

Abstract
Pittsburgh compound B (PIB) is a neutral derivative of the fluorescent dye Thioflavin T (ThT), which displays enhanced hydrophobicity and binding affinity to amyloid fibrils. We present molecular dynamics simulations of binding of PIB and ThT to a common cross-β-subunit of the Alzheimer Amyloid-β peptide (Aβ). Our simulations of binding to Aβ9-40 protofibrils show that PIB, like ThT, selectively binds to the hydrophobic or aromatic surface grooves on the β-sheet surface along the fibril axis. The lack of two methyl groups and charge in PIB not only improves its hydrophobicity but also leads to a deeper insertion of PIB compared to ThT into the surface grooves. This significantly increases the steric, aromatic, and hydrophobic interactions, and hence leads to stronger binding. Simulations on protofibrils consisting of the more-toxic Aβ17-42 revealed an additional binding mode in which PIB and ThT insert into the channel that forms in the loop region of the protofibril, sandwiched between two sheet layers. Our simulations indicate that the rotation between the two ring parts of the dyes is significantly more restricted when the dyes are bound to the surface of the cross-β-subunits or to the channel inside the Aβ17-42 cross-β-subunit, compared with free solution. The specific conformations of the dyes are influenced by small chemical modifications (ThT versus PIB) and by the environment in which the dye is placed.
Introduction
Alzheimer's disease (AD) is a progressive and fatal brain disease, and currently is the most common form of dementia worldwide with no known cure. The incidence of AD is increasing exponentially (1) as the lifespan of the global population continues to lengthen, and thus there is an urgent need for the development of accurate diagnostic tools (2) and therapies (3) for this disease.
The presence of amyloid plaques in the brain that consist primarily of fibrils of the Alzheimer Amyloid-β (Aβ) peptide is a major hallmark of AD (4). Aβ peptides are produced by proteolytic cleavage of the APP precursor protein in two main forms: the 40-residue-long Aβ1-40 and the 42-residue-long Aβ1-42. The Aβ1-42 peptide aggregates more rapidly than Aβ1-40 and is more cytotoxic (5). Biomarkers of the presence of amyloid fibrils in the brain are invaluable diagnostic tools for identifying AD and determining the efficacy of new anti-amyloid therapies for eliminating fibril formation (2). Thioflavin-T (ThT) is one of most commonly used fluorescent dyes to detect the presence of amyloid fibrils in posthumous brain samples and to monitor fibril formation in vitro (6). ThT exhibits a red shift in its excitation spectrum and an emission enhancement upon binding to β-sheet-rich amyloid fibrils. These phenomena stem from two effects upon binding to the fibril: 1), steric and electronic stabilization (via charge transfer) of the ground-state charge distribution (7,8); and 2), restriction in the rotation of the aromatic rings of the dye (see Fig. 1 A) in its electronically excited state (9,10). Although ThT offers a reliable means of visualizing amyloid aggregates in vitro, it suffers from two major drawbacks that hinder its clinical use in living beings. First, ThT is weakly hydrophobic and does not readily enter the brain, and second, its binding affinity to fibrils is low. Many derivatives of ThT have been developed over the past few years (11), with the Pittsburgh compound B (PIB) developed by Mathis et al. (12,13) emerging as the most promising candidate. This dye, a derivative of ThT, is currently being evaluated clinically in ∼40 positron emission tomography (PET) centers around the world (11) for direct visualization of amyloid plaques in the brains of AD patients.
Figure 1.

Structures of two amyloid dyes. (A) ThT. (B) The neutral analog PIB.
Two types of structural modifications were introduced in ThT to generate PIB. First, two methyl groups and the positive charge that is present in ThT were removed, leading to a compound known as BTA-1. These modifications relative to ThT were shown to increase both hydrophobicity/lipophilicity and the binding affinity, which increase the probability of crossing the brain-blood barrier and binding amyloid fibrils in brain tissue (14). Second, a methyl group was replaced by a hydroxyl group to improve brain clearance properties. The resulting PIB molecule (see Fig. 1) shows good brain clearance abilities, increased binding affinity by 135-fold to Kd = 4.3 nM and increased lipophilicity by ∼300-fold over ThT (12). It is interesting to note that the modifications introduced to develop this neutral derivative led to a slight change in the fluorescence properties. Although the excitation and emission spectra of PIB are similar to those of ThT, the uncharged derivative does not demonstrate the red shift in excitation and the emission enhancement when bound to amyloid fibrils (14,15). The experimentally observed higher binding affinity of PIB over ThT compensates for the latter and explains why PIB is a successful amyloid dye.
The location of the binding sites of PIB on Aβ amyloid fibrils is not known at atomically detailed resolution (16). The Aβ fibrils are not crystalline, which prevents x-ray determination of a dye-fibril complex. NMR studies of binding are equally challenging. The lack of available high-resolution structural data regarding precisely where PIB binds on the fibrils poses a real challenge for understanding the dye-fibril recognition mechanisms.
Our goal in this study was to use molecular dynamics simulations to provide an atomically detailed picture of the binding of PIB and ThT to fibril models of the Aβ40 and Aβ42 peptides, and to identify β-sheet-specific binding modes. Although fibrils of the Aβ peptides come in a variety of morphologies, depending on the details of the sample preparation conditions (17,18), they appear to share a common U-shape building block: a single cross-β subunit in which each Aβ peptide has β-strand-loop-β-strand conformation and registers to form parallel β-sheets. Even though both Aβ1-40 and Aβ1-42 have similar β-strand-loop-β-strand conformations, the loop region differs significantly in the terms of the location and the width. Whereas the loop of Aβ1-40 is tight and located at V24-A30, the Aβ1-42 loop is wide and located at S26-I31 (see Fig. 2). Two or more cross-β subunits can stack together to form higher-order fibrils, and several models of such higher-order fibrils were recently proposed based on this common motif (17,19,20). These models are consistent with mass-per-length studies showing that Aβ fibrils can be formed from a single subunit, a double subunit (typically seen in agitated samples), or a triple subunit (seen in quiescently grown samples of Aβ1-40) (21). Our simulations focused on the single cross-β subunit, as this subunit encompasses the maximum number of binding sites available to the dyes (in fibrils with multiple subunits, some binding sites are not available for binding because they are buried inside the subunit-subunit interface).
Figure 2.

Initial structure of an Aβ cross-β-subunit from solid-state NMR studies. (A) A single Aβ9-40 peptide in the cross-β-subunit. (B) An Aβ9-40 cross-β subunit containing six peptides. (C) A single Aβ17-42 peptide in the cross-β-subunit. (D) An Aβ17-42 cross-β subunit containing six peptides. Negatively charged, positively charged, polar, and hydrophobic residues are colored in red, blue, green, and black, respectively.
A single cross-β subunit consisting of six Aβ9-40/Aβ17-42 peptides, based on solid-state NMR studies (see Materials and Methods section), is shown in Fig. 2 (residues 1–8 in fibrils of Aβ1-40 and residues 1–16 in fibrils of Aβ1-42 are disordered and are omitted here). For the Aβ9-40 cross-β subunit, the peptides in the U-shape are registered in parallel, with seven grooves on the first β-sheet formed by residues 10–24 and two grooves on the second β-sheet formed by residues 31–39. We will refer to the surface grooves by specifying the exposed residues as follows: Y10_V12_H14_K16_V18_F20_E22_V24 for the N-terminal β-sheet layer, and I31_M35_V39 for the C-terminal β-sheet layer. For the Aβ17-42 cross-β subunit, there are four grooves on the first β-sheet formed by residues 17–26 and three grooves on the second β-sheet formed by residues 31–42. The grooves are specified as V18_F20_E22_V24_S26 for the N-terminal β-sheet layer, and I31_M35_V39_I41 for the C-terminal β-sheet layer. Both cross-β subunits contain a water/ion channel formed in the loop region (20,22,23). Our simulations enable us to identify the binding sites and binding modes of the dyes, and our solvation free-energy and binding-energy analyses shed light on the origin of the experimentally observed enhancement of lipophilicity and binding affinity of PIB over ThT. Finally, an analysis of the rotation of the central torsion angle of the dyes in their electronic ground states offers a glimpse into how conformational restriction affects the different binding modes in their excited electronic states.
Materials and Methods
System preparation
Each of our four binding simulation system contains a single cross-β-subunit (6 Aβ9-40/Aβ17-42 peptides), two molecules of each dye (ThT/PIB), ∼11000 water molecules, and an additional four/six sodium ions in the case of the positively charged ThT/neutral PIB to neutralize the system. The initial structure of the Aβ9-40 cross-β-subunit (see Fig. 2 B) is taken from the study by Petkova and co-workers (17), and the initial structure of the Aβ17-42 cross-β-subunit (Fig. 2 D) is taken from the study by Lührs and co-workers (18). Two dye molecules are initially placed ∼10 Å away from the N-/C-terminal sheet layers (see Fig. S22 of the Supporting Material). This allows simultaneous sampling of two sheet layers in a single trajectory and thus enhances sampling in comparison to systems with a single dye molecule (i.e., five simulations on a system with two dye molecules are equivalent to 10 simulations on a system with one dye molecule). For ThT, four positive sodium ions (Na+) plus two positive charges carried by two ThT molecules are added to neutralize the six peptide negative charges. For PIB, six positive sodium ions (Na+) are added to neutralize the peptide charges. The solute molecules are immersed into a truncated octahedral box (a = b = c = ∼80 Å, α = β = γ = 109.47°) of ∼11,000 water molecules. The solute is at least ∼12 Å away from the box surface, and thus the minimum distance between the solute and the image is ∼24 Å.
To investigate the rotation of the aromatic rings of the dyes in bulk waters, we constructed two systems containing one dye (ThT/PIB) molecule, ∼1600 water molecules, and an additional sodium ion in the case of the positively charged ThT.
The all-atom point-charge force field (AMBER ff03) developed by Duan et al. (24), which shows a good balance in the balance between helix and sheet (25), was chosen to represent the peptides. The water solvent is explicitly represented by the TIP3P model (26). The parameters for ThT were obtained from a previous study (27). Following the same protocol, the parameters for PIB are developed as follows: the electrostatic potential of PIB is obtained at the HF/6-31G∗∗ level after a geometry optimization at the same level. The partial charges are derived by fitting the gas-phase electrostatic potential using the restrained electrostatic potential (RESP) method (28), and other force parameters of the PIB molecule are taken from the AMBER GAFF (29) parameter set. The PIB parameter file in AMBER format is provided in the Supporting Material.
Details on the simulations and analysis methods are given in the Supporting Material.
Results
Binding to the Aβ9-40 cross-β-subunit
We constructed a system consisting of two dye molecules and an Aβ9-40 cross-β-subunit built from two parallel β-sheets as described in the Methods section of the Supporting Material. We omitted residues 1–9 because they are disordered (based on solid-state NMR studies). The overall sheet-loop-sheet fold was stable at 320 K over the course of all 10 60-ns binding simulations (see the root mean-square deviation (RMSD) over time plots in Fig. S1 and Fig. S4). Nonetheless, the structures showed some deformations (Cα RMSD = ∼4 Å) as a result of the small size of the protofibril and the binding of the dyes. The overall convergence of the binding simulations appears to be confirmed by many reversible events of dye molecules binding to the cross-β-subunit (Fig. S2 and Fig. S5). The simulations were initiated with two copies of the same dye (ThT or PIB) placed above and below the protofibrils. We found that the two copies bound to opposite sides of the cross-β-subunit, resulting in statistically independent simulations of each copy, in all trajectories except for one trajectory in which two PIB monomers formed a dimer and then bound to the cross-β-subunit (Fig. S13 and Fig. S14). In a recent study of binding of naproxen to the Aβ9-40 cross-β-subunit, Takeda et al. (30) observed that its strong ligand-fibril binding affinity can be partially attributed to the favorable interligand ring-ring stacking interactions due to aligning multiple ligands along the fibril axis in the grooves. In other words, multiple ligands can bind to the same site. This effect should also be applicable to ThT/PIB at a high ligand concentration.
To visualize the dye binding, we superimposed the cross-β-subunit of the bound complexes identified from the trajectories (Fig. S15). For ThT, we identified seven populated dye clusters: six clusters along the grooves perpendicular to the β-strands, and one cluster at one end of the β-sheet and oriented parallel to the β-strands (Fig. S15 A). It is interesting that the positively charged ThT molecules did not bind to the negatively charged E22, indicating that charge-charge interaction does not play an important role in binding. The binding pattern for PIB is quite similar to that of ThT: four clusters along the grooves perpendicular to the β-strands, two clusters at the loop region, and two clusters at one end of the β-sheet (Fig. S15 B). In other words, removing the charge on ThT (as was done to create the neutral analog PIB) does not significantly alter the binding pattern. This again indicates that charge-charge interaction is not necessary for dye binding to the fibril.
To gain more insight into the binding modes, we classified the bound complexes into different structural families based on a clustering analysis using the RMSD of the dye molecule (cutoff of 2.5 Å), after aligning the cross-β-subunit. The centroids of the top structural families (>1% of the population) for each dye are shown in Fig. S16 and Fig. S17. For ThT, the top 20 structural families can be further merged into the six binding modes shown in Fig. S18, labeled ThT_A–F (A: binding on the top of F20 of the N-terminal sheet-layer; B: binding in the M35_V39 groove of the C-terminal sheet layer; C: binding in the E11_H13 inner groove of the N-terminal sheet layer; D: binding in the V12_H14 groove of the N-terminal sheet layer (we note that this mode may be due to thermal deformations of the cross-β-subunit); E: binding at the loop region of the cross-β-subunit; and F: binding at the edge of the β-sheet). For PIB, the top eight structural families could also be grouped into six binding modes (PIB_A–F; Fig. S18). The last four binding modes, C–F, are identical to binding modes C–F of ThT. The first two have subtle differences. In the case of PIB, binding modes A and B correspond to binding in the V18_H20 groove of the N-terminal sheet layer and binding in the I31_M35 groove of the C-terminal sheet layer, respectively. Of interest, although nine grooves are available for binding, the dyes do not bind to all nine grooves and instead selectively bind to hydrophobic/aromatic grooves.
To gain a quantitative understanding of the binding differences between ThT and PIB, we evaluated the binding energy for each binding mode using the molecular mechanics-generalized Born surface area (MM-GBSA) method as described in the Supporting Material. The six binding modes for ThT have binding energies ranging from −5.2 to −14.5 kcal/mol (see Table 1), whereas those for PIB range from −9.0 to −30.6 kcal/mol. The most favorable binding site for ThT (−14.5 kcal/mol) is on top of the F20 rings of the N-terminal sheet layer. This binding mode is consistent with our earlier simulations on Aβ16-22 and a peptide self-assembly mimic (31,32), and suggests that ThT preferentially binds to grooves formed by aromatic or hydrophobic residues (Phe and His). The site with the most favorable binding energy for PIB is in the M35_V39 hydrophobic groove of the C-terminal β-sheet. On the basis of the fact that ThT and PIB do not share the same lowest binding-energy site, we predict that PIB, like its analog BTA-1 (in which the hydroxyl group of PIB is replaced by a methyl group) (15,33), binds to fibrils noncompetitively with ThT. For both ThT and PIB, binding in the grooves is more favorable than binding at the loop and the β-sheet edges, suggesting that binding in the grooves is the primary fibril recognition mode. It is interesting to note that the primary binding mode seen here for the amyloidogenic dyes differs from the main binding mode observed for other types of ligands, such as the aggregation inhibitor ibuprofen. In the case of ibuprofen, it binds most favorably to the concave edges of the Aβ40 protofibrils, possibly blocking β-sheet extension in this manner (34).
Table 1.
Locations and MM-GBSA binding energies (kcal/mol) of the different binding modes of dyes on the Aβ protofibrils (Fig. S18 and Fig. S21)
| Unit (kcal/mol) | Site A | Site B | Site C | Site D | Site E | Site F |
|---|---|---|---|---|---|---|
| Aβ9-40 + ThT | F20 | M35_V39 | E11_H13 | V12_H14 | loop | edge |
| −14.5 ± 4.5 | −12.2 ± 3.1 | −9.9 | −5.2 ± 3.3 | −8.6 ± 3.4 | −11.6 ± 5.5 | |
| Aβ9-40 + PIB | V18_F20 | 131_M35 | E11_H13 | V12_H14 | loop | edge |
| −20.0 ± 3.5 | −30.6 ± 4.8 | −9.0 | −16.1 | −10.5 | −12.4 | |
| Aβ17-42 + ThT | V18_F20 | 131_M35 | V24_S26 | inside loop | edge | parallel |
| −12.4 ± 4.1 | −17.5 | −8.2 ± 2.4 | −33.9 | −7.8 ± 4.8 | −12.3 ± 3.7 | |
| Aβ17-42 + PIB | V18_F20 | 131_M35 | V24_S26 | inside loop | edge | parallel |
| −11.2 ± 0.1 | −19.4 ± 6.4 | −14.5 | −34.3 ± 0.9 | −7.6 ± 0.9 | −12.0 ± 0.9 |
Decomposition of the total binding energy (see Table 2), obtained from averaging over all modes, reveals that the van der Waals and surface term (apolar solvation term) favor binding, whereas the electrostatic contributions (GB desolvation + gas phase electrostatics) are unfavorable. This explains why ThT and PIB bind only weakly or not at all to the charged grooves (H14_K16_V18, F20_E22_V24 and H13_E11), but bind strongly to the other hydrophobic or aromatic grooves (I31_M35, V18_F20, F20, V12_H14 and M35_V39). Our calculations support a picture in which hydrophobic or aromatic and steric interactions are the stabilizing forces for the dye binding, and explain the experimental observation that ThT does not appreciably bind to highly charged fibrils (35). The relative binding free-energy difference between PIB and ThT is −7.3 kcal/mol (assuming similar binding entropy), which is qualitatively consistent with the experimental number of ∼−3 kcal/mol. Removal of the charge and the two methyl groups from ThT to form PIB leads to a deeper insertion of PIB into the hydrophobic or aromatic grooves (M35_V39, V12_H14 and V18_F20) compared with ThT, as indicated by the ∼20% increase in surface burial of PIB over ThT into the protofibrils (Table 3). This insertion significantly increases the van der Waals and hydrophobic interactions.
Table 2.
Averaged binding energies (kcal/mol) and surface burial of ThT and its neutral analog, PIB, to the Aβ protofibrils
| Unit (kcal/mol) | ΔEVDW | ΔESUR | ΔEGBELE | ΔETOT | Surface burial |
|---|---|---|---|---|---|
| Aβ9-40 + ThT | −11.1 ± 8.9 | −0.9 ± 0.7 | 2.1 ± 2.2 | −9.9 ± 7.8 | 35 ± 19% |
| Aβ9-40 + PIB | −21.1 ± 7.5 | −2.2 ± 0.8 | 6.1 ± 0.7 | −17.2 ± 7.9 | 56 ± 17% |
| Change (ΔΔE) | −10.0 | −1.3 | 4.0 | −7.3 | – |
| Aβ17-42 + ThT | −16.4 ± 6.3 | −1.6 ± 0.6 | 2.7 ± 1.3 | −15.3 ± 5.8 | 43 ± 13% |
| Aβ17-42 + PIB | −20.5 ± 10.7 | −2.3 ± 1.0 | 5.9 ± 2.4 | −16.9 ± 9.5 | 52 ± 20% |
| Change (ΔΔE) | −4.1 | −0.7 | 3.2 | −1.6 | – |
ΔEVDW: The change of VDW energy in gas phase upon complex formation.
ΔESUR: The change of nonelectrostatic solvation energy due to surface area change upon complex formation.
ΔEGBELE: The change of GB electrostatic solvation energy + gas phase electrostatic energy upon complex formation.
ΔETOT = ΔEVDW +ΔESUR + ΔEGBELE: The change of potential energy in water upon complex formation.
GB: Generalized Born reaction field energy.
Table 3.
Calculated solvation free energies of the dyes
| Ligand | Electrostatics | ΔGsolv (kcal/mol)VDE | Total∗ |
|---|---|---|---|
| ThT+ + C1−† | −84.6 | 6.3 | −78.3 ± 0.3 |
| PIB | −9.6 | 0.2 | −9.3 ± 0.2 |
Errors were calculated by using three blocks of trajectories (3–4 ns, 4–5 ns, and 5–6 ns).
From Wu et al. (31).
We calculated the absolute solvation free energies of PIB using the same thermodynamic integration protocol we employed for ThT+Cl− in our earlier work on Aβ16-22 (31). The hydrophobicity of PIB is significantly improved compared with that of ThT, as indicated by the change in solvation free energy in water from −78.3 kcal/mol for ThT+Cl− to −9.4 kcal/mol for PIB (Table 3). Although the absolute value of the solvation free energy in water is not equal to the solvation free-energy difference in a lipid, the trend will be the same because hydrophobicity is positively correlated with lipophilicity. Thus, our results regarding the water difference (68.9 kcal/mol) are consistent with the experimental observation that PIB is ∼300-fold (3.4 kcal/mol) more lipophilic than ThT (33).
Binding to Aβ17-42 cross-β-subunit
In addition to investigating the binding of ThT/PIB to Aβ40 fibrils, we also considered the binding of these dyes to the fibrils of the more-toxic Aβ42 peptides. We constructed a system consisting of two dye molecules and an Aβ17-42 cross-β-subunit built from two parallel β-sheets as described in Materials and Methods. We omitted residues 1–16 because they are disordered (based on solid-state NMR studies). The protofibril was stable at 320 K over the course of all five 60-ns binding simulations of each dye (Fig. S7 and Fig. S10). The overall convergence of the binding simulations appears to be confirmed by many reversible events of dye molecules binding to the cross-β-subunit (Fig. S8 and Fig. S11).
Following the clustering analysis, the top 24 and 12 structural families for ThT and PIB (Fig. S19 and Fig. S20) were be grouped into six binding modes (Fig. S21). With the exception of mode D, the other five modes (A–C and E-F) are very similar to the corresponding binding modes (A–C and E-F) seen in the case of the Aβ9-40 cross-β-subunit. In particular, binding modes A–C correspond to binding in the V18_H20 groove of the N-terminal sheet layer, binding in the I31_M35 groove of the C-terminal sheet layer, and binding in the V24_S26 groove of the N-terminal sheet layer, respectively, whereas binding modes E and F are located close to the β-sheet edge. Mode D is not seen in the case of Aβ9-40. In this mode, ThT/PIB inserts into the loop channel and binds under the salt bridge K28–D23 and above the I32–L34 side chains. This insertion mode can be clearly seen from selected snapshots from an ThT insertion trajectory shown in Fig. S22. We did not observe this insertion mode for the Aβ9-40 cross-β-subunit, probably as a result of the smaller channel formed by the disordered loop of Aβ9-40 or the limited simulation time.
The six binding modes of ThT and PIB on the Aβ17-42 cross-β-subunit have binding energies ranging from −7.6 to −34.3 kcal/mol (Table 2). It is interesting to note that the most favorable binding site (∼−34 kcal/mol) for both dyes is located inside the loop channel (mode D). The overall binding energy of PIB to the Aβ17-42 cross-β-subunit (−16.9 ± 9.5 kcal/mol) is slightly more favorable than that of ThT (−15.3 ± 5.9 kcal/mol), by −1.6 kcal/mol (Table 3). The surface burial of PIB (52 ± 20%) upon binding is again larger than that of ThT (43 ± 13%) by ∼10%, which may explain the slightly more favorable van der Waals and hydrophobic interactions between PIB and the Aβ17-42 cross-β-subunit compared with those between ThT and the Aβ17-42 cross-β-subunit (Table 3).
Rotation of the central torsion angle in dyes
It has been proposed that restriction of the rotation of the central bond linking the two ring parts of ThT is an important component of the observed fluorescence enhancement upon binding to fibrils (9,10). To characterize the preferential orientation of the central torsion angle of the dyes under different environments, we calculated the probability distributions of the torsion angle (Fig. S23). Because the dyes have roughly a C2 rotation symmetry, the torsion angle is 0–90°. In water, the most probable torsion angles for ThT and PIB are ∼60° and ∼30°, respectively. The less-planar conformation for ThT over PIB is caused by the steric effect of a CH3 linked to the five-member ring in ThT. We note that our minimum free-energy torsion angle (∼60°) of ThT in water is larger than the minimum energy torsion angle of ThT in the gas phase (45.6°) obtained from a quantum mechanism calculation at a level of MP2/RHF/6-31G∗∗ (9). This small difference may be due to the solvent effect or imprecision in the force field. Nonetheless, the possible shift in the minimum free-energy torsion angle for the two dyes should not affect relative trends under different environments. When ThT is bound to the surface of the AβB17-42B cross-β-subunit, its distribution becomes narrower and it shifts to a more planar conformation than observed in water. When ThT is inserted into the loop channel, its distribution becomes broader and more rugged than in water, which may be indicative of conformational trapping and reflect a slow relaxation of the torsion angles when ThT interacts with the fibril side chains rather than water molecules. Similar effects were seen for PIB, highlighting the fact that the environment in which the dye is placed influences its conformation.
To further characterize the rotation relaxation of the central torsion angle of the two dyes under different environments, we calculated the autocorrelation of each dye molecule in each binding trajectory (Fig. S3, Fig. S6, Fig. S9, and Fig. S12). The fitted rotation relaxation time for each dye trajectory is listed in Table S1. A summary of the results is provided in Table 4. Clearly, the rotation relaxation time of the two dyes increases from 0.4 to 0.7 ps in bulk water, to 6.1–10.5 ps on the surface, and to 39.4–1962.0 ps inside the channel of the cross-β-subunit. The increase of the rotation relaxation time (τ) indicates larger friction forces/viscosity(η) due to stronger rotational restriction imposed by the more rigid environment on dye molecules, as reflected by the Debye-Einstein-Stokes equation (, where v is volume, Kb is Boltzmann's constant, and T is temperature) (36). Although our simulations deal with the ground-state conformation and not the excited state, there will likely be similarities between the rotation restriction effects in the ground and electronically excited states. In this sense, our simulations support the proposal that the environment of the fibril is responsible for the rotational restriction of the dye molecules (9).
Table 4.
Relaxation times in ps of the central torsion angle of dyes under different environments
| Environment τ(ps) |
ThT |
PIB |
|---|---|---|
| In water | 0.4 | 0.7 |
| Aβ9-40 surface binding | 10.2 ± 3.2 | 6.1 ± 2.0 |
| Aβ9-40 channel inserting | – | – |
| Aβ17-42 surface binding | 10.5 ± 5.5 | 7.7 ± 3.4 |
| Aβ17-42 channel inserting | 1962.0 | 39.4 |
Discussion and Conclusions
When ThT binds to amyloid fibrils, a net fluorescence enhancement is observed. Recent work by Wolfe and co-workers (7) illustrates this point in a compelling manner. When ThT binds to amyloid-like β2m crystal or insulin amyloid fibrils, the enhancements are 28- and 15-fold, but when it binds to monomeric or nonamyloid-like protein aggregates, the enhancements are only six- and eightfold. The induced fluorescence enhancement includes two steps: 1), β-sheet-specific binding, involving such factors as binding sites, binding affinity, protein/dye ratio, and dye conformation dynamics; and 2), the steric and electronic (via charge transfer) stabilization of the ground-state charge distribution (7,8) coupled with a restriction in the rotation of the aromatic rings of the dye at the electronically excited state (10). In this study, we focused on elucidating the nature of the binding modes of ThT and its neutral derivative, PIB, to cross-β structures, as well as determining the conformational preferences of the dyes in their electronic ground states. A study of the excited state would necessitate a quantum mechanical analysis, which would be beyond the scope of this work. The ground-state conformations are of particular importance when nonfluorescence detection techniques (such as nuclear radiation) are employed to detect amyloid fibrils.
Experiments on ThT binding to amyloid fibrils suggest the presence of multiple distinct binding sites with different stoichiometries and binding affinities (37,38). Krebs et al. (39) proposed that ThT binds along the fibril axis in the regular channels between the side chains of residues n and n + 2 of the registered β-strands on the surface of the β-sheet, and that steric interactions are responsible for binding. Although this model paints a general picture of ThT β-sheet binding, it does not predict any binding preferences for particular types of side chains. In other words, in this model the dyes bind to any grooves formed by any type of side chain (charged, polar, hydrophobic, or aromatic). However, recent studies by Biancalana et al. (35) indicated that ThT does not appreciably bind to designed, highly charged fibrils with only charged side chains at the β-sheet surface, whereas its neutral derivatives bind with high affinities to such fibrils (12). We note that ThT has been observed to bind to fibrils that are charged (e.g.,, amylin or β2 microglobulin at low pH), the critical difference being that these fibrils have mixed types of side chains at the β-sheet surface, rather than presenting only charged side chains as in the Koide construct. We recently performed molecular dynamics simulations on two model fibril systems consisting of a small fragment of the AD Aβ peptide (the Aβ16-22 fragment (31)) and a peptide self-assembly mimic (32). These studies explained for the first time (to our knowledge) the binding specificity of ThT to different possible binding sites on the fibrils. Furthermore, we showed that the primary binding mode of ThT is located in the grooves created by the hydrophobic and aromatic residues (e.g., Phe, His, and Val) on the lateral face of the fibrils, and that the hydrophobic or aromatic interactions play an important role in determining this binding preference. For ThT to bind, the linear grooves must consist of at least five β-strands registered in a planar sheet (32). In this respect, these grooves are specific to fibrils, as most proteins or small oligomers do not possess such linear grooves. β-barrel proteins, for example, contain only curved grooves, which weakens the fit of linear dyes to these grooves. We note that ThT-fibril binding is less specific than typical protein-ligand binding, in which only a few binding pockets are involved.
In this study, we investigated the binding of both ThT and PIB to fibrils of the AD Aβ9-40 peptide and the more-toxic AD Aβ17-42 peptide. These systems offer the closest models of fibrils of the full-length Aβ1-40 and Aβ1-42 peptides (residues 1–8/1–16 are disordered in solid-state NMR models of Aβ1-40/Aβ1-42 fibrils). They also provide a more general platform for studying dye binding than the simpler systems we studied previously. For example, the single cross-β subunit of the full-length Aβ9-40 has a much larger surface available for potential dye binding, with a total of nine surface grooves formed by the solvent-exposed side chains of several different types of residues (i.e., Y10_V12_H14_K16_V18_F20_E22_V24 for the N-terminal β-sheet layer and I31_M35_V39 for the C-terminal β-sheet layer). This allows a more in-depth study of binding selectivity. By using a single cross-β subunit rather than a higher-order fibril construct, we can monitor all possible binding sites. Indeed, some grooves in multi-unit assemblies (such as the two-layer system seen in agitated fibrils) will be buried inside the subunit-subunit interface.
Our simulations on the Aβ9-40 cross-β-subunit reveal that ThT and PIB bind to multiple aromatic or hydrophobic grooves, loops, and edges with different binding ratios and affinities, in agreement with experimental observations of multiple binding sites and binding affinities (37,38). Our energetic analysis over all modes shows that the stabilizing forces for fibril binding are the van der Waals term (including steric and aromatic interactions) and surface term (hydrophobic interaction), whereas the electrostatic term (gas phase electrostatics + GB electrostatic salvation) is unfavorable (Table 2). This finding not only explains the selective binding of the dyes to aromatic/hydrophobic grooves, it also provides insights into the improved binding that can be obtained by modifying ThT to PIB. In particular, removal of the charge and two methyl groups from ThT improves the steric, aromatic, and hydrophobic interactions of PIB by allowing it to insert more deeply into aromatic/hydrophobic grooves than ThT would be able to.
Our binding simulations of ThT and PIB to the more-toxic Aβ17-42 cross-β-subunit reveal that the overall binding sites and the trend of overall binding energies between ThT and PIB on the Aβ17-42 cross-β-subunit are quite similar to those on the Aβ9-40 cross-β-subunit, likely because the two systems have a similar β-strand-loop-β-strand U-shape. However, we observed an intriguing difference: ThT and PIB inserted into the channel located at the loop of the Aβ17-42 cross-β-subunit, but not into the channel at the loop of the Aβ9-40 cross-β-subunit. In previous simulation studies, small water molecules and ions were observed to pass through the loop channels of both the Aβ17-42 (23) and Aβ9-40 (20,22) cross-β-subunits. Our simulations appear to indicate that the two channels are different in size, and thus they are selectively accessible to large ligands such as ThT and PIB. In other words, the Aβ9-42 loop appears to be wider and more ordered than the Aβ9-40 loop, and offers a larger channel. This mode may be useful for distinguishing Aβ42 fibrils from Aβ40 fibrils.
Our population analysis on the central torsion angle reveals that the conformational preference for ThT and PIB depends on both the chemical modification used and the environment. ThT and PIB have a rugged distribution of the central torsion angle in the two fibril binding modes (i.e., bound to the surface and inserted into the channel) but a smooth distribution in water. Furthermore, our rotation relaxation time analysis shows that the rotation restriction significantly increases when the dyes move from bulk water to the surfaces of the cross-β-subunits, and to the loop channel of the Aβ17-42 cross-β-subunit.
On the basis of our binding information (i.e., binding sites, binding energies, and conformational and rotational restriction data), we suggest that binding to the aromatic or hydrophobic grooves of the fibril is the mode of recognition that leads to ThT fluorescence enhancement. The insertion mode observed for the Aβ17-42 cross-β-subunit may further enhance fluorescence emission. Intriguingly, this mode is reminiscent of the ThT binding mode seen in the amyloid-like β2m crystal. In this case, ThT intercalates between β-sheets orthogonal to the β-strands, boosting the fluorescence emission (7). In the case of PIB, although the chemical modifications remove the red shift excitation and the emission enhancement, the intrinsic fluorescence and the elevated binding affinity to the grooves or the channel appears to make it a better fluorescence dye. Our simulations indicate that the PIB/peptide ratio may be high (perhaps up to ∼1:1). This is in contrast to other amyloid imaging tracers such as FDDNP (40), with a ratio of ∼3–7:10,000, which may indicate that some of the PIB binding modes are different from those of FDDNP. Our simulations provide new (to our knowledge) insight into how to further optimize dyes for routine clinical use. In particular, our simulations suggest that reducing disordered binding to the loops and edges (type II of Fig. 3 and type III of Fig. 4), and decreasing the size of linear ligands would lead to improved fibril recognition ability by enhancing the specific binding to hydrophobic and aromatic grooves (type I of Figs. 3 and 4) as well as the insertion of dyes into the specific loop channel (type II of Fig. 4).
Figure 3.
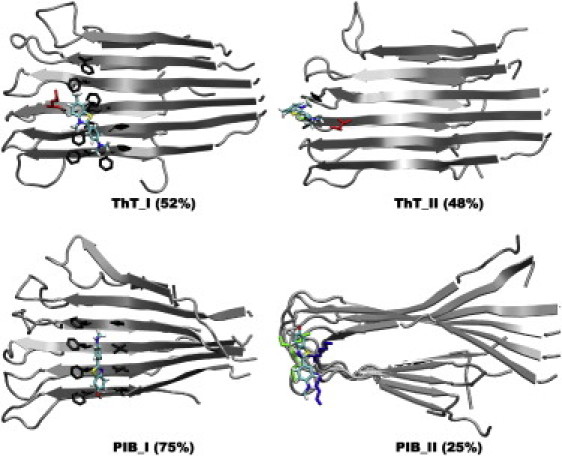
Ordered and disordered binding modes of ThT and PIB to the Aβ9-40 cross-β-subunit. (I) Binding in the grooves (modes A–D in Fig. S18). (II) Binding modes E and F and others. The aggregated abundance is noted in parentheses; for clarity, only the side chains in contact with a ThT/PIB molecule are shown.
Figure 4.

Ordered and disordered binding modes of ThT and PIB to the Aβ17-42 cross-β-subunit. (I) Binding in the grooves (modes A–C in Fig. S21). (II) Insertion into the channel (mode D in Fig. S21). (III) Binding modes E and F and others. The aggregated abundance is noted in parentheses; for clarity, only the side chains in contact with a ThT/PIB molecule are shown.
Acknowledgments
This study was supported by the David and Lucile Packard Foundation, the National Science Foundation (MCB 0642086 to J.E.S.), and the National Institutes of Health (AG027818 to M.T.B.). Computer time was provided by the Lonestar and Ranger clusters in the Texas Advanced Computing Center (LRAC MCA 05S027).
Supporting Material
References
- 1.Jorm A.F., Jolley D. The incidence of dementia: a meta-analysis. Neurology. 1998;51:728–733. doi: 10.1212/wnl.51.3.728. [DOI] [PubMed] [Google Scholar]
- 2.Drzezga A. Amyloid-plaque imaging in early and differential diagnosis of dementia. Ann. Nucl. Med. 2010;24:55–66. doi: 10.1007/s12149-009-0330-9. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 4.Selkoe D.J. Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 5.Dahlgren K.N., Manelli A.M., LaDu M.J. Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J. Biol. Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 6.Furumoto S., Okamura N., Kudo Y. Recent advances in the development of amyloid imaging agents. Curr. Top. Med. Chem. 2007;7:1773–1789. doi: 10.2174/156802607782507402. [DOI] [PubMed] [Google Scholar]
- 7.Wolfe L.S., Calabrese M.F., Xiong Y. Protein-induced photophysical changes to the amyloid indicator dye thioflavin T. Proc. Natl. Acad. Sci. USA. 2010;107:16863–16868. doi: 10.1073/pnas.1002867107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sulatskaya A.I., Maskevich A.A., Turoverov K.K. Fluorescence quantum yield of thioflavin T in rigid isotropic solution and incorporated into the amyloid fibrils. PLoS ONE. 2010;5:e15385. doi: 10.1371/journal.pone.0015385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stsiapura V.I., Maskevich A.A., Kuznetsova I.M. Computational study of thioflavin T torsional relaxation in the excited state. J. Phys. Chem. A. 2007;111:4829–4835. doi: 10.1021/jp070590o. [DOI] [PubMed] [Google Scholar]
- 10.Stsiapura V.I., Maskevich A.A., Buganov O.V. Charge transfer process determines ultrafast excited state deactivation of thioflavin T in low-viscosity solvents. J. Phys. Chem. A. 2010;114:8345–8350. doi: 10.1021/jp105186z. [DOI] [PubMed] [Google Scholar]
- 11.Raji C.A., Becker J.T., Klunk W.E. Characterizing regional correlation, laterality and symmetry of amyloid deposition in mild cognitive impairment and Alzheimer's disease with Pittsburgh compound B. J. Neurosci. Methods. 2008;172:277–282. doi: 10.1016/j.jneumeth.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mathis C.A., Wang Y.M., Klunk W.E. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J. Med. Chem. 2003;46:2740–2754. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- 13.Mathis C., Holt D., Klunk W. Evaluation of a potent thioflavin-T analog for in vivo imaging of amyloid with PET. Neurobiol. Aging. 2004;25:248–249. [Google Scholar]
- 14.Klunk W.E., Wang Y.M., Mathis C.A. Uncharged thioflavin-T derivatives bind to amyloid-β protein with high affinity and readily enter the brain. Life Sci. 2001;69:1471–1484. doi: 10.1016/s0024-3205(01)01232-2. [DOI] [PubMed] [Google Scholar]
- 15.Rosen R.F., Ciliax B.J., Walker L.C. Deficient high-affinity binding of Pittsburgh compound B in a case of Alzheimer's disease. Acta Neuropathol. 2010;119:221–233. doi: 10.1007/s00401-009-0583-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Groenning M. Binding mode of Thioflavin T and other molecular probes in the context of amyloid fibrils-current status. J. Chem. Biol. 2009;3:1–18. doi: 10.1007/s12154-009-0027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petkova A.T., Yau W.M., Tycko R. Experimental constraints on quaternary structure in Alzheimer's β-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lührs T., Ritter C., Riek R. 3D structure of Alzheimer's amyloid-β(1-42) fibrils. Proc. Natl. Acad. Sci. USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma B.Y., Nussinov R. Stabilities and conformations of Alzheimer's β -amyloid peptide oligomers (Aβ 16-22, Aβ 16-35, and Aβ 10-35): sequence effects. Proc. Natl. Acad. Sci. USA. 2002;99:14126–14131. doi: 10.1073/pnas.212206899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buchete N.V., Hummer G. Structure and dynamics of parallel β-sheets, hydrophobic core, and loops in Alzheimer's A β fibrils. Biophys. J. 2007;92:3032–3039. doi: 10.1529/biophysj.106.100404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen B., Thurber K.R., Tycko R. Measurement of amyloid fibril mass-per-length by tilted-beam transmission electron microscopy. Proc. Natl. Acad. Sci. USA. 2009;106:14339–14344. doi: 10.1073/pnas.0907821106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buchete N.V., Tycko R., Hummer G. Molecular dynamics simulations of Alzheimer's β-amyloid protofilaments. J. Mol. Biol. 2005;353:804–821. doi: 10.1016/j.jmb.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 23.Zheng J., Jang H., Nussinov R. Modeling the Alzheimer Aβ 17-42 fibril architecture: tight intermolecular sheet-sheet association and intramolecular hydrated cavities. Biophys. J. 2007;93:3046–3057. doi: 10.1529/biophysj.107.110700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duan Y., Wu C., Kollman P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 25.Hornak V., Abel R., Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparisons of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 27.Wu C., Wang Z.X., Duan Y. Dual binding modes of Congo red to amyloid protofibril surface observed in molecular dynamics simulations. J. Am. Chem. Soc. 2007;129:1225–1232. doi: 10.1021/ja0662772. [DOI] [PubMed] [Google Scholar]
- 28.Bayly C.I., Cieplak P., Kollman P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges—the RESP model. J. Phys. Chem. 1993;97:10269–10280. [Google Scholar]
- 29.Wang J.M., Wolf R.M., Case D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 30.Takeda T., Chang W.L.E., Klimov D.K. Binding of nonsteroidal anti-inflammatory drugs to Aβ fibril. Proteins. 2010;78:2849–2860. doi: 10.1002/prot.22804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu C., Wang Z.X., Shea J.E. The binding of thioflavin T and its neutral analog BTA-1 to protofibrils of the Alzheimer's disease Aβ (16-22) peptide probed by molecular dynamics simulations. J. Mol. Biol. 2008;384:718–729. doi: 10.1016/j.jmb.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu C., Biancalana M., Shea J.E. Binding modes of thioflavin-T to the single-layer β-sheet of the peptide self-assembly mimics. J. Mol. Biol. 2009;394:627–633. doi: 10.1016/j.jmb.2009.09.056. [DOI] [PubMed] [Google Scholar]
- 33.Mathis C.A., Holt D.P., Klunk W.E. A lipophilic C-11-labeled derivative of thioflavin-T for amyloid assessments in Alzheimer's disease. J. Nucl. Med. 2001;42:113P. [Google Scholar]
- 34.Raman E.P., Takeda T., Klimov D.K. Molecular dynamics simulations of ibuprofen binding to Aβ peptides. Biophys. J. 2009;97:2070–2079. doi: 10.1016/j.bpj.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biancalana M., Makabe K., Koide S. Molecular mechanism of thioflavin-T binding to the surface of β-rich peptide self-assemblies. J. Mol. Biol. 2009;385:1052–1063. doi: 10.1016/j.jmb.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dote J., Kivelson D., Schwartz R.N. A molecular quasi-hydrodynamic free-space model for molecular rotational relaxation in liquids. J. Phys. Chem. 1981;85:2169–2180. [Google Scholar]
- 37.Lockhart A., Ye L., Brown J. Evidence for the presence of three distinct binding sites for the thioflavin T class of Alzheimer's disease PET imaging agents on β-amyloid peptide fibrils. J. Biol. Chem. 2005;280:7677–7684. doi: 10.1074/jbc.M412056200. [DOI] [PubMed] [Google Scholar]
- 38.Lockhart A., Lamb J.R., Beach T.G. PIB is a non-specific imaging marker of amyloid-β (Aβ) peptide-related cerebral amyloidosis. Brain. 2007;130:2607–2615. doi: 10.1093/brain/awm191. [DOI] [PubMed] [Google Scholar]
- 39.Krebs M.R.H., Bromley E.H.C., Donald A.M. The binding of thioflavin-T to amyloid fibrils: localisation and implications. J. Struct. Biol. 2005;149:30–37. doi: 10.1016/j.jsb.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 40.Agdeppa E.D., Kepe V., Barrio J.R. Binding characteristics of radiofluorinated 6-dialkylamino-2-naphthylethylidene derivatives as positron emission tomography imaging probes for β-amyloid plaques in Alzheimer's disease. J. Neurosci. 2001;21:RC189. doi: 10.1523/JNEUROSCI.21-24-j0004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.