

Abstract
Kir1.1 inactivation, associated with transient internal acidification, is strongly dependent on external K, Ca, and Mg. Here, we show that in 1 mM K, a 15 min internal acidification (pH 6.3) followed by a 30 min recovery (pH 8.0) produced 84 ± 3% inactivation in 2 mM Ca but only 18 ± 4% inactivation in the absence of external Ca and Mg. In 100 mM external K, the same acidification protocol produced 29 ± 4% inactivation in 10 mM external Ca but no inactivation when extracellular Ca was reduced below 2 mM (with 0 Mg). However, chelation of external K with 15 mM of 18-Crown-6 (a crown ether) restored inactivation even in the absence of external divalents. External Ca was more effective than external Mg at producing inactivation, but Mg caused a greater degree of open channel block than Ca, making it unlikely that Kir1.1 inactivation arises from divalent block per se. Because the Ca sensitivity of inactivation persisted in 100 mM external K, it is also unlikely that Ca enhanced Kir1.1 inactivation by reducing the local K concentration at the outer mouth of the channel. The removal of four surface, negative side chains at E92, D97, E104, and E132 (Kir1.1b) increased the sensitivity of inactivation to external Ca (and Mg), whereas addition of a negative surface charge (N105E-Kir1.1b) decreased the sensitivity of inactivation to Ca and Mg. This result is consistent with the notion that negative surface charges stabilize external K in the selectivity filter or at the S0-K binding site just outside the filter. Extracellular Ca and Mg probably potentiate the slow, K-dependent inactivation of Kir1.1 by decreasing the affinity of the channel for external K independently of divalent block. The removal of external Ca and Mg largely eliminated both Kir1.1 inactivation and the K-dependence of pH gating, thereby uncoupling the selectivity filter gate from the cytoplasmic-side bundle-crossing gate.
Introduction
Gating of Kir1.1 by internal pH is believed to occur via hinged motion of the inner transmembrane helix (1–5). In addition, a secondary gate, responsible for channel inactivation, may also exist at the Kir1.1 selectivity filter (6–9), in analogy to the C-type inactivation gate of Kv channels (10–12). Both Kir1.1 inactivation and C-type inactivation depend on the occupancy of a K-selective site at the outer mouth of the channel, and both types of inactivation are modulated by permeant ions and channel blockers (1,3,6,8,9,11–18). However, closure of this Kir1.1 inactivation gate requires prior closure of the pH gate at the bundle-crossing of inner transmembrane helices, which is not the case for C-type inactivation.
Coupling between the K-channel activation gate at the bundle-crossing of inner helices and the selectivity filter gate has been proposed for KcsA (19–21) and Kv channels (22–24). However, the allosteric mechanism of this coupling (which can also be bidirectional) is only beginning to be defined. Furthermore, cross talk between the bundle-crossing activation gate and the selectivity filter gate may be different for Kir channels (25).
However, the K dependence of Kir1.1 inactivation is similar to the K dependence of KcsA slow inactivation, which has been linked to structural changes at the selectivity filter in which external K appears to stabilize the active state (20,21,26). This suggests that Kir1.1 inactivation probably involves some conformational change at the selectivity filter and/or the outer mouth of the channel (7). Functional studies with different extracellular permeant ions and extracellular blockers have also implicated the selectivity filter and the outer mouth of the channel as the locus of Kir1.1 inactivation (6–9). However, little is known about the specific role of external Ca and Mg on Kir1.1 inactivation.
Recently, Chang et al. (27) showed that external Ca modulated the effects of K on Kir2.1. Since Kir1.1 inactivation is reduced by elevating extracellular K, it is possible that external divalents such as Ca and Mg might affect inactivation by competing with K at the outer mouth of the channel. In addition, Kir1.1b has four negatively charged side chains at its outer surface (E132, E104, D97, and E92) that may stabilize extracellular K in the selectivity filter or act as a locus for external divalent binding and modulation of Kir1.1 inactivation. In this study, we examined the effect of extracellular Ca and Mg on K-dependent Kir1.1 inactivation.
Materials and Methods
Mutant construction and expression of channels
We engineered point mutations in Kir1.1b (ROMK2; EMBL/GenBank/DDBJ accession No. L29403) using a polymerase chain reaction QuickChange mutagenesis kit (Stratagene) and primers synthesized by Integrated Data Technologies (Coralville, IA). Nucleotide sequences were checked on an Applied Biosystems 3100 DNA sequencing machine at the University of Chicago Cancer Research Center.
Plasmids were linearized with Not I restriction enzyme and transcribed in vitro with T7 RNA polymerase in the presence of the GpppG cap by means of an mMESSAGE mMACHINE kit (Ambion, Austin, TX). Synthetic cRNA was dissolved in water and stored at −70°C before use. Stage V-VI oocytes were obtained by partial ovariectomy of female Xenopus laevis (NASCO, Ft. Atkinson, WI) anesthetized with tricaine methanesulfonate (1.5 g/L, adjusted to pH 7.0). Oocytes were defolliculated by incubation (on a Vari-Mix rocker) in Ca-free modified Barth's solution (82.5 mM NaCl, 2 mM KCl, 1 mM MgCl2, and 5 mM HEPES, adjusted to pH 7.5 with NaOH) containing 2 mg/ml collagenase type IA (catalog No. C9891; Sigma Chemical, St. Louis, MO) for 90 min, and (if necessary) for another 90 min in a fresh enzyme solution at 23°C. Oocytes were injected with 5–10 ng of cRNA and incubated at 19°C in 2× diluted Leibovitz medium (Life Technologies, Grand Island, NY) for 1–3 days before measurements were made.
Whole-cell experiments
We measured whole-cell currents and conductances in intact oocytes using a two-electrode voltage clamp (model CA-1; Dagan, Minneapolis, MN) with 16 command pulses of 30 ms duration between −160 mV and +100 mV, centered around the resting potential.
Oocytes expressing Kir1.1 or mutants of Kir1.1 were bathed in permeant acetate buffers to control their internal pH as previously described (28). The relation between intracellular (i) and extracellular (o) pH was calculated from a previous calibration with Kir1.1 oocytes: pHi = 0.595 × pHo + 2.4 (28). There was no external pH dependence of single-channel conductance or gating for the channels used in this study, as determined in separate experiments with impermeant external buffers.
Whole-cell conductances measured with the two-electrode voltage clamp provided a simple measure of aggregate channel activity at constant [K], because the internal pH had no effect on single-channel conductance or channel kinetics until the channel abruptly closed (Po = 0) as a result of intracellular acidification (28). All whole-cell conductances were normalized to the maximum whole-cell conductance for that oocyte to compensate for differences in expression efficiency between wild-type (wt) and mutant channels.
The bath for the whole-cell experiments consisted of 50 mM KCl, 50 mM K acetate, 1 mM MgCl2, 2 mM CaCl2, and 5 mM HEPES. In low-K solutions, NaCl and NaAcetate replaced KCl and KAcetate, respectively. All solutions had the same ionic strength, except for the 10 mM and 20 mM Ca solutions. The 0 Ca, Mg solutions contained 1 mM EGTA. The 0 K external solutions had no added K and 15 mM of the K chelator 18-Crown-6 (crown ether, catalog No. 07673; Sigma). None of the oocytes in these experiments had significant chloride currents, even with external Ca concentrations as high as 20 mM. Oocytes that exhibited chloride conductances > 2 μS were not used.
We determined the rate of exchange of the external bath by measuring the membrane potential during a rapid increase in external K (16). These results demonstrated that 98% of the bath solution could be replaced within 10 s.
Single-channel methods
Single-channel currents were sampled at 5 kHz, filtered at 1 kHz, and recorded with pClamp (version 6) on oocytes expressing either wt-Kir1.1b or the mutant E132Q-Kir1.1b. Voltage commands were generated in 10 mV steps between −160 mV and +100 mV.
The pipette solutions consisted of 100 mM KCl, 5 mM HEPES, and either a), 0 or 10 mM Ca in 0 Mg; or b), 0 or 10 mM Mg in 0 Ca. The potential difference across the patch of membrane was taken as the negative of the pipette holding potential because the oocyte was completely depolarized by the 100 mM K bath solution.
Homology modeling
To facilitate interpretation of the electrophysiological results, we constructed a homology-based model of Kir1.1b. The closed-state 3.1 Å crystal structure of the chicken inward rectifier Kir2.2 (3JYC) (29) was used as the template (49% sequence homology to Kir1.1b). Modeling was done with Molecular Operating Environment, version 2009.10 (Chemical Computing Group, Montreal, Quebec, Canada). The initial sequence alignment was checked manually to ensure that insertions and deletions were confined to surface residues.
Results
Kir1.1 inactivation depends on external Ca and Mg
An example of the Ca dependence of Kir1.1b inactivation is shown in Fig. 1. Oocytes incubated in 1 mM external K at an internal pH of 8 were subjected to a 15 min internal acidification to pHi = 6.3, followed by realkalization to pHi = 8. This pH protocol was repeated at different extracellular Ca concentrations using separate oocytes expressing wt-Kir1.1b. Representative raw data used to construct Fig. 1 are shown in Fig. S1 and Fig. S2 of the Supporting Material. The family of whole-cell conductance curves in 1 mM K (Fig. 1) indicates a strong dependence on external Ca, with 82 ± 4% recovery after 30 min in 0 mM Ca (red curve, Fig. 1), compared with only 8 ± 2% recovery in 10 mM external Ca (blue curve, Fig. 1). Decreased levels of recovery correspond to greater levels of inactivation.
Figure 1.
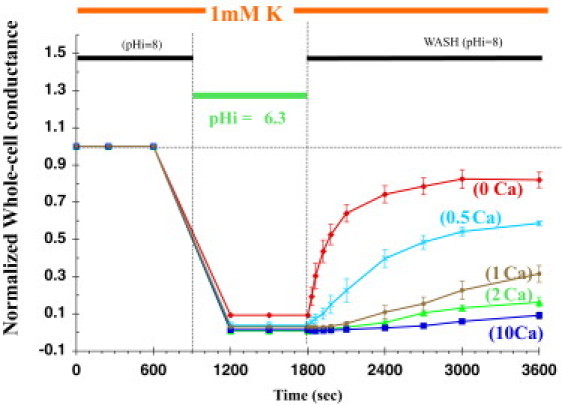
Ca-dependent recovery of wt-Kir1.1b channel activity after a 15 min internal acidification in 1 mM external K, 0 mM Mg. Separate oocytes were exposed to different external Ca levels (mM), and conductance was normalized to its initial value in each oocyte (n = 3 for each Ca concentration).
The dependence of Kir1.1 inactivation on external Ca was weaker in 100 mM K (Fig. 2) than in 1 mM K (Fig. 1), with 10 mM external Ca producing a maximal inactivation of only 29 ± 4% (71 ± 4% channel recovery; blue curve in Fig. 2). Repeating the pH protocol in the absence of Ca and Mg completely eliminated inactivation in 100 mM K media (red curve in Fig. 2). In both 1 mM and 100 mM extracellular K, the rate of recovery of channel activity exhibited a fast time course at the start of realkalization (time = 1800 s, Figs. 1 and 2) and a slower time course at times > 3000 s. The fast recovery rates for the pH protocols in 1 mM and 100 mM extracellular K depended on external Ca as indicated in Fig. 3. Low extracellular K enhanced the sensitivity of inactivation to Ca (and Mg).
Figure 2.
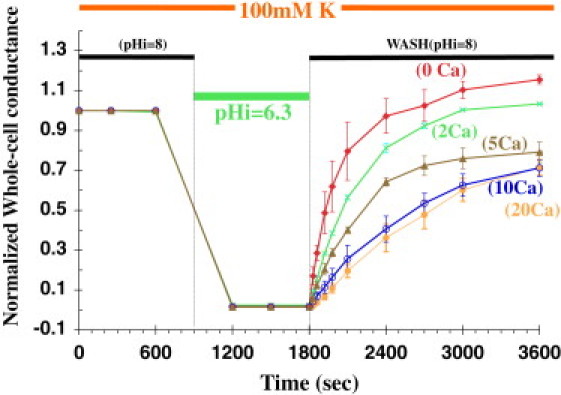
Ca-dependent recovery of wt-Kir1.1b channel activity after a 15 min internal acidification in 100 mM external K, 0 mM Mg. Separate oocytes were exposed to different external Ca levels (mM), and conductance was normalized to its initial value in each oocyte (n = 3 for each Ca concentration).
Figure 3.
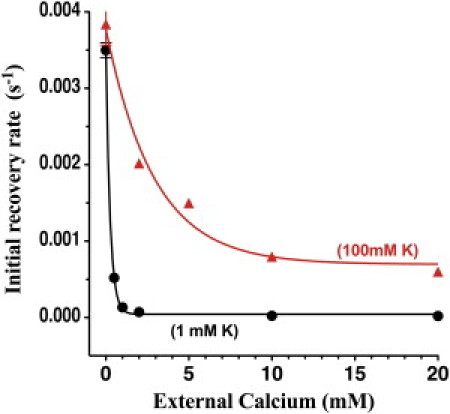
Initial (fast) rate constants for recovery of wt-Kir1.1b channel activity at pHi = 8, after the 15 min internal acidifications of Figs. 1 and 2.
As observed in a previous study (7), the pH-dependent whole-cell Kir1.1 conductances reflected channel activity (gating) rather than single-channel conductance, since pH gating in Kir1.1 occurred via an abrupt reduction of open probability (Po) from >0.9 to zero at low pH (28). In addition, there was no indication that pH-related changes in whole-cell conductance resulted from channel internalization or trafficking to the surface (7).
Calcium is more effective than Mg at sustaining inactivation
To facilitate a comparison of the effects of Ca and Mg on Kir1.1 inactivation, we plotted the 30 min recovery of channel activity at different levels of either Ca or Mg, based on pH protocols similar to those shown in Figs. 1 and 2. Raising external Ca (in the absence of Mg) reduced recovery of wt-Kir1.1 activity (increased inactivation) in 100 mM, 10 mM, and 1 mM external K (solid lines, Fig. 4). At the three different K levels, the steepest Ca dependence occurred near the physiological range of extracellular Ca (2 mM). At external [Ca] > 2 mM, larger extracellular K levels were associated with greater channel recovery (less inactivation).
Figure 4.
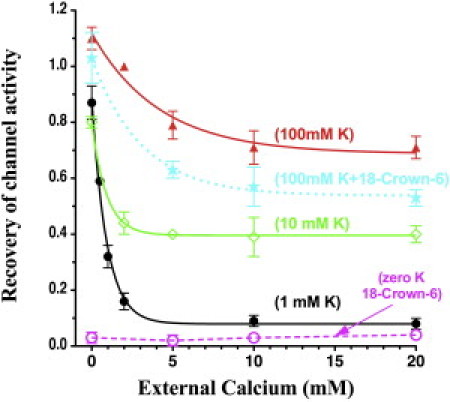
Recovery of wt-Kir1.1 channel activity as a function of external Ca at 0 Mg, measured at 30 min of realkalization, after a 15 min internal acidification from pH 8.0 to 6.3. External K is indicated for each curve (n = 3 for each Ca concentration). The Ca dependence of channel recovery was also assessed in 0 K +15 mM 18-crown ether (dashed magenta line) and in 100 mM K +15 mM 18-crown ether (dotted cyan line). Data for each oocyte were normalized to its initial conductance before acidification. Normalized whole-cell conductances were fit as a single exponential function of Ca concentration.
An external [Ca] of 0 (with 0 Mg) essentially blocked the inactivation process (Fig. 4), except when external K was chelated by 15 mM 18-Crown-6 (dashed magenta line, Fig. 4). Chelation of external K inactivated wt-Kir1.1b independently of external Ca. In contrast, the same concentration of 18-Crown-6 had only a small effect on Ca-dependent inactivation in 100 mM external K (dotted cyan line, Fig. 4), and this could be largely attributed to 18-Crown-6 reducing the effective [K] below 100 mM.
The effect of external Mg (0 Ca) on channel recovery was similar, but smaller, than the effect of external Ca (0 Mg) and occurred over a larger concentration range. In 1 mM extracellular K solutions, only 9 ±2% of wt-Kir1.1b (black curve, Fig. 4) recovered their original activity in 10 mM Ca (0 Mg), whereas in the same 1 mM extracellular K, 49 ±4% of wt-Kir1.1b (black curve, Fig. 5) recovered their original activity level in 10 mM Mg (0 Ca). The Ca- and Mg-induced increase in Kir1.1 inactivation is fundamentally different from effects of other extracellular blockers such as Ba, Cs, and TPNQ, all of which decrease the level of inactivation (7).
Figure 5.
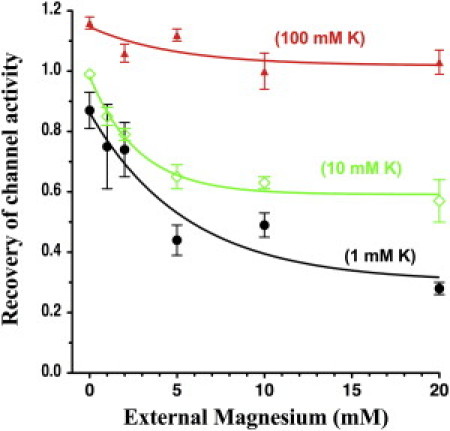
Recovery of wt-Kir1.1 channel activity as a function of external Mg at 0 Ca, measured at 30 min of realkalization, after a 15 min internal acidification from pH 8.0 to 6.3. External K is indicated for each curve (n = 3 for each Mg concentration). Data for each oocyte were normalized to its initial conductance before acidification, and conductances were fit to a single exponential function of Mg concentration.
Negative surface charges affect the divalent sensitivity of Kir inactivation
Kir1.1b has four negatively charged side chains (E132, E104, D97, and E92) at the outer mouth of each monomer that are possible candidates for interaction with external divalents as well as extracellular K (Fig. 6). Replacing all four negative surface charges with neutral side chains greatly increased the sensitivity of the channel to inactivation by external Ca. In 10 mM external K, the quadruple mutant (E132Q, E104S, D97V, and E92A) recovered 100% of its activity in 0 Ca (0 Mg) external solutions (red curve, Fig. 7). However, as little as 0.1 mM external Ca produced an 85% inactivation of the quadruple mutant, compared with <15% inactivation of wt-Kir1.1b channel activity for the same acidification-realkalization protocol in 0.1 mM Ca (green curve, Fig. 7).
Figure 6.
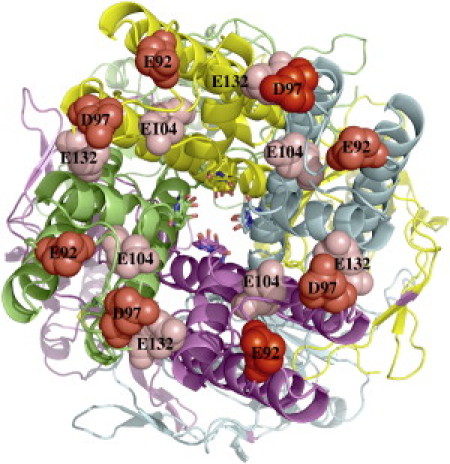
View of outer mouth of Kir1.1b, showing four negatively charged side chains (space-filled residues) on each of the four subunits.
Figure 7.
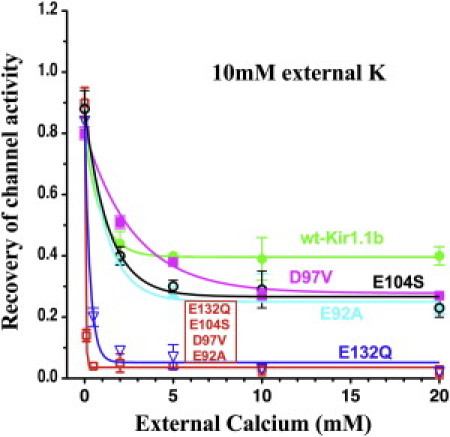
Effect of mutating individual negative surface charges of Kir1.1b on Ca-dependent recovery of channel activity after a 15 min internal acidification. All oocytes were bathed in 10 mM K, 0 Mg. Data for each oocyte were normalized to its initial conductance before acidification.
Individual removal of negative surface charges at E104, D97, and E92 produced intermediate sensitivities of inactivation to external Ca (Fig. 7). The greatest sensitivity occurred for removal of the negative Glu side chain at E132, which showed a Ca dependence similar to that of the quadruple mutant (E132Q, E104S, D97V, and E92A).
In contrast to negative surface charge removal, introduction of an additional negative surface charge by the N105E-Kir1.1b mutation greatly diminished the sensitivity of Kir1.1 inactivation to both Ca (solid lines, Fig. 8) and Mg (dashed lines, Fig. 8) while preserving the K dependence of inactivation that characterizes the Kir1.1 family. The initial recovery rates for N105E-Kir1.1b were strongly dependent on external K, even in the presence of 0 Ca and Mg, which was not the case for wt-Kir1.1b (Fig. S3). We also removed a negative residue (E104) adjacent to N105E to form a double mutant, E104S-N105E, which has the same net side-chain charge in this region as wt-Kir1.1b. This not only restored Ca sensitivity to the channel but also made it hypersensitive to external Ca, for reasons that are not completely clear (green curve, Fig. 8). (see Fig. 13 for the locations of E104 and N105E).
Figure 8.
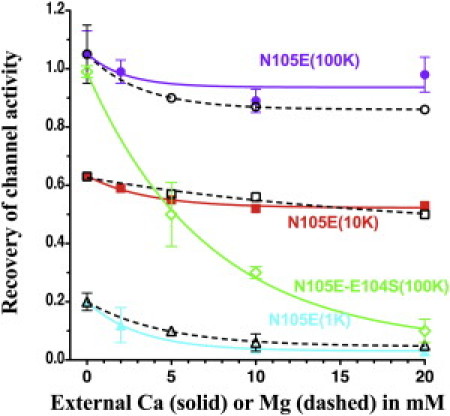
Addition of a negatively charged surface side chain (N105E-Kir1.1b) greatly reduced the sensitivity of Kir1.1 inactivation to external Ca and Mg but not to external K. Oocytes were bathed in either 0 Mg (solid colored curves) or zero Ca (dashed curves) and 100 mM, 10 mM, or 1 mM external K. Recovery of channel activity was measured after 30 min of realkalization using the protocol of Fig. 1 (n = 3 for each Ca, Mg concentration). Data for each oocyte were normalized to its initial conductance before acidification. The double mutant N105E-E104S (green curve) has the same net side-chain charge as wt-Kir1.1b but actually has a greater sensitivity to Ca than wt-Kir1.1b.
Figure 13.
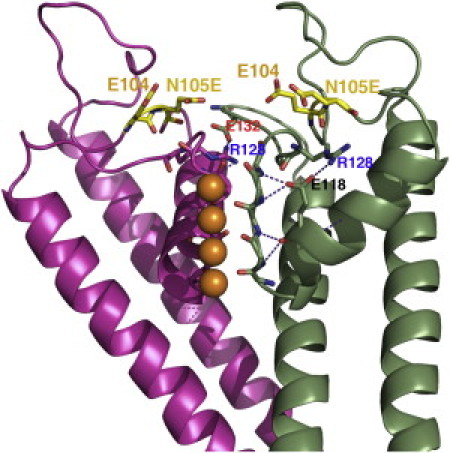
Homology model of N105E-Kir1.1b, showing two adjacent subunits and a region of enhanced negative surface charge resulting from the proximity of the wt-E104 and the mutant N105E. Salt bridging between E118, R128, and E132 has been suggested to have a role in stabilizing the selectivity filter (7).
Disruption of H-bonding between inner and outer helices
A critical lysine (K61-Kir1.1b) in the outer helix is essential for Kir1.1 inactivation (6,7,9). Consequently, we examined the effect of removing this lysine by the mutation K61M-Kir1.1b(K80M-Kir1.1a). As indicated in Fig. 9, K61M-Kir1.1b prevented inactivation at extracellular K concentrations of 1 mM, 10 mM, and 100 mM. At all three of these K concentrations, K61M completely recovered activity, independently of external Ca (and Mg), after a 15 min closure of the bundle-crossing pH gate by internal acidification to pH 4. K61M required a lower internal pH to close the bundle-crossing pH gate than wt-Kir1.1, but the protocol was similar to that shown in Fig. 1. However, chelation of extracellular K with 18-Crown-6 inactivated K61M-Kir1.1b by 30% (green dashed line, Fig. 9) independently of extracellular Ca.
Figure 9.
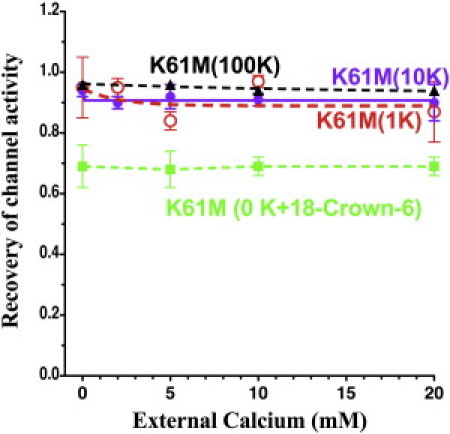
K61M-Kir1.1b abolished both inactivation and Ca sensitivity at external [K] of 100 mM (black dashed), 10 mM (purple), and 1 mM (red, dashed), using a protocol similar to that shown in Fig. 1 (n = 3 for each Ca concentration). Chelation of external K with 15 mM 18-Crown-6 restored inactivation by 30% but not its Ca dependence (dashed green line). Recovery of channel activity was measured 30 min after a 15 min acidification from pHi = 8 to pHi = 4.0, since K61M has a pKa of 5.4 ± 0.1 (33).
External Ca has a minimal effect on Kir single-channel conductance
The single-channel current-voltage relations indicate that 10 mM external Ca has a minimal effect on K channel conductance (gK) near the reversal potential (of 0 mV) in 100 mM external K for both wt-Kir1.1b and the E132Q-Kir1.1b surface charge mutant (Fig. 10). For wt channels in 100 mM K, the single-channel conductance was 44 ±1 pS (n = 3) in the absence of divalents and 40 ± 1 pS (n = 3) with 10 mM external Ca. For the E132Q-Kir1.1b mutant in 100 mM K, there was no measurable difference in single-channel conductance (41 ± 1 pS, n = 3) between zero divalents and 10 mM external Ca near the reversal potential (0 mV). At large negative potentials, the reduction in current became stronger, consistent with a weak block in the permeation pathway near the outer part of the electric field. This effect of Ca was similar to but weaker than that of Mg. At a potential of −120 mV, 10 mM Ca reduced the inward K current by 17%, whereas under similar conditions a lower concentration of Mg (3 mM) resulted in a larger block (30%) of the current (30).
Figure 10.
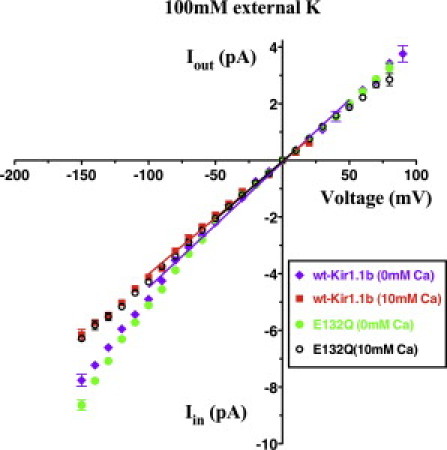
Single-channel current-voltage relations for wt-Kir1.1b and E132Q-Kir1.1b. In 100 mM extracellular K, 10 mM external Ca (0 Mg) decreased gK(wt-Kir1.1b) from 44 ± 1 pS to 40 ± 1 pS, near the reversal potential (0 mV), but produced no significant change in gK(E132Q) = 41 ± 1 pS.
Ca hyperpolarizes the membrane potential at very low external K
Ca screening of the negative surface charges (E132, E104, D97, and E92) may well decrease the local K concentration near the outside of the channel, as indicated by the Ca dependence of the reversal potential (Vrev) in wt-Kir1.1 expressing oocytes (Fig. 11). This effect on local [K] is more pronounced at low external K and is only significant for extracellular K ≤ 1 mM. At external [K] > 1 mM, Ca had a negligible effect on the local [K] adjacent to the membrane (as determined from Vrev), whereas Ca still had a large effect on Kir1.1 inactivation (Fig. 4).
Figure 11.
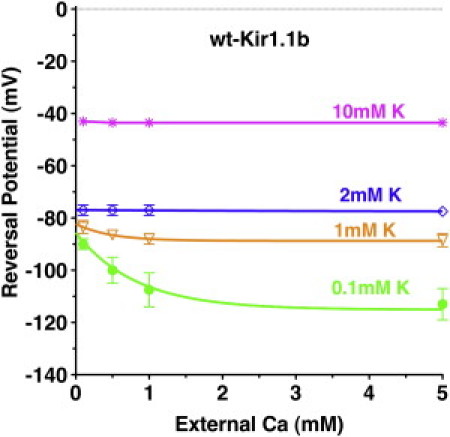
Reversal potential of wt-Kir1.1b in unclamped oocytes as a function of extracellular Ca (0 Mg) at different external K concentrations (n = 3 for each Ca concentration). The potential reflects the K concentration adjacent to the outside of the membrane.
K dependence of Kir1.1 pKa is abolished by Ca removal
Results from a previous study (8) indicated that in 2 mM Ca and 1 mM Mg, the pHi titration of Kir1.1 is a function of extracellular K, with an apparent pKa that varies from 7.2 in 1 mM external K to a pKa of 6.6 in 100 mM external K. This macroscopic pKa reflects the composite pH dependence of all gating mechanisms in the permeation path.
In this study, we remeasured the K dependence of macroscopic pHi gating in Kir1.1 at different levels of extracellular Ca. Complete removal of both external Ca and Mg abolished the K dependence of pHi gating (dashed lines, Fig. 12), whereas raising external Ca to 5 mM (1 mM Mg) increased the K dependence of the apparent pKa, with the largest shift occurring at the lowest extracellular K (solid lines, Fig. 12). The inset of Fig. 12 summarizes pKa as a function of extracellular K at external Ca concentrations of 0, 2 mM, 5 mM, and 10 mM. The observed dependence of pKa on external K and Ca implicates changes at or near the selectivity filter as contributing to the global pHi dependence of Kir1.1
Figure 12.
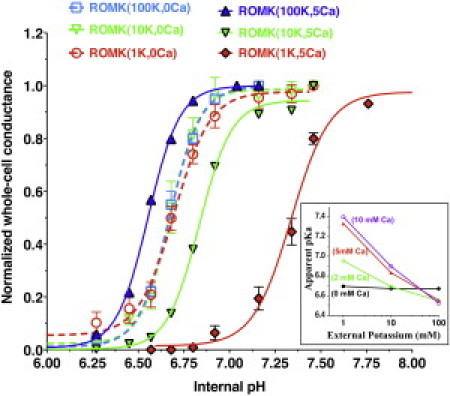
Effect of external Ca on pH titration of Kir1.1b. Solid lines depict the K dependence of pKa in 5 mM external Ca (1 mM Mg) at an external K of 1 mM (red), 10 mM (green), and 100 mM (blue). Removal of external divalents eliminated the K dependence of the Kir1.1-pKa (dashed lines). Inset shows the apparent pKa as a function of external [K] at four different external Ca concentrations. All data were obtained by progressive internal acidification of the oocytes and were normalized to the value at the highest pH tested (n = 4 oocytes at each K).
Discussion
Inactivation of Kir1.1, associated with transient internal acidification, depends not only on the permeant (K) ion concentration (1,3,6,8,9,11–18) but also on the external concentrations of Ca and Mg. At physiological Ca levels of 2 mM, transient internal acidification inactivated wt-Kir1.1 by 84 ± 3% in 1 mM K (green curve, Fig. 1) but produced no significant inactivation in 100 mM external K (Fig. 2). Raising external Ca to 10 mM inactivated Kir1.1 by 92 ± 2% in 1 mM K (blue curve, Fig. 1) but only by 29 ± 5% in 100 mM K (Fig. 2). Further elevation of Ca above 10 mM did not increase inactivation.
Complete removal of both Ca and Mg abolished >80% of Kir1.1 inactivation at external K concentrations of ≥1 mM. At very low external Ca, the initial recovery rate (Fig. 3) and the 30 min recovery level (Figs. 1 and 2) were largely independent of extracellular K. However, at higher external Ca, both the initial recovery rate (Fig. 3) and the 30 min recovery level (Figs. 4 and 5) were strong functions of extracellular K.
The dependence of Kir1.1 inactivation on extracellular K was similar to external K stabilization of the selectivity filter in C-type inactivation of Kv channels (11,12,15–18,27). However, the mechanism underlying the dependence of Kir1.1 inactivation on external divalents is harder to understand. Neither Ca nor Mg appears to be inhibiting the channels by simple open-channel block, since such an effect would be rapidly reversible. Furthermore, in oocytes depolarized to 0 mV by 100 mM external K, addition of 10 mM external Ca decreased single-channel conductance (gK) by 7% but decreased Kir1.1b recovery by 29 ± 5% (Figs. 4 and 10).
In principle, Ca and Mg could be acting by screening negative surface charges. since wt-Kir1.1b has four negatively charged side chains (E92, D97, E104, and E132) at the surface of each monomer. Normally, these charges would be expected to electrostatically enhance the local K concentration near the outer mouth of the channel. Ca binding and screening of these surface charges could decrease the local K concentration at the outer mouth of the pore, thereby increasing inactivation. This predicts a shift in the reversal potential for currents through the pore. Although raising external Ca hyperpolarized the reversal potential (Vrev) at extracellular [K] below 1 mM, no significant change in Vrev could be detected above 2 mM K (Fig. 11). Therefore, this mechanism could not account for the observed Ca dependence of inactivation in either 10 or 100 mM K (Fig. 4).
An additional argument against a simple screening of surface charge by Ca (and Mg) is that large increases in ionic strength, with consequent charge screening, would be expected to occur at high divalent concentrations. In fact, the predominant effect of Ca and Mg on inactivation occurred at submillimolar concentrations (Figs. 4 and 5) that are more consistent with divalent binding than with electrostatic screening.
Ca and Mg may also be exerting their effects by directly displacing K from the selectivity filter and thus destabilizing the open configuration. This also seems unlikely since occupancy of the filter would result in block, which, as discussed above, occurred at higher concentrations than were required for inactivation. In addition, the preferential Ca > Mg sensitivity of inactivation was opposite to the preferential Mg > Ca channel block. External Mg, with a smaller Pauling radius than Ca, decreased gK more than Ca, consistent with a higher affinity of the selectivity filter for Mg. Finally, the E132Q mutant was more susceptible to Ca-dependent inactivation, although there was no significant difference in Ca block of this mutant near the reversal potential, relative to the wt channel (Fig. 10).
Rather than a simple charge screening or block, it seems more likely that Ca and Mg bind to an external site and allosterically decrease the stability of the selectivity filter or decrease the affinity of the filter for extracellular K. Either of these effects would enhance Kir1.1 inactivation. In fact, complete removal of external K using zero K media and 15 mM of 18-Crown-6 (a K chelator) prevented recovery of channel activity, even with zero external Ca and Mg. This suggests that Ca removal increases the affinity of the filter for extracellular K, allowing recovery of channel activity as long as there is some extracellular K in the neighborhood of the filter. Chelation of extracellular K did not significantly reduce the baseline outward whole-cell K conductance, but probably caused inactivation by depriving the outer mouth of the channel of enough K to stabilize the filter during acidification. This occurred independently of extracellular Ca and confirms that external divalents are not the sole cause of Kir1.1 inactivation.
Individual removal of E92, D97, and E104 (Kir1.1b) slightly increased the sensitivity of inactivation to external Ca above wt-Kir1.1b (Fig. 7), but the largest increase in Ca sensitivity to inactivation occurred after the Glu at E132 was removed. This raises the possibility that the E132Q-Kir1.1b mutation itself destabilizes the filter, making the channel more susceptible to inactivation by extracellular divalents. This would be consistent with our previous finding that E132 (see Fig. 13) stabilizes an inactivation gate at the Kir1.1b selectivity filter by forming a salt bridge with R128 of an adjacent subunit (7). Alternatively, E132 could stabilize K binding at the S0, S2, S4 sites of the selectivity filter. Replacing this negative Glu by Gln (E132Q-Kir1.1b) might destabilize K within the filter (or at S0), making the filter more susceptible to inactivation by extracellular divalents.
The heightened Ca sensitivity of mutants lacking negative surface charge (E92A, D97V, E104S, and E132Q) argues against the idea that these negative residues are binding sites for external Ca. Rather, the negative surface charges may act to sequester extracellular K close to the outer surface of the channel.
Finally, the Kir1.1 inactivation associated with elevated (5–20 mM) external Ca was prevented by the K61M-Kir1.1b mutation, which abolished the inactivation due to low extracellular K, possibly by disrupting H-bonding between inner and outer helices at the cytoplasmic side of the pore (6). The time course of K61M-Kir1.1b channel recovery was similar to the 2 mM Ca data of Fig. 2, but independent of Ca. However, complete removal of external K by chelation with 18-Crown-6 reduced K61M channel recovery by 30% (partial inactivation). This suggests that inactivation is influenced by a diverse set of signals involving interaction between the bundle-crossing gate, the transmembrane helices (at K61), and the extracellular concentrations of K, Ca, and Mg. The allosteric basis of inactivation is further supported by the observation that chelation of extracellular K did not by itself cause inactivation unless there was transient closure of the helix-bundle-crossing gate.
A number of studies have addressed the coupling between the bundle-crossing and selectivity filter gates in Kv channels (11,12,19,21), KcsA (21,31,32), and eukaryotic Kir channels (6,7,16). In Kv channels, there is evidence linking the activation gate with C-type inactivation (18–20), perhaps through long-range allosteric communication (22–24). In KcsA, there is good evidence that opening of the inner helical bundle gate initiates conformational changes at the selectivity filter (21,26,32) in analogy to the C-type inactivated state of Kv channels. This would be similar to the link between Kir1.1 pH-gate closure and inactivation reported here, except that with Kir1.1 the inactivated state is associated with the closed rather than the open state.
On the basis of 11 different crystallographic snapshots of KirBac3.1, Clarke et al. (25) suggested that coupling between the bundle-crossing gate and the selectivity filter may not be obligatory, and that gating at the filter may include but not be limited to C-type inactivation. This differs from our observations with Kir1.1, which suggest that closure of the bundle-crossing gate is a necessary but insufficient condition for selectivity filter inactivation. In addition to bundle-crossing closure, Kir1.1 inactivation also required either extracellular Ca > 1 mM when external K was 1 mM, or chelation of all external K by 18-Crown-6.
In summary, physiological concentrations of extracellular Ca and Mg potentiate the slow K-dependent inactivation of Kir1.1 associated with transient internal acidification. Since Kir1.1 is present at the apical membrane of the thick ascending limb, the connecting tubule, and the cortical collecting duct, Ca modulation of Kir1.1 inactivation may have significant effects on renal K secretion and recycling in these nephron segments. The precise mechanism of divalent modulation of inactivation is not understood, although inactivation is significantly more sensitive to Ca than Mg. Modulation of inactivation by divalents does not seem to result from either direct block of the permeation pathway, simple screening of surface charges, or direct displacement of K from the filter. Chelation of extracellular K produced a sustained inactivation that was independent of external Ca, implying that external Ca and Mg act allosterically to decrease the affinity of the outer part of the channel for extracellular K, thereby destabilizing the conducting conformation of the selectivity filter. Conversely, removal of external Ca may keep the filter gate constitutively open, as long as there are trace amounts of extracellular K.
Acknowledgments
We thank D. E. Walters for expert assistance with the Kir1.1b homology model.
This work was supported by National Institutes of Health grants DK46950 (H.S.) and DK27847 (L.G.P.).
Supporting Material
References
- 1.Doi T., Fakler B., Ruppersberg J.P. Extracellular K+ and intracellular pH allosterically regulate renal Kir1.1 channels. J. Biol. Chem. 1996;271:17261–17266. doi: 10.1074/jbc.271.29.17261. [DOI] [PubMed] [Google Scholar]
- 2.Kuo A., Gulbis J.M., Doyle D.A. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 2003;300:1922–1926. doi: 10.1126/science.1085028. [DOI] [PubMed] [Google Scholar]
- 3.Rapedius M., Haider S., Tucker S.J. Structural and functional analysis of the putative pH sensor in the Kir1.1 (ROMK) potassium channel. EMBO Rep. 2006;7:611–616. doi: 10.1038/sj.embor.7400678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sackin H., Nanazashvili M., Walters D.E. Structural locus of the pH gate in the Kir1.1 inward rectifier channel. Biophys. J. 2005;88:2597–2606. doi: 10.1529/biophysj.104.051474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y.Y., Sackin H., Palmer L.G. Localization of the pH gate in Kir1.1 channels. Biophys. J. 2006;91:2901–2909. doi: 10.1529/biophysj.106.087700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rapedius M., Fowler P.W., Baukrowitz T. H bonding at the helix-bundle crossing controls gating in Kir potassium channels. Neuron. 2007;55:602–614. doi: 10.1016/j.neuron.2007.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sackin H., Nanazashvili M., Walters D.E. An intersubunit salt bridge near the selectivity filter stabilizes the active state of Kir1.1. Biophys. J. 2009;97:1058–1066. doi: 10.1016/j.bpj.2009.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sackin H., Vasilyev A., Krambis M. Permeant cations and blockers modulate pH gating of ROMK channels. Biophys. J. 2003;84:910–921. doi: 10.1016/S0006-3495(03)74908-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schulte U., Weidemann S., Fakler B. K(+)-dependent gating of K(ir)1.1 channels is linked to pH gating through a conformational change in the pore. J. Physiol. 2001;534:49–58. doi: 10.1111/j.1469-7793.2001.t01-1-00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoshi T., Zagotta W.N., Aldrich R.W. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- 11.Kurata H.T., Fedida D. A structural interpretation of voltage-gated potassium channel inactivation. Prog. Biophys. Mol. Biol. 2006;92:185–208. doi: 10.1016/j.pbiomolbio.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y., Jurman M.E., Yellen G. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- 13.Kiss L., Korn S.J. Modulation of C-type inactivation by K+ at the potassium channel selectivity filter. Biophys. J. 1998;74:1840–1849. doi: 10.1016/S0006-3495(98)77894-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiss L., LoTurco J., Korn S.J. Contribution of the selectivity filter to inactivation in potassium channels. Biophys. J. 1999;76:253–263. doi: 10.1016/S0006-3495(99)77194-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.López-Barneo J., Hoshi T., Aldrich R.W. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- 16.Sackin H., Nanazashvili M., Walters D.E. External K activation of Kir1.1 depends on the pH gate. Biophys. J. 2007;93:L14–L16. doi: 10.1529/biophysj.107.110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sackin H., Syn S., Walters D.E. Regulation of ROMK by extracellular cations. Biophys. J. 2001;80:683–697. doi: 10.1016/S0006-3495(01)76048-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yellen G. The moving parts of voltage-gated ion channels. Q. Rev. Biophys. 1998;31:239–295. doi: 10.1017/s0033583598003448. [DOI] [PubMed] [Google Scholar]
- 19.Ader C., Schneider R., Baldus M. Coupling of activation and inactivation gate in a K+-channel: potassium and ligand sensitivity. EMBO J. 2009;28:2825–2834. doi: 10.1038/emboj.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cordero-Morales J.F., Jogini V., Perozo E. Molecular driving forces determining potassium channel slow inactivation. Nat. Struct. Mol. Biol. 2007;14:1062–1069. doi: 10.1038/nsmb1309. [DOI] [PubMed] [Google Scholar]
- 21.Cuello L.G., Jogini V., Perozo E. Structural basis for the coupling between activation and inactivation gates in K(+) channels. Nature. 2010;466:272–275. doi: 10.1038/nature09136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Azaria R., Irit O., Yifrach O. Probing the transition state of the allosteric pathway of the Shaker Kv channel pore by linear free-energy relations. J. Mol. Biol. 2010;403:167–173. doi: 10.1016/j.jmb.2010.08.041. [DOI] [PubMed] [Google Scholar]
- 23.Panyi G., Deutsch C. Cross talk between activation and slow inactivation gates of Shaker potassium channels. J. Gen. Physiol. 2006;128:547–559. doi: 10.1085/jgp.200609644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sadovsky E., Yifrach O. Principles underlying energetic coupling along an allosteric communication trajectory of a voltage-activated K+ channel. Proc. Natl. Acad. Sci. USA. 2007;104:19813–19818. doi: 10.1073/pnas.0708120104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clarke O.B., Caputo A.T., Gulbis J.M. Domain reorientation and rotation of an intracellular assembly regulate conduction in Kir potassium channels. Cell. 2010;141:1018–1029. doi: 10.1016/j.cell.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 26.Cordero-Morales J.F., Cuello L.G., Perozo E. Molecular determinants of gating at the potassium-channel selectivity filter. Nat. Struct. Mol. Biol. 2006;13:311–318. doi: 10.1038/nsmb1069. [DOI] [PubMed] [Google Scholar]
- 27.Chang H.-K., Lee J.-R., Shieh R. The extracellular K+ concentration dependence of outward currents through Kir2.1 channels is regulated by extracellular Na+ and Ca+ J. Biol. Chem. 2010;285:23115–23125. doi: 10.1074/jbc.M110.121186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choe H., Zhou H., Sackin H. A conserved cytoplasmic region of ROMK modulates pH sensitivity, conductance, and gating. Am. J. Physiol. 1997;273:F516–F529. doi: 10.1152/ajprenal.1997.273.4.F516. [DOI] [PubMed] [Google Scholar]
- 29.Tao X., Avalos J.L., MacKinnon R. Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 A resolution. Science. 2009;326:1668–1674. doi: 10.1126/science.1180310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang L., Frindt G., Palmer L.G. Magnesium modulates ROMK channel-mediated potassium secretion. J. Am. Soc. Nephrol. 2010;21:2109–2116. doi: 10.1681/ASN.2010060617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blunck R., Cordero-Morales J.F., Bezanilla F. Detection of the opening of the bundle crossing in KcsA with fluorescence lifetime spectroscopy reveals the existence of two gates for ion conduction. J. Gen. Physiol. 2006;128:569–581. doi: 10.1085/jgp.200609638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cuello L.G., Jogini V., Perozo E. Structural mechanism of C-type inactivation in K(+) channels. Nature. 2010;466:203–208. doi: 10.1038/nature09153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sackin H., Nanazashvili M., Li H. Role of conserved glycines in pH gating of Kir1.1 (ROMK) Biophys. J. 2006;90:3582–3589. doi: 10.1529/biophysj.105.076653. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.