

Abstract
Photosystem I (PSI) plays a major role in the light reactions of photosynthesis. In higher plants, PSI is composed of a core complex and four outer antennas that are assembled as two dimers, Lhca1/4 and Lhca2/3. Time-resolved fluorescence measurements on the isolated dimers show very similar kinetics. The intermonomer transfer processes are resolved using target analysis. They occur at rates similar to those observed in transfer to the PSI core, suggesting competition between the two transfer pathways. It appears that each dimer is adopting various conformations that correspond to different lifetimes and emission spectra. A special feature of the Lhca complexes is the presence of an absorption band at low energy, originating from an excitonic state of a chlorophyll dimer, mixed with a charge-transfer state. These low-energy bands have high oscillator strengths and they are superradiant in both Lhca1/4 and Lhca2/3. This challenges the view that the low-energy charge-transfer state always functions as a quencher in plant Lhc's and it also challenges previous interpretations of PSI kinetics. The very similar properties of the low-energy states of both dimers indicate that the organization of the involved chlorophylls should also be similar, in disagreement with the available structural data.
Introduction
The driving force of photosynthesis is light, which is harvested by membrane-embedded photosystems (PSs). In oxygen-evolving photosynthesis, two photosystems, PSII and PSI, work in series to drive electrons from water to NADP+. The PSs are pigment-protein supercomplexes, consisting of a core complex and a peripheral light-harvesting system. The core complex of PSI harbors the reaction center, the electron-transport chain, ∼100 chlorophylls a (Chls a) and ∼20 β-carotene pigments (1,2). In higher plants, another ∼70 Chls a and b and ∼15 carotenoids are coordinated by the peripheral light-harvesting complex of PSI (LHCI) (1,3). Their function is to absorb light and transfer the excitation energy to the core complex where it can be used for photochemistry by the reaction center.
LHCI is composed of four Lhca complexes, located at one side of the core and assembled as two heterodimers, Lhca1/4 and Lhca2/3 (1,4,5). Lhca proteins are encoded by nuclear genes belonging to the Lhc multigene family, which also encodes the Lhcb proteins of Photosystem II (PSII) (6). A special feature of the Lhca complexes is the presence of red forms, Chls with extremely red-shifted and broad absorption and fluorescence spectra. It has been found that the red forms of LHCI have absorption bands around 705–712 nm (7,8). This is ∼30 nm red-shifted, and thus ∼3 kT lower in energy, compared to bulk Lhc Chls a.
Red forms are conserved in plants, algae, and bacteria. Still, their function is not fully understood. It has been suggested that they 1), focus the energy to the primary electron donor; 2), have a role in protection against light stress; or 3), absorb light efficiently in a dense vegetation system where light is enriched in wavelengths >690 nm (9).
In higher plants, the red forms are only responsible for a small part of the absorption of PSI, but due to their low energy they have a strong effect on excitation-energy transfer and trapping of the whole PSI complex (10–15). It has been shown that at room temperature (RT), excitations reside 80% of the time on the red forms (11). Thus, to be used for photochemistry (12), these excitations must be transferred energetically uphill, from the red forms to P700.
It has been believed for a long time that LHCI is composed of two fractions: LHCI-680 and LHCI-730, named after their 77 K fluorescence emission maxima, and that the first fraction consisted mainly of monomeric Lhca2 and Lhca3, whereas the second was highly enriched in the Lhca1/4 heterodimer (16–18). Therefore, it was assumed that only the Lhca1/4 dimer possessed red forms, but we have shown recently that Lhca2 and Lhca3 also form a red-emitting heterodimer, and that LHCI-680 is not a native state of LHCI (4).
Due to the dimeric nature and the similar biochemical properties of the native Lhca complexes it has so far been impossible to purify the individual Lhcas. Therefore, it was difficult to acquire information about their properties. This problem was partially solved by the use of in vitro reconstituted Lhca complexes (17,19–21). It was found that the low-temperature (LT) fluorescence emission maxima were located at: 690 nm (Lhca1), 702 nm (Lhca2), 725 nm (Lhca3), and 733 nm (Lhca4) (17,19–21). Furthermore, using in vitro reconstitution, it has been shown that the red forms in the Lhca complexes originate from a strongly excitonically coupled Chl dimer involving Chl603 and Chl609 (nomenclature as in Liu et al. (22)) (23–27). To account for the extremely broad and red-shifted spectra, it was proposed that the lowest exciton state of the dimer mixes with a charge-transfer (CT) state (28); this was recently proven to be indeed the case for the Lhca4 monomer (29). It was suggested by Ihalainen et al. (30) that this CT state is responsible for the low fluorescence quantum yield and emitting dipole strength of Lhca3 and Lhca4, but a recent study has shown that the fluorescence quenching of Lhca4 is not related to the presence of the red forms and, thus, the CT state (31).
Although several studies have analyzed the energy transfer and trapping kinetics in PSI in higher plants, no general agreement has been reached about their kinetics (15,32,33). This is mainly due to the fact that PSI is a large and complex system. To be able to disentangle the contribution of the individual complexes from the analysis of the whole system, information is needed about the excitation energy transfer in and the spectroscopic properties of the PSI building blocks (Lhca1/4, Lhca2/3, and the PSI core).
The steady-state spectroscopic properties of the Lhca1/4 complex have been thoroughly investigated by studying both the reconstituted complex (17,19–21) and the LHCI-730 fraction (16–18). Time-resolved fluorescence studies were also performed. The reconstituted Lhca1/4 complex showed two main decay components of 0.7 and 2.9 ns at RT (30) and of 3.2 and 7 ns at 77 K (34). Time-resolved studies have also been reported for the LHCI-730 fraction (35,36), but the measured fluorescence decay lifetimes were very short, on the subnanosecond timescale. This can be explained by the absence of detergent in the sample, which is known to induce aggregation and a shortening of the lifetimes (37).
The Lhca2/3 dimer could not be obtained either by reconstitution or purification from wild-type PSI. Therefore, only a mixture of Lhca1/4 and Lhca2/3 (henceforth called LHCI) could be studied, giving information averaged over the two dimers (8,13,38). Time-resolved fluorescence studies on this preparation have revealed that the fluorescence decay is multiexponential, with a 2.7- to 3.0-ns lifetime being the major decay component (13,38).
So far, accurate information about the excitation-energy decay pathways of the native Lhca1/4 and Lhca2/3 dimers is lacking. Recently, the use of Lhca-lacking mutant Arabidopsis thaliana plants allowed us to purify both native dimers (4). In this work, we study the dimers by time-resolved fluorescence. We elucidate the intermonomer energy-transfer rates and show that the fluorescence decays multiexponentially for both dimers. The emitting dipole strengths of the dimers are determined, to answer the question whether the CT state is indeed responsible for the low fluorescence quantum yield, as proposed earlier (30).
Materials and Methods
Lhca1/4 and Lhca2/3 isolation
Samples were isolated and characterized, as in Wientjes and Croce (4). In short, PSI from Lhca2- and Lhca1-lacking A. thaliana plants that only contained the Lhca1/4 or Lhca2/3 dimer, respectively (39), were solubilized and fractionated by sucrose density ultracentrifugation. Using this method, Lhca1/4 was obtained without any Lhca2/3 contamination, and the Lhca2/3 dimer was contaminated with only ∼5% Lhca1/4 (4).
Steady-state spectroscopy
Absorption spectra were recorded on a Cary 4000 UV-Vis spectrophotometer (Varian, Palo Alto, CA). For 77 K measurements a homebuilt liquid-N2-cooled low-temperature device was used. Fluorescence spectra were recorded at 77 K and 283 K on a Fluorolog 3.22 spectrofluorimeter (HORIBA Jobin Yvon, Longjumeau, France). Samples were diluted to an OD of 0.04 cm−1 at the Qy maximum. All measurements were performed in 10 mM tricine, pH 7.8, 0.03% α-DM and 0.5 M sucrose (room temperature (RT) and 283 K) or 67% (w/v) glycerol for 77 K measurements.
Streak camera measurements and data analysis
Streak-camera fluorescence measurements were performed with a set of lasers and a synchroscan streak-camera detection system, as described in van Oort et al. (40). In short, vertically polarized excitation pulses (wavelength, 475 nm; duration, 200 fs; power, 0.2 mW) with a repetition rate of 253 kHz were focused to a spot 150 μm in diameter in a static cuvette of 4 × 10 mm at RT. The sample (absorption of 0.2 cm−1 at Qy maximum) was pumped through the cuvette with a flow rate of 4 ml/min. Fluorescence light was collected at 90° with respect to the excitation direction through a 630-nm long-pass filter and a polarizer set at magic-angle orientation, and dispersed as a function of wavelength and time using a spectrograph and a synchroscan streak camera, respectively. Streak images were recorded on a CCD camera. The detection wavelength ranged from 560 to 810 nm. Time windows of 160 ps and 2.1 ns were used. Images were corrected for background signal and sensitivity nonlinearities of the detection system. The experimental data were globally analyzed using the R package TIMP (41), from 650 to 780 nm, with a spectral resolution of 5 nm, and the decay-associated spectra (DAS) were estimated. The instrument response function was modeled as a Gaussian, the location in the time window and the full width at half maximum (FWHM) were free fit parameters. FWHM values of ∼3 and 13 ps were found for the 160-ps and 2.1-ns time windows, respectively.
Target analysis yielded the species associated spectra (SAS) of the individual Lhcas and the excitation energy transfer rates between the Lhcas. To solve the model, it was necessary to constrain the spectra of the Lhca1 and Lhca2 compartments to zero for λ > 760, where they are known to have little emission (19,21). The ratio of forward and backward energy transfer between the monomers was estimated by requiring reasonable relative amplitudes of the SAS. For details on target analysis, see van Stokkum et al. (42).
For the Lhca1/4 data, the 160-ps and 2.1-ns time windows were linked in the analysis. For the Lhca2/3 data there was some difference in the kinetics obtained with the two time windows; therefore, the data sets were analyzed separately. The results obtained on the 2.1-ns data set are shown (Figs. 3 and 4). It should be noted that the transfer component, which is important for solving the intermonomer energy transfer, is nearly the same for the two time windows. The difference is found in the spectrum of the nanosecond component.
TCSPC
Time-correlated single-photon counting (TCSPC) of fluorescence was performed with a homebuilt setup, as described previously (43). The samples were diluted to an OD of 0.1 cm−1 at the Qy maximum, stirred in a 3.5-ml cuvette with a path length of 1 cm, and kept at 283 K. Excitation was performed with a light pulse at 475 nm and a repetition rate of 3.8 MHz. Pulse energies of subpicojoules were used with a pulse duration of 0.2 ps and a spot diameter of ∼1 mm. The instrument response function (∼54 ps FWHM) was obtained with pinacyanol iodide in methanol, with a 6-ps fluorescence lifetime (33). Fluorescence was collected at 90° with respect to the excitation direction through a long-pass (530 nm) and interference filter with FWHM of 10–15 nm and transmission maxima (see Fig. 4, C and D) (Schott, Mainz, Germany, or Balzer B-40, Rolyn Optics, Covina, CA). Individual photons were detected with magic-angle polarization by a microchannel plate photomultiplier, and arrival times were stored in 4096 channels of a multichannel analyzer, with channel time spacing of 5 ps, resulting in a 20-ns time window. Excitation intensity was reduced to obtain count rates of 30,000 s−1, and care was taken to minimize data distortion.
Figure 4.
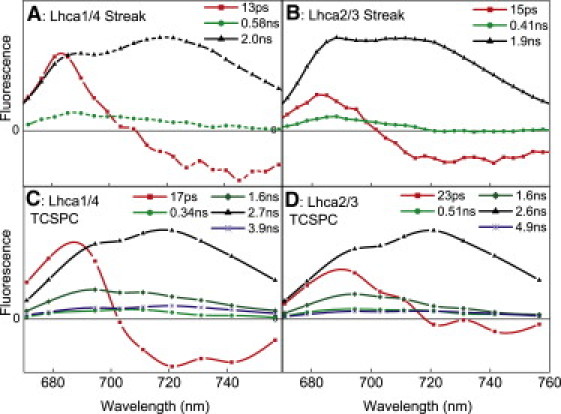
DAS of Lhca1/4 (A and C) and Lhca2/3 (B and D) estimated from streak- (A and B) and TCSPC measurements (C and D). Streak measurements were performed at RT, TCSPC at 283 K, and excitation was at 475 nm.
The experimental data were globally analyzed using homebuilt software (44). The fit quality was evaluated from the χ2, and from plots of the weighted residuals and the autocorrelation thereof. The steady-state fluorescence emission spectra were used to calculate the DAS.
Results
Steady-state absorption and fluorescence
The RT absorption properties of Lhca1/4 and Lhca2/3 are similar (Fig. 1 A), in agreement with the presence of red forms in both dimers and their similar pigment composition (4). The red forms have a large effect on the fluorescence spectra, as apparent from the strong emission at wavelengths >700 nm for both dimers (Fig. 1 B).
Figure 1.
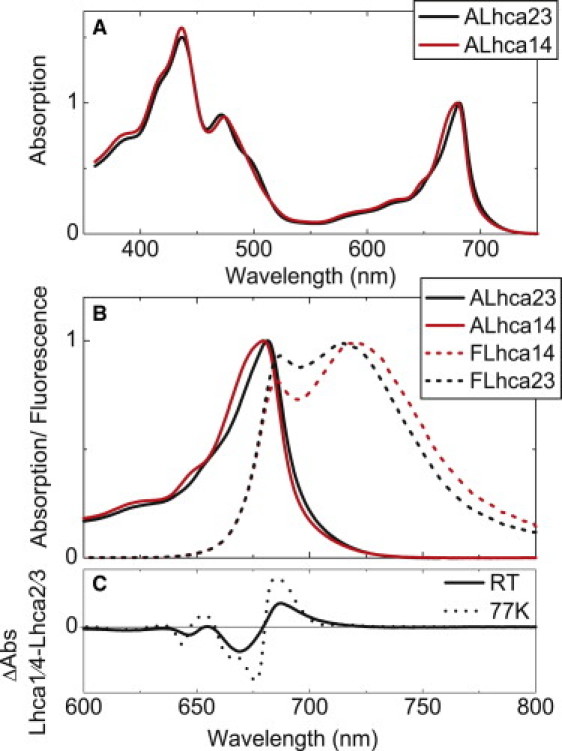
Steady-state absorption and fluorescence of Lhca1/4 and Lhca2/3. (A) RT absorption. (B) RT absorption and 283 K fluorescence. (C) Difference between absorption spectra of Lhca1/4 and Lhca2/3 (normalized to the same area in the Qy region) at RT and 77 K.
Zooming in on the Qy region of the absorption spectra, clear differences can also be appreciated (Fig. 1 B). Lhca1/4 shows stronger absorption at wavelengths <680 nm, whereas Lhca2/3 absorbs more strongly at longer wavelengths (Fig. 1, B and C). However, at wavelengths >705 nm where the red forms are mainly responsible for absorption, the difference is small, especially at 77 K (Fig. 1 C). To get more detailed information about the absorption of the Qy region, which in principle also defines the fluorescence emission, the spectra were described in terms of Gaussians. At 77 K, the redmost bands show maxima at 706–707 nm and a FWHM of 25 nm, and they represent 8.5% and 8.9% of the total oscillator strength in the Qy region (630–750 nm) for Lhca1/4 and Lhca2/3, respectively (Fig. 2). A second red band with maxima at 689 nm for Lhca2/3 and 684 nm for Lhca1/4 is needed to describe the spectra. Although the uncertainty is large due to the lack of structure in this spectral region, it is clear that the oscillator strength around 690 nm is far stronger for Lhca2/3 than for Lhca1/4. This additional absorption can be assigned to Lhca2, which was shown to have a band around 690 nm (23), whereas Lhca1 has almost no absorption at this wavelength (25). The stronger absorption around 690 nm is reflected in an increased fluorescence emission at this wavelength for the Lhca2/3 dimer, as compared to the Lhca1/4 dimer (Fig. 1 B), and this can thus be assigned to a relatively stronger excitation population of Lhca2 compared to Lhca1.
Figure 2.
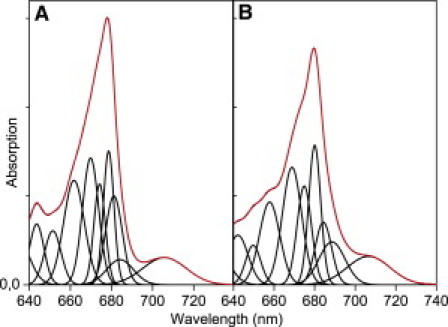
The Qy region of the LT absorption spectra (red) of Lhca1/4 (A) and Lhca2/3 (B) described by Gaussian bands (black), see online version for color. Note that the fitting is meant to extract the characteristics of the red bands, using the restrictions as explained in the text.
Time-resolved fluorescence
To get a better insight into the excitation energy transfer dynamics of the two dimers, their fluorescence dynamics was studied by streak-camera and TCSPC measurements. Due to its narrow instrument response function (FWHM 3 ps), the streak camera has a high time resolution, whereas one measurement provides information over a broad spectral range of 570–830 nm. However, due to the short time window (2.1 ns for the longest time range), the method is not very accurate for discriminating between fluorescence lifetimes on the nanosecond timescale. To better cover the entire decay process, we also performed TCSPC measurements with a 20-ns time window.
Lhca1/4 and Lhca2/3 show similar fluorescence kinetics
Fig. 3 A shows examples of the spectral evolution from the Lhca1/4 and Lhca2/3 dimers measured with the streak camera at RT. On the 160-ps timescale (Fig. 3 A, left), a rapid decrease of fluorescence can be observed at ∼690 nm, whereas there is a concomitant increase of fluorescence at longer wavelengths, thus reflecting energy transfer. On the 2.1-ns timescale (Fig. 3 A, right), the overall fluorescence decay can be observed. To solve in detail the fluorescence kinetics on the picosecond-to-nanosecond scale, global analysis was performed (see Materials and Methods), providing the DAS. The DAS (Fig. 4, A and B) of both dimers are very similar. The spectra associated with a lifetime of 13–15 ps have a positive amplitude peaking around 685 nm and a negative amplitude in the red, thus representing energy equilibration between bulk and low-energy Chls. It has been shown that in monomeric (m)Lhca4, bulk-red equilibration occurs in <5 ps (45). This indicates that the 13–15 ps component observed in the dimers mainly represents excited-state energy equilibration between Lhca1 and Lhca2, which do not have low-energy Chls, and Lhca4 and Lhca3, respectively. The fluorescence decay is largely described with a 2-ns lifetime, but a second subnanosecond component is also resolved.
Figure 3.
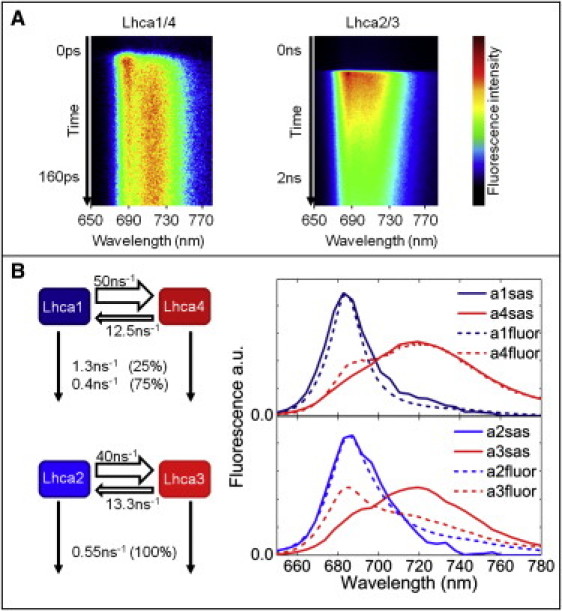
(A) Example of streak images for the 160-ps and 2.1-ns time windows of Lhca1/4 and Lhca2/3, respectively, recorded at RT. (B) Excitation energy transfer and decay model, with rates (left) and SAS (right) obtained by target analysis. Fluorescence spectra of reconstituted Lhcas are also reported. Spectra of Lhca1–Lhca3 were recorded at RT, and spectra of Lhca4 were recorded at 283 K, which can slightly enhance the red emission due to a higher population of the red forms at lower temperature. Initial excitations at 475 nm were estimated, for the Lhca1/4 dimer Lhca1 (56%) and Lhca4 (44%), and in Lhca2/3 Lhca2 (60%) and Lhca3 (40%). These values are in good agreement with the expected initial excitations based on the absorption and excitation spectra of the reconstituted Lhcas.
Resolving the intermonomer energy transfer
The DAS are a mathematical description of the fluorescence decay, and they do not necessarily represent a physical part (for instance a pigment pool) of the sample. To extract more information about the physical origin of the observed fluorescence kinetics, target analysis was performed. Using this analysis, the data is fitted to a compartmental model where the compartments represent the different physical parts of the system (see van Stokkum et al. (42) for a detailed description). Our model (Fig. 3 B, left) aims to resolve the excitation energy transfer rates between the individual Lhca complexes of each dimer. Solving such a model also gives the emission spectra of the different compartments (the SAS). It should be noted that the aim of the model is to solve the intermonomer transfer rates. For details on the constraints used to solve the model, see Materials and Methods.
Fig. 3 B shows the result of the fit. In this case, the SAS are the emission spectra of Lhca1–4, which should thus be similar to the steady-state spectra of the reconstituted Lhcas. This is indeed the case for Lhca1, Lhca2, and Lhca4 (Fig. 3 B, right), only for Lhca3, the spectrum of the reconstituted complex shows less ∼720 nm emission than the SAS. This is probably related to the lower stability of reconstituted Lhca3 as compared to the other Lhcas, as is apparent, for instance, from its lower denaturation temperature (24).
In the Lhca1/4 dimer, energy transfer from Lhca1 to Lhca4 occurs at a rate of 50 ns−1, whereas the backward transfer is four times slower (Fig. 3 B). In the Lhca2/3 dimer, the rate of transfer from Lhca2 to Lhca3 is 40 ns−1, whereas the backward rate is three times slower (Fig. 3 B). Thus, after equilibration, only 25% of the emission arises from Lhca1 in Lhca1/4, whereas this is 33% for Lhca2 in the Lhca2/3 dimer. This is in agreement with the analysis of the steady-state spectra, which show stronger Lhca2 fluorescence emission compared to Lhca1 emission (Fig. 1). The high excitation probability of Lhca3 and Lhca4 clearly shows the large effect of the low-energy Chls present in these complexes on the energy-transfer kinetics.
Multiexponential decay
The fluorescence kinetics of Lhca1/4 and Lhca2/3 measured with the streak camera shows a biexponential decay with time constants of ∼0.5 and 2 ns. This is puzzling, because from an equilibrated system, the fluorescence decay is expected in principle to be monoexponential. To study this in more detail, TCSPC measurements were performed, which allow more accurate estimation of longer lifetimes. Fluorescence decay traces were recorded at nine emission wavelengths and were globally analyzed. The calculated DAS are presented in Fig. 4, C and D. The streak and TCSCP measurements gave qualitatively similar results, but the TCSPC allowed resolution of five components—one transfer component and four decay components—in the (sub)nanosecond range, thus confirming the multiexponential fluorescence decay. The main component has a lifetime of 2.6–2.7 ns for both dimers, in agreement with previous studies on LHCI (13,38). The DAS obtained by TCSPC allow for an accurate calculation of the average fluorescence lifetimes (Table 1), which are 2.45 ns for both dimers. This value is far lower than those of Chl in solution (5.9 ns) and LHCII (3.9 ns) (Table 2), supporting the idea of partially quenched states of the dimers.
Table 1.
Relative area of the DAS obtained by TCSPC
| Lhca2/3 | Lhca1/4 | ||
|---|---|---|---|
| 0.5 ns | 8% | 0.3 ns | 6% |
| 1.6 ns | 16% | 1.6 ns | 20% |
| 2.6 ns | 70% | 2.7 ns | 65% |
| 4.9 ns | 7% | 3.9 ns | 10% |
| <τ> | 2.45 ns | 2.45 ns | |
Relative areas (A) of DAS (Fig. 4, C and D) are presented, as well as the averaged lifetimes, given by <τ> = ΣAiτi.
Table 2.
Fluorescence quantum yield, average lifetime, and calculated average emitting dipole strength
| Sample | ΦF | <τ> (ns) | <ν3>∗(cm−3) | Radiative rate(ns−1)† | Refractive index | Emitting dipole strength (relative to Chl a‡) |
|---|---|---|---|---|---|---|
| Chl a | 0.30§ | 5.9§ | 3.19e12 | 0.051 | 1.36 | 1.00 |
| Lhca1/4 | 0.14¶ | 2.5‖ | 2.73e12 | 0.057 | 1.33 | 1.37 |
| Lhca2/3 | 0.15¶ | 2.5‖ | 2.76e12 | 0.061 | 1.33 | 1.46 |
| LHCII | 0.21¶ | 3.9‖ | 3.05e12 | 0.054 | 1.33 | 1.16 |
The average value <ν3> is obtained as described in Palacios and de Weerd (47).
The radiative rate is calculated according to krad = ΦF/τfl.
The emitting dipole moment is calculated according to krad = (16nπ3ν3/3ɛ0hc3)|μ|2, the emitting dipole moment of Chl a is corrected for the somewhat different value of the refractive index (1.36 vs. 1.33) according to the empirical relation D = 20.2 + 23.6(n − 1)(D2) (64).
Values taken from Weber and Teale (65).
Fluorescence quantum yields were obtained by the method reported in Wientjes and Croce (4).
Fluorescence lifetimes were obtained by TCSPC. For further details, see Palacios et al. (47).
Are the red forms superradiant?
Analysis of the LT absorption spectrum of the dimeric Lhca complexes indicates that the red-most bands account for 8.5% and 8.9% of the LT absorption in Lhca1/4 and Lhca2/3, respectively. Assuming a number of 24 Chls per dimer (4), the absorption of the red forms corresponds to a dipole strength of ∼2 Chls per dimer. It has been shown that the red forms represent the lowest-energy state of a dimer of two excitonically coupled Chls a (603–609) in both Lhca3 and Lhca4 (24,27). Taken together, this indicates that most of the oscillator strength of the exciton state of the coupled Chl dimer corresponds to the low-energy band. Because the low-energy state is heavily populated, it can be expected that the Lhca dimers have relatively large emitting dipole strengths and are thus superradiant. In agreement with this, we found that the area under the SAS (on a wavenumber scale), which correlates with the emitting dipole strength, is 1.7 times higher for Lhca4 than for Lhca1. However, it has also been proposed that the CT state quenches the fluorescence in LHCII aggregates (46) and Lhcas (30). To investigate this further, the emitting dipole strengths were calculated according to Eqs. 1 and 2 (Table 2):
| (1) |
| (2) |
where krad is the radiative rate (in s−1), ΦF is the fluorescence quantum yield (taken from Wientjes and Croce (4)), <τF> is the average fluorescence lifetime, |μ|2 is the emitting dipole strength (C2 m2), ɛ0 is the vacuum dielectric constant (C2/Jm), h is Planck's constant (J), c is the speed of light in vacuum (m/s), ν is the emission frequency (s−1), and n is the refractive index. Equation 2 was taken from Palacios et al. (47).
The emitting dipole strengths of both dimers are higher than that of LHCII and 1.4–1.5 times that of Chl a in solution, meaning that both dimers are indeed superradiant. One should realize that this value of 1.5 is (even) a lower limit for the red band because of the contribution of nondimeric Chl bands to the fluorescence. It was suggested that in Lhca3 and Lhca4, mixing of the excitonic state with a dark charge-transfer state reduces the emitting dipole moment to a value of <1.0 (30). It is clear that the data presented here are in disagreement with that finding.
Discussion
The domain harboring the Chl responsible for the red forms is conserved in the two dimers
A remarkable feature of the antenna complexes of PSI is the presence of Chls that absorb above 700 nm and are associated with Lhca3 and Lhca4 (7). The red forms represent the lowest-energy state of a dimer of two excitonically coupled Chls a (24,27). In an excitonically coupled dimer, the sum of the electronic oscillator strengths is identical to the sum of the oscillator strengths of the isolated molecules, but the oscillator strengths can be redistributed over the two transitions. The redistribution depends on the energy levels of the involved Chls, the distance between them, and the geometric arrangement of their transition dipoles (48).
Analysis of the absorption spectra of Lhca1/4 and Lhca2/3 shows that the oscillator strength of the lowest-energy band corresponds to ∼2 Chls, indicating that most of the oscillator strength belongs to this low-energy band of the excitonically coupled Chl a dimer. This means that the transition dipole moments of the two interacting Chls must be more or less parallel and in line (48). At the moment there are no Lhca structures available where the orientation of the transition dipole moments are assigned; thus, a comparison is not possible. However, the relative geometric organization of the transition dipole moments is expected to be similar to those in LH1 and the special pair of purple bacteria (thus parallel and in line) (49,50), where indeed almost all the oscillator strength is on the low-energy bands of excitonically coupled bacteriochlorophyll dimers (51,52).
The red forms have nearly identical spectroscopic properties in both dimers (Fig. 1 C) (4), suggesting that their environment and organization must be very similar. This is at variance with the current structural data of PSI-LHCI (1,53), which show a very different pigment organization (both orientation and distance) of the domains responsible for the red forms in the two complexes, thus indicating that there is room for improvement in the LHCI structure.
Multiexponential fluorescence decay of the Lhca dimers
The fluorescence decay of a homogeneous preparation of a light-harvesting antenna, where the excitation equilibration has been reached, is expected to be monoexponential. However, in the case of the dimeric Lhca complexes, four rates are needed to get a satisfactory description of the TCSPC decay traces. Heterogeneity in the fluorescence decay of Lhca dimers has been observed previously (13,30,35,38,54), and several explanations have been provided: 1), the presence of three different dimeric Lhca complexes in the sample (38); 2), the presence of four Lhca complexes in the native LHCI preparation (13); 3), slow excitation equilibration between the bulk and low-energy pigments (55); and 4), a structural rearrangement in the excited state (30). The first two explanations can be discarded, because we analyzed one dimer at a time and still observed mutiexponential decay kinetics. The third option would require slow downhill and uphill energy transfer, i.e., from the bulk pigments to the low-energy red forms and vice versa. The authors suggest (55) that the uphill energy transfer is slow due to the large energy difference. However, according to the detailed balance relationship, the downhill energy transfer should still be very fast, which would mean that the equilibration rate is even faster. Therefore, slow uphill energy transfer cannot explain the multiple slow decay components. Ihalainen et al. (30) propose a structural rearrangement in the excited state of the Lhca complexes, to account for the 45- to 190-ps DAS, which deviates from a typical Chl a emission spectrum in the sense that it lacks the vibronic wing. However, in our TCSPC experiments, such a deformed spectrum could not be observed. Thus, none of the proposed explanations can account for our observations.
Lhca dimers adopt different conformations
The occurrence of a multiexponential fluorescence decay also has been found for the antenna of PSII, especially for the minor Lhcb complexes (56) and for monomeric LHCII (57), and it has been proposed that such a decay arises from the presence of various distinct conformations of the pigment-protein complex (56). The fluorescence lifetime can be affected by pigment-pigment and pigment-protein interactions (58,59). Because these interactions are strongly dependent on the distances between the chromophores and molecular groups in their direct environment, small conformational variations can have a substantial effect on the fluorescence decay (see, e.g., van Oort et al. (60)). In addition, in Lhca complexes, small conformational differences can have a strong effect on the emission spectra, because the red forms, which are the low-energy states of the system, originate from interactions (excitonic and charge-transfer) that are strongly distance- and orientation-dependent. The spectra associated with the two main decay components of both dimers differ substantially (Fig. 5), with the 2.6–2.7 ns component emitting more to the red than the 1.6-ns (and the 0.34–0.51 ns) component. Similar observations were made for reconstituted Lhca4 (31). This can be explained by different conformations having different emission spectra and lifetimes. It should be noted that a protein is not a static scaffold, but a rather dynamic system. For example, spontaneous switching between conformations with different emission spectra has been observed for the homologous major light-harvesting antenna of PSII in a single-molecule study (61). It can be concluded that the possibility of assuming different conformations and existing in different quenching states is a property of all Lhc family members. It is interesting to note that in the case of the Lhcas, the different quenching states have different emission spectra. The redder spectra observed for the longer lifetimes indicate that the quenched conformation and the red conformation are mutually exclusive, as is also the case for reconstituted Lhca4 (31).
Figure 5.
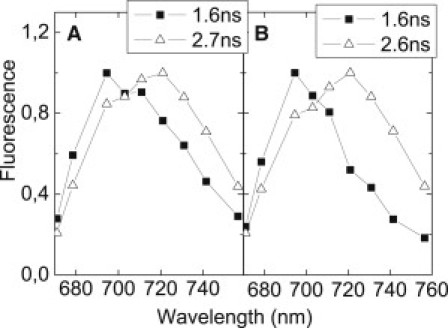
Spectra estimated from TCSPC data of Lhca1/4 (A) and Lhca2/3 (B) associated with lifetimes of 1.6 ns and 2.6–2.7 ns, respectively. Spectra are normalized to each other in their maxima.
The presence of various emitting states can also explain why the RT fluorescence emission spectra of LHCI are different upon excitation of the bulk pigments compared to excitation of the red band (720 nm) (62). In the first case, both red and nonred conformations are excited, whereas at 720 nm, only the red conformations are excited, thus resulting in a relatively stronger red emission.
Energy transfer in PSI
The study of excitation energy transfer and trapping in PSI is extremely complex, because the system is composed of ∼170 Chl molecules, making the modeling of the fluorescence kinetics very challenging. The system is usually described using compartmental models in which the major building blocks (Lhca complexes, core complex, and reaction center) are considered. Knowledge about the spectroscopic properties of the compartments is required for evaluation of these models (32). Until now, this information has not been available for the native Lhca dimers. Modeling of time-resolved fluorescence data of PSI complexes led to the assignment of two spectra with very different fluorescence quantum yields and emission maxima to the red states of Lhca3 and Lhca4 (32). However, our experimental work shows that the fluorescence quantum yield of both dimers (4) and the emission maxima of Lhca3 and Lhca4 are very similar, thus suggesting that the model of Slavov et al. (32) can be improved by taking these new constraints into account.
The complete excitation energy transfer of PSI can be described by transfer between Lhcas, transfer between Lhcas and the core, and trapping of excitation energy by the reaction center. In this study, we were able to resolve the intermonomer energy transfer rates for both dimers. Transfer from Lhca1 and Lhca2 to Lhca4 and Lhca3, respectively, occurs at a rate of 40–50/ns. The backward transfer is three to four times slower, resulting in an equilibration time of 16–18 ps. From time-resolved fluorescence measurements on PSI-LHCI, it was concluded that the average excited-state lifetime is 10 ± 5 ps longer when LHCI is excited than when the PSI core is excited (33). This difference was mainly ascribed to the extra time needed to reach the reaction center when LHCI instead of the core is excited. This implies that the time of transfer from LHCI to core is in the same order of magnitude as the equilibration time within the Lhca1/4 and Lhca2/3 dimer, meaning that Lhca1 and Lhca2 transfer a considerable part of their absorbed energy directly to the core and not to their dimeric partners.
Based on the highly similar absorption spectra and fluorescence kinetics, it can be concluded that the basic light-harvesting properties are practically identical for both dimers, and thus the difference in energy transfer to the PSI core, if any, would be mainly determined by the connections between the Lhca dimers and the core complex.
The information of the isolated dimers will thus be helpful to finally understand the energy-transfer pathways in the complete PSI complex, which with a quantum yield of ∼100% is to our knowledge the most efficiently operating photoelectric nanomachine known to date (63).
Acknowledgments
The authors thank Arie van Hoek and Rob Koehorst for technical support with the time-resolved fluorescence measurements, Sergey Laptenok for help with the use of TIMP for the streak-data analysis, and Stefan Jansson for kindly providing the seeds used in this work.
This work was supported by De Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO), Earth and Life Science (ALW), through a Vidi grant (to R.C.).
References
- 1.Amunts A., Drory O., Nelson N. The structure of a plant photosystem I supercomplex at 3.4 Å resolution. Nature. 2007;447:58–63. doi: 10.1038/nature05687. [DOI] [PubMed] [Google Scholar]
- 2.Jordan P., Fromme P., Krauss N. Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution. Nature. 2001;411:909–917. doi: 10.1038/35082000. [DOI] [PubMed] [Google Scholar]
- 3.Croce R., Morosinotto T., Bassi R. Springer; Dordrecht, The Netherlands: 2006. Photosystem I: The Light-Driven Plastocyanin: Ferredoxin Oxidoreductase. [Google Scholar]
- 4.Wientjes E., Croce R. The light-harvesting complexes of higher plant Photosystem I: Lhca1/4 and Lhca2/3 form two red-emitting heterodimers. Biochem. J. 2010;433:447–485. doi: 10.1042/BJ20101538. [DOI] [PubMed] [Google Scholar]
- 5.Boekema E.J., Jensen P.E., Dekker J.P. Green plant photosystem I binds light-harvesting complex I on one side of the complex. Biochemistry. 2001;40:1029–1036. doi: 10.1021/bi0015358. [DOI] [PubMed] [Google Scholar]
- 6.Jansson S. A guide to the Lhc genes and their relatives in Arabidopsis/IT>. Trends Plant Sci. 1999;4:236–240. doi: 10.1016/s1360-1385(99)01419-3. [DOI] [PubMed] [Google Scholar]
- 7.Croce R., Chojnicka A., van Grondelle R. The low-energy forms of photosystem I light-harvesting complexes: spectroscopic properties and pigment-pigment interaction characteristics. Biophys. J. 2007;93:2418–2428. doi: 10.1529/biophysj.107.106955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ihalainen J.A., Gobets B., Dekker J.P. Evidence for two spectroscopically different dimers of light-harvesting complex I from green plants. Biochemistry. 2000;39:8625–8631. doi: 10.1021/bi0007369. [DOI] [PubMed] [Google Scholar]
- 9.Rivadossi A., Zucchelli G., Jennings R.C. The importance of PSI chlorophyll red forms in light-harvesting by leaves. Photosynth. Res. 1999;60:209–215. [Google Scholar]
- 10.Croce R., Dorra D., Jennings R.C. Fluorescence decay and spectral evolution in intact photosystem I of higher plants. Biochemistry. 2000;39:6341–6348. doi: 10.1021/bi992659r. [DOI] [PubMed] [Google Scholar]
- 11.Croce R., Zucchelli G., Jennings R.C. Excited state equilibration in the photosystem I-light-harvesting I complex: P700 is almost isoenergetic with its antenna. Biochemistry. 1996;35:8572–8579. doi: 10.1021/bi960214m. [DOI] [PubMed] [Google Scholar]
- 12.Jennings R.C., Zucchelli G., Garlaschi F.M. The photochemical trapping rate from red spectral states in PSI-LHCI is determined by thermal activation of energy transfer to bulk chlorophylls. Biochim. Biophys. Acta. 2003;1557:91–98. doi: 10.1016/s0005-2728(02)00399-7. [DOI] [PubMed] [Google Scholar]
- 13.Engelmann E., Zucchelli G., Jennings R.C. Influence of the photosystem I-light harvesting complex I antenna domains on fluorescence decay. Biochemistry. 2006;45:6947–6955. doi: 10.1021/bi060243p. [DOI] [PubMed] [Google Scholar]
- 14.Gobets B., van Stokkum I.H.M., van Grondelle R. Time-resolved fluorescence emission measurements of photosystem I particles of various cyanobacteria: a unified compartmental model. Biophys. J. 2001;81:407–424. doi: 10.1016/S0006-3495(01)75709-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ihalainen J.A., van Stokkum I.H.M., Dekker J.P. Kinetics of excitation trapping in intact Photosystem I of Chlamydomonas reinhardtii and Arabidopsis thaliana. Biochim. Biophys. Acta. 2005;1706:267–275. doi: 10.1016/j.bbabio.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 16.Knoetzel J., Svendsen I., Simpson D.J. Identification of the photosystem I antenna polypeptides in barley. Isolation of three pigment-binding antenna complexes. Eur. J. Biochem. 1992;206:209–215. doi: 10.1111/j.1432-1033.1992.tb16918.x. [DOI] [PubMed] [Google Scholar]
- 17.Schmid V.H.R., Potthast S., Storf S. Pigment binding of photosystem I light-harvesting proteins. J. Biol. Chem. 2002;277:37307–37314. doi: 10.1074/jbc.M205889200. [DOI] [PubMed] [Google Scholar]
- 18.Tjus S.E., Roobolboza M., Andersson B. Rapid isolation of photosystem-I chlorophyll-binding proteins by anion-exchange perfusion chromatography. Photosynth. Res. 1995;45:41–49. doi: 10.1007/BF00032234. [DOI] [PubMed] [Google Scholar]
- 19.Castelletti S., Morosinotto T., Croce R. Recombinant Lhca2 and Lhca3 subunits of the photosystem I antenna system. Biochemistry. 2003;42:4226–4234. doi: 10.1021/bi027398r. [DOI] [PubMed] [Google Scholar]
- 20.Croce R., Morosinotto T., Bassi R. The Lhca antenna complexes of higher plants photosystem I. Biochim. Biophys. Acta. 2002;1556:29–40. doi: 10.1016/s0005-2728(02)00304-3. [DOI] [PubMed] [Google Scholar]
- 21.Schmid V.H.R., Cammarata K.V., Schmidt G.W. In vitro reconstitution of the photosystem I light-harvesting complex LHCI-730: heterodimerization is required for antenna pigment organization. Proc. Natl. Acad. Sci. USA. 1997;94:7667–7672. doi: 10.1073/pnas.94.14.7667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Z., Yan H., Chang W. Crystal structure of spinach major light-harvesting complex at 2.72 Å resolution. Nature. 2004;428:287–292. doi: 10.1038/nature02373. [DOI] [PubMed] [Google Scholar]
- 23.Croce R., Morosinotto T., Bassi R. Origin of the 701-nm fluorescence emission of the Lhca2 subunit of higher plant photosystem I. J. Biol. Chem. 2004;279:48543–48549. doi: 10.1074/jbc.M408908200. [DOI] [PubMed] [Google Scholar]
- 24.Morosinotto T., Breton J., Croce R. The nature of a chlorophyll ligand in Lhca proteins determines the far red fluorescence emission typical of photosystem I. J. Biol. Chem. 2003;278:49223–49229. doi: 10.1074/jbc.M309203200. [DOI] [PubMed] [Google Scholar]
- 25.Morosinotto T., Castelletti S., Croce R. Mutation analysis of Lhca1 antenna complex. Low energy absorption forms originate from pigment-pigment interactions. J. Biol. Chem. 2002;277:36253–36261. doi: 10.1074/jbc.M205062200. [DOI] [PubMed] [Google Scholar]
- 26.Morosinotto T., Mozzo M., Croce R. Pigment-pigment interactions in Lhca4 antenna complex of higher plants photosystem I. J. Biol. Chem. 2005;280:20612–20619. doi: 10.1074/jbc.M500705200. [DOI] [PubMed] [Google Scholar]
- 27.Mozzo M., Morosinotto T., Croce R. Probing the structure of Lhca3 by mutation analysis. Biochim. Biophys. Acta. 2006;1757:1607–1613. doi: 10.1016/j.bbabio.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 28.Ihalainen J.A., Ratsep M., Freiberg A. Red spectral forms of chlorophylls in green plant PSI - a site-selective and high-pressure spectroscopy study. J. Phys. Chem. B. 2003;107:9086–9093. [Google Scholar]
- 29.Romero E., Mozzo M., Croce R. The origin of the low-energy form of photosystem I light-harvesting complex Lhca4: mixing of the lowest exciton with a charge-transfer state. Biophys. J. 2009;96:L35–L37. doi: 10.1016/j.bpj.2008.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ihalainen J.A., Croce R., van Grondelle R. Excitation decay pathways of Lhca proteins: a time-resolved fluorescence study. J. Phys. Chem. B. 2005;109:21150–21158. doi: 10.1021/jp0519316. [DOI] [PubMed] [Google Scholar]
- 31.Passarini F., Wientjes E., Croce R. Photosystem I light-harvesting complex Lhca4 adopts multiple conformations: red forms and excited-state quenching are mutually exclusive. Biochim. Biophys. Acta. 2010;1797:501–508. doi: 10.1016/j.bbabio.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 32.Slavov C., Ballottari M., Holzwarth A.R. Trap-limited charge separation kinetics in higher plant photosystem I complexes. Biophys. J. 2008;94:3601–3612. doi: 10.1529/biophysj.107.117101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Oort B., Amunts A., Croce R. Picosecond fluorescence of intact and dissolved PSI-LHCI crystals. Biophys. J. 2008;95:5851–5861. doi: 10.1529/biophysj.108.140467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melkozernov A.N., Lin S., Blankenship R.E. Ultrafast excitation dynamics of low energy pigments in reconstituted peripheral light-harvesting complexes of photosystem I. FEBS Lett. 2000;471:89–92. doi: 10.1016/s0014-5793(00)01370-3. [DOI] [PubMed] [Google Scholar]
- 35.Melkozernov A.N., Schmid V.H.R., Blankenship R.E. Energy redistribution in heterodimeric light-harvesting complex LHCI-730 of photosystem I. J. Phys. Chem. B. 1998;102:8183–8189. [Google Scholar]
- 36.Palsson L.O., Tjus S.E., Gillbro T. Ultrafast energy-transfer dynamics resolved in isolated spinach light-harvesting complex-I and the Lhc-I-730 subpopulation. Biochim. Biophys. Acta. 1995;1230:1–9. [Google Scholar]
- 37.Horton P., Ruban A.V., Walters R.G. Regulation of light harvesting in green plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996;47:655–684. doi: 10.1146/annurev.arplant.47.1.655. [DOI] [PubMed] [Google Scholar]
- 38.Gobets B., Kennis J.T.M., van Grondelle R. Excitation energy transfer in dimeric light harvesting complex I: a combined streak-camera/fluorescence upconversion study. J. Phys. Chem. B. 2001;105:10132–10139. [Google Scholar]
- 39.Wientjes E., Oostergetel G.T., Croce R. The role of Lhca complexes in the supramolecular organization of higher plant photosystem I. J. Biol. Chem. 2009;284:7803–7810. doi: 10.1074/jbc.M808395200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Oort B., Murali S., van Amerongen H. Ultrafast resonance energy transfer from a site-specifically attached fluorescent chromophore reveals the folding of the N-terminal domain of CP29. Chem. Phys. 2009;357:113–119. [Google Scholar]
- 41.Mullen K.M., van Stokkum I.H.M. TIMP: an R package for modeling multi-way spectroscopic measurements. J. Stat. Softw. 2007;18:1–46. [Google Scholar]
- 42.van Stokkum I.H., Larsen D.S., van Grondelle R. Global and target analysis of time-resolved spectra. Biochim. Biophys. Acta. 2004;1657:82–104. doi: 10.1016/j.bbabio.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 43.Somsen O.J., Keukens L.B., van Amerongen H. Structural heterogeneity in DNA: temperature dependence of 2-aminopurine fluorescence in dinucleotides. ChemPhysChem. 2005;6:1622–1627. doi: 10.1002/cphc.200400648. [DOI] [PubMed] [Google Scholar]
- 44.Digris A.V., Skakoun V.V., Visser A.J. Thermal stability of a flavoprotein assessed from associative analysis of polarized time-resolved fluorescence spectroscopy. Eur. Biophys. J. 1999;28:526–531. doi: 10.1007/s002490050235. [DOI] [PubMed] [Google Scholar]
- 45.Gibasiewicz K., Croce R., van Grondelle R. Excitation energy transfer pathways in Lhca4. Biophys. J. 2005;88:1959–1969. doi: 10.1529/biophysj.104.049916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miloslavina Y., Wehner A., Holzwarth A.R. Far-red fluorescence: a direct spectroscopic marker for LHCII oligomer formation in non-photochemical quenching. FEBS Lett. 2008;582:3625–3631. doi: 10.1016/j.febslet.2008.09.044. [DOI] [PubMed] [Google Scholar]
- 47.Palacios M.A., de Weerd F.L., van Amerongen H. Superradiance and exciton (de)localization in light-harvesting complex II from green plants? J. Phys. Chem. B. 2002;106:5782–5787. [Google Scholar]
- 48.van Amerongen H., Valkunas L., van Grondelle R. World Scientific; Singapore: 2000. Photosynthetic Excitons. [Google Scholar]
- 49.Koepke J., Krammer E.M., Fritzsch G. pH modulates the quinone position in the photosynthetic reaction center from Rhodobacter sphaeroides in the neutral and charge separated states. J. Mol. Biol. 2007;371:396–409. doi: 10.1016/j.jmb.2007.04.082. [DOI] [PubMed] [Google Scholar]
- 50.Roszak A.W., Howard T.D., Cogdell R.J. Crystal structure of the RC-LH1 core complex from Rhodopseudomonas palustris. Science. 2003;302:1969–1972. doi: 10.1126/science.1088892. [DOI] [PubMed] [Google Scholar]
- 51.Knapp E.W., Fischer S.F., Michel H. Analysis of optical spectra from single crystals of Rhodopseudomonas viridis reaction centers. Proc. Natl. Acad. Sci. USA. 1985;82:8463–8467. doi: 10.1073/pnas.82.24.8463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Visschers R.W., Chang M.C., van Grondelle R. Fluorescence polarization and low-temperature absorption spectroscopy of a subunit form of light-harvesting complex I from purple photosynthetic bacteria. Biochemistry. 1991;30:5734–5742. doi: 10.1021/bi00237a015. [DOI] [PubMed] [Google Scholar]
- 53.Amunts A., Toporik H., Nelson N. Structure determination and improved model of plant photosystem I. J. Biol. Chem. 2010;285:3478–3486. doi: 10.1074/jbc.M109.072645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mukerji I., Sauer K. Energy-transfer dynamics of an isolated light-harvesting complex of Photosystem I from spinach: time-resolved fluorescence measurements at 295 K and 77 K. Biochim. Biophys. Acta. 1993;1142:311–320. [Google Scholar]
- 55.Jennings R.C., Zucchelli G., Garlaschi F.M. The long-wavelength chlorophyll states of plant LHCI at room temperature: a comparison with PSI-LHCI. Biophys. J. 2004;87:488–497. doi: 10.1529/biophysj.103.038117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moya I., Silvestri M., Bassi R. Time-resolved fluorescence analysis of the photosystem II antenna proteins in detergent micelles and liposomes. Biochemistry. 2001;40:12552–12561. doi: 10.1021/bi010342x. [DOI] [PubMed] [Google Scholar]
- 57.van Oort B., van Hoek A., van Amerongen H. Aggregation of light-harvesting complex II leads to formation of efficient excitation energy traps in monomeric and trimeric complexes. FEBS Lett. 2007;581:3528–3532. doi: 10.1016/j.febslet.2007.06.070. [DOI] [PubMed] [Google Scholar]
- 58.van Amerongen H., van Grondelle R. Understanding the energy transfer function of LHCII, the major light-harvesting complex of green plants. J. Phys. Chem. B. 2001;105:604–617. [Google Scholar]
- 59.Naqvi R. Kluwer Academic; Dordrecht, The Netherlands: 1998. Photosynthesis Mechanisms and Effects. [Google Scholar]
- 60.van Oort B., van Hoek A., van Amerongen H. Equilibrium between quenched and nonquenched conformations of the major plant light-harvesting complex studied with high-pressure time-resolved fluorescence. J. Phys. Chem. B. 2007;111:7631–7637. doi: 10.1021/jp070573z. [DOI] [PubMed] [Google Scholar]
- 61.Krüger T.P., Novoderezhkin V.I., van Grondelle R. Fluorescence spectral dynamics of single LHCII trimers. Biophys. J. 2010;98:3093–3101. doi: 10.1016/j.bpj.2010.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jennings R.C., Garlaschi F.M., Zucchelli G. The room temperature emission band shape of the lowest energy chlorophyll spectral form of LHCI. FEBS Lett. 2003;547:107–110. doi: 10.1016/s0014-5793(03)00687-2. [DOI] [PubMed] [Google Scholar]
- 63.Nelson N., Yocum C.F. Structure and function of photosystems I and II. Annu. Rev. Plant Biol. 2006;57:521–565. doi: 10.1146/annurev.arplant.57.032905.105350. [DOI] [PubMed] [Google Scholar]
- 64.Knox R.S. Dipole and oscillator strengths of chromophores in solution. Photochem. Photobiol. 2003;77:492–496. doi: 10.1562/0031-8655(2003)077<0492:DAOSOC>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 65.Weber G., Teale F.W.J. Determination of the absolute quantum yield of fluorescent solutions. Trans. Faraday Soc. 1957;53:646–655. [Google Scholar]