J Am Soc Nephrol. 2009;20:2556–2569. doi: 10.1681/ASN.2009030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romaker D, Puetz M, Teschner S, Donauer J, Geyer M, Gerke P, Rumberger B, Dworniczak B, Pennekamp P, Buchholz B, Neumann HP, Kumar R, Gloy J, Eckardt KU, Walz G. Increased expression of secreted frizzled-related protein 4 in polycystic kidneys. J Am Soc Nephrol. 2009;20:48–56. doi: 10.1681/ASN.2008040345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto S, Aiba A, Saito Y, Nakao K, Nakamura K, Tomita K, Kitamura T, Kurabayashi M, Nagai R, Higashihara E, Harris PC, Katsuki M, Horie S. Pioglitazone improves the phenotype and molecular defects of a targeted Pkd1 mutant. Human molecular genetics. 2002;11:1731–1742. doi: 10.1093/hmg/11.15.1731. [DOI] [PubMed] [Google Scholar]
- Roitbak T, Ward CJ, Harris PC, Bacallao R, Ness SA, Wandinger-Ness A. A polycystin-1 multiprotein complex is disrupted in polycystic kidney disease cells. Molecular biology of the cell. 2004;15:1334–1346. doi: 10.1091/mbc.E03-05-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorenson CM. Nuclear localization of beta-catenin and loss of apical brush border actin in cystic tubules of bcl-2 -/-mice. The American journal of physiology. 1999;276:F210–217. doi: 10.1152/ajprenal.1999.276.2.F210. [DOI] [PubMed] [Google Scholar]
- Fischer E, Legue E, Doyen A, Nato F, Nicolas JF, Torres V, Yaniv M, Pontoglio M. Defective planar cell polarity in polycystic kidney disease. Nature genetics. 2006;38:21–23. doi: 10.1038/ng1701. [DOI] [PubMed] [Google Scholar]
- Jonassen JA, San Agustin J, Follit JA, Pazour GJ. Deletion of IFT20 in the mouse kidney causes misorientation of the mitotic spindle and cystic kidney disease. The Journal of cell biology. 2008;183:377–384. doi: 10.1083/jcb.200808137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karner CM, Chirumamilla R, Aoki S, Igarashi P, Wallingford JB, Carroll TJ. Wnt9b signaling regulates planar cell polarity and kidney tubule morphogenesis. Nature genetics. 2009;41:793–799. doi: 10.1038/ng.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel V, Li L, Cobo-Stark P, Shao X, Somlo S, Lin F, Igarashi P. Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Human molecular genetics. 2008;17:1578–1590. doi: 10.1093/hmg/ddn045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saburi S, Hester I, Fischer E, Pontoglio M, Eremina V, Gessler M, Quaggin SE, Harrison R, Mount R, McNeill H. Loss of Fat4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nature genetics. 2008;40:1010–1015. doi: 10.1038/ng.179. [DOI] [PubMed] [Google Scholar]
- Nishio S, Tian X, Gallagher AR, Yu Z, Patel V, Igarashi P, Somlo S. Loss of oriented cell division does not initiate cyst formation. J Am Soc Nephrol. 2010;21:295–302. doi: 10.1681/ASN.2009060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Turkel N, Hemati N, Fuller MT, Hunt AJ, Yamashita YM. Centrosome misorientation reduces stem cell division during ageing. Nature. 2008;456:599–604. doi: 10.1038/nature07386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnould T, Kim E, Tsiokas L, Jochimsen F, Gruning W, Chang JD, Walz G. The polycystic kidney disease 1 gene product mediates protein kinase C alpha-dependent and c-Jun N-terminal kinase-dependent activation of the transcription factor AP-1. The Journal of biological chemistry. 1998;273:6013–6018. doi: 10.1074/jbc.273.11.6013. [DOI] [PubMed] [Google Scholar]
- Nishio S, Hatano M, Nagata M, Horie S, Koike T, Tokuhisa T, Mochizuki T. Pkd1 regulates immortalized proliferation of renal tubular epithelial cells through p53 induction and JNK activation. The Journal of clinical investigation. 2005;115:910–918. doi: 10.1172/JCI22850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori S, Hida M, Fujita H, Takahashi H, Tanimura S, Kohno M, Awazu M. Extracellular signal-regulated kinase inhibition slows disease progression in mice with polycystic kidney disease. J Am Soc Nephrol. 2006;17:1604–1614. doi: 10.1681/ASN.2004090800. [DOI] [PubMed] [Google Scholar]
- Parnell SC, Magenheimer BS, Maser RL, Zien CA, Frischauf AM, Calvet JP. Polycystin-1 activation of c-Jun N-terminal kinase and AP-1 is mediated by heterotrimeric G proteins. The Journal of biological chemistry. 2002;277:19566–19572. doi: 10.1074/jbc.M201875200. [DOI] [PubMed] [Google Scholar]
- Yu W, Kong T, Beaudry S, Tran M, Negoro H, Yanamadala V, Denker BM. Polycystin-1 protein level determines activity of the G{alpha}12/JNK apoptosis pathway. The Journal of biological chemistry. 2010 doi: 10.1074/jbc.M109.070821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togawa A, Miyoshi J, Ishizaki H, Tanaka M, Takakura A, Nishioka H, Yoshida H, Doi T, Mizoguchi A, Matsuura N, Niho Y, Nishimune Y, Nishikawa S, Takai Y. Progressive impairment of kidneys and reproductive organs in mice lacking Rho GDIalpha. Oncogene. 1999;18:5373–5380. doi: 10.1038/sj.onc.1202921. [DOI] [PubMed] [Google Scholar]
- Belibi FA, Reif G, Wallace DP, Yamaguchi T, Olsen L, Li H, Helmkamp GM, Jr, Grantham JJ. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney international. 2004;66:964–973. doi: 10.1111/j.1523-1755.2004.00843.x. [DOI] [PubMed] [Google Scholar]
- Hanaoka K, Guggino WB. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J Am Soc Nephrol. 2000;11:1179–1187. doi: 10.1681/ASN.V1171179. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Nagao S, Kasahara M, Takahashi H, Grantham JJ. Renal accumulation and excretion of cyclic adenosine monophosphate in a murine model of slowly progressive polycystic kidney disease. Am J Kidney Dis. 1997;30:703–709. doi: 10.1016/s0272-6386(97)90496-0. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Pelling JC, Ramaswamy NT, Eppler JW, Wallace DP, Nagao S, Rome LA, Sullivan LP, Grantham JJ. cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney international. 2000;57:1460–1471. doi: 10.1046/j.1523-1755.2000.00991.x. [DOI] [PubMed] [Google Scholar]
- Banales JM, Masyuk TV, Gradilone SA, Masyuk AI, Medina JF, LaRusso NF. Hepatology. Vol. 49. Baltimore, Md: 2009. The cAMP effectors Epac and protein kinase a (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD) pp. 160–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kip SN, Hunter LW, Ren Q, Harris PC, Somlo S, Torres VE, Sieck GC, Qian Q. [Ca2+]i reduction increases cellular proliferation and apoptosis in vascular smooth muscle cells: relevance to the ADPKD phenotype. Circulation research. 2005;96:873–880. doi: 10.1161/01.RES.0000163278.68142.8a. [DOI] [PubMed] [Google Scholar]
- Wang X, Ward CJ, Harris PC, Torres VE. Cyclic nucleotide signaling in polycystic kidney disease. Kidney international. 2010;77:129–140. doi: 10.1038/ki.2009.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortora G, Ciardiello F, Pepe S, Tagliaferri P, Ruggiero A, Bianco C, Guarrasi R, Miki K, Bianco AR. Phase I clinical study with 8-chloro-cAMP and evaluation of immunological effects in cancer patients. Clin Cancer Res. 1995;1:377–384. [PubMed] [Google Scholar]
- Yu SM, Cheng ZJ, Kuo SC. Antiproliferative effects of A02011-1, an adenylyl cyclase activator, in cultured vascular smooth muscle cells of rat. British journal of pharmacology. 1995;114:1227–1235. doi: 10.1111/j.1476-5381.1995.tb13337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutters M, Yamaguchi T, Maser RL, Magenheimer BS, St John PL, Abrahamson DR, Grantham JJ, Calvet JP. Polycystin-1 transforms the cAMP growth-responsive phenotype of M-1 cells. Kidney international. 2001;60:484–494. doi: 10.1046/j.1523-1755.2001.060002484.x. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. The Journal of biological chemistry. 2004;279:40419–40430. doi: 10.1074/jbc.M405079200. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Hempson SJ, Reif GA, Hedge AM, Wallace DP. Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol. 2006;17:178–187. doi: 10.1681/ASN.2005060645. [DOI] [PubMed] [Google Scholar]
- Wang X, Harris PC, Somlo S, Batlle D, Torres VE. Effect of calcium-sensing receptor activation in models of autosomal recessive or dominant polycystic kidney disease. Nephrol Dial Transplant. 2009;24:526–534. doi: 10.1093/ndt/gfn527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley BD., Jr Calcium, cyclic AMP, and MAP kinases: dysregulation in polycystic kidney disease. Kidney international. 2008;73:251–253. doi: 10.1038/sj.ki.5002695. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Nagao S, Wallace DP, Belibi FA, Cowley BD, Pelling JC, Grantham JJ. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney international. 2003;63:1983–1994. doi: 10.1046/j.1523-1755.2003.00023.x. [DOI] [PubMed] [Google Scholar]
- Boca M, D’Amato L, Distefano G, Polishchuk RS, Germino GG, Boletta A. Polycystin-1 induces cell migration by regulating phosphatidylinositol 3-kinase-dependent cytoskeletal rearrangements and GSK3beta-dependent cell cell mechanical adhesion. Molecular biology of the cell. 2007;18:4050–4061. doi: 10.1091/mbc.E07-02-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EY, Sung YH, Yang MH, Noh JY, Park SY, Lee TY, Yook YJ, Yoo KH, Roh KJ, Kim I, Hwang YH, Oh GT, Seong JK, Ahn C, Lee HW, Park JH. Cyst formation in kidney via B-Raf signaling in the PKD2 transgenic mice. The Journal of biological chemistry. 2009;284:7214–7222. doi: 10.1074/jbc.M805890200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boletta A. Emerging evidence of a link between the polycystins and the mTOR pathways. PathoGenetics. 2009;2:6. doi: 10.1186/1755-8417-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberthal W, Levine JS. The role of the mammalian target of rapamycin (mTOR) in renal disease. J Am Soc Nephrol. 2009;20:2493–2502. doi: 10.1681/ASN.2008111186. [DOI] [PubMed] [Google Scholar]
- Weimbs T. Cell cycle. Vol. 5. Georgetown, Tex: 2006. Regulation of mTOR by polycystin-1: is polycystic kidney disease a case of futile repair? pp. 2425–2429. [DOI] [PubMed] [Google Scholar]
- Bonnet CS, Aldred M, von Ruhland C, Harris R, Sandford R, Cheadle JP. Defects in cell polarity underlie TSC and ADPKD-associated cystogenesis. Human molecular genetics. 2009;18:2166–2176. doi: 10.1093/hmg/ddp149. [DOI] [PubMed] [Google Scholar]
- Hartman TR, Liu D, Zilfou JT, Robb V, Morrison T, Watnick T, Henske EP. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Human molecular genetics. 2009;18:151–163. doi: 10.1093/hmg/ddn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, Flask CA, Novick AC, Goldfarb DA, Kramer-Zucker A, Walz G, Piontek KB, Germino GG, Weimbs T. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proceedings of the National Academy of Sciences of the United States of America; 2006. pp. 5466–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattone VH, 2, Sinders RM, Hornberger TA, Robling AG. Late progression of renal pathology and cyst enlargement is reduced by rapamycin in a mouse model of nephronophthisis. Kidney international. 2009;76:178–182. doi: 10.1038/ki.2009.147. [DOI] [PubMed] [Google Scholar]
- Shillingford JM, Piontek KB, Germino GG, Weimbs T. Rapamycin Ameliorates PKD Resulting from Conditional Inactivation of Pkd1. J Am Soc Nephrol. 2010 doi: 10.1681/ASN.2009040421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Kim J, Schrier RW, Edelstein CL. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J Am Soc Nephrol. 2005;16:46–51. doi: 10.1681/ASN.2004080660. [DOI] [PubMed] [Google Scholar]
- Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD) Nephrol Dial Transplant. 2006;21:598–604. doi: 10.1093/ndt/gfi181. [DOI] [PubMed] [Google Scholar]
- Wu M, Arcaro A, Varga Z, Vogetseder A, Le Hir M, Wuthrich RP, Serra AL. Pulse mTOR inhibitor treatment effectively controls cyst growth but leads to severe parenchymal and glomerular hypertrophy in rat polycystic kidney disease. American journal of physiology. 2009;297:F1597–1605. doi: 10.1152/ajprenal.00430.2009. [DOI] [PubMed] [Google Scholar]
- Zafar I, Belibi FA, He Z, Edelstein CL. Long-term rapamycin therapy in the Han:SPRD ra model of polycystic kidney disease (PKD) Nephrol Dial Transplant. 2009;24:2349–2353. doi: 10.1093/ndt/gfp129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dere R, Wilson PD, Sandford RN, Walker CL. Carboxy Terminal Tail of Polycystin-1 Regulates Localization of TSC2 to Repress mTOR. PloS one. 2010;5:e9239. doi: 10.1371/journal.pone.0009239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleymenova E, Ibraghimov-Beskrovnaya O, Kugoh H, Everitt J, Xu H, Kiguchi K, Landes G, Harris P, Walker C. Tuberin-dependent membrane localization of polycystin-1: a functional link between polycystic kidney disease and the TSC2 tumor suppressor gene. Molecular cell. 2001;7:823–832. doi: 10.1016/s1097-2765(01)00226-x. [DOI] [PubMed] [Google Scholar]
- Distefano G, Boca M, Rowe I, Wodarczyk C, Ma L, Piontek KB, Germino GG, Pandolfi PP, Boletta A. Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Molecular and cellular biology. 2009;29:2359–2371. doi: 10.1128/MCB.01259-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer DC, Jacoby U, Pape L, Ward CJ, Kuwertz-Broeking E, Renken C, Nizze H, Querfeld U, Rudolph B, Mueller-Wiefel DE, Bergmann C, Haffner D. Activation of the AKT/mTOR pathway in autosomal recessive polycystic kidney disease (ARPKD) Nephrol Dial Transplant. 2009;24:1819–1827. doi: 10.1093/ndt/gfn744. [DOI] [PubMed] [Google Scholar]
- Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, Rentsch KM, Spanaus KS, Senn O, Kristanto P, Scheffel H, Weishaupt D, Wuthrich RP. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. The New England journal of medicine. 2010;363:820–829. doi: 10.1056/NEJMoa0907419. [DOI] [PubMed] [Google Scholar]
- Walz G, Budde K, Mannaa M, Nurnberger J, Wanner C, Sommerer C, Kunzendorf U, Banas B, Horl WH, Obermuller N, Arns W, Pavenstadt H, Gaedeke J, Buchert M, May C, Gschaidmeier H, Kramer S, Eckardt KU. Everolimus in patients with autosomal dominant polycystic kidney disease. The New England journal of medicine. 2010;363:830–840. doi: 10.1056/NEJMoa1003491. [DOI] [PubMed] [Google Scholar]
- Watnick T, Germino GG. mTOR inhibitors in polycystic kidney disease. The New England journal of medicine. 2010;363:879–881. doi: 10.1056/NEJMe1006925. [DOI] [PubMed] [Google Scholar]