

Abstract
Size, weight and position are three of the most important parameters that describe a molecule in a biological system. Ion mobility spectrometry is capable of separating molecules on the basis of their size or shape, whereas imaging mass spectrometry is an effective tool to measure the molecular weight and spatial distribution of molecules. Recent developments in both fields enabled the combination of the two technologies. As a result, ion-mobility-based imaging mass spectrometry is gaining more and more popularity as a (bio-)analytical tool enabling the determination of the size, weight and position of several molecules simultaneously on biological surfaces. This paper reviews the evolution of ion-mobility-based imaging mass spectrometry and provides examples of its application in analytical studies of biological surfaces.
Keywords: Mass spectrometry, Ion chromatography, Imaging, Genomics/proteomics
Introduction
Key questions in analytical biology and biochemistry evolve around the determination of three basic molecular properties: size, weight and position. The complex interplay between molecules in living systems results in a continuous alteration of these properties at the molecular level. The need for analytical approaches that provide better insight into these processes is growing, as understanding of these systems is increasing. A cell, a tissue section, a tissue extract or a body fluid contains a huge amount of chemical and biological information. A single analytical method is usually insufficient when a better understanding of the function of these different biological systems is targeted. This is caused by the complexity of these systems. For that reason a combination of different instruments is extensively used in biochemical research. Ion mobility spectrometry (IMS) coupled with imaging mass spectrometry (MS) is one of these combined approaches that have the potential to provide direct insight into the shape, structure and position of biomolecules. The combination of MS with IMS has already proven to be an extremely successful technique for determining the structures of ions in the gas phase as it allows the separation of different structural isomers. The addition of a molecular imaging component enhances the molecular detail provided.
The basics of IMS have been known for a long time. Rutherford [1] was one of the first researchers who performed measurements based on the mobility of ions when he measured the velocity of different gas ions. Still, the first ion mobility spectrometers that enabled the separation of ions on the basis of their physical collision cross-section were built only in the 1960s. Ions were simply forced to drift in a gas cell. The first ion mobility mass spectrometers coupled with magnetic sector instruments were also built in the 1960s [2], almost immediately after the development of the first ion mobility spectrometers. Since then the combination of ion mobility cells with both continuous and pulsed ion sources has been solved and ion mobility spectrometers have been coupled with many different kinds of mass analysers, including time-of-flight (TOF) analysers [3, 4] magnetic sector instruments [2], quadrupoles [5, 6] ion traps [7–9] and Fourier transform ion cyclotron resonance instruments [10–12]. The first ion mobility instruments were pure drift-time instruments. Two other types of ion mobility spectrometers evolved, differential mobility analysers (also called high-field asymmetric ion mobility spectrometers) and recently travelling-wave instruments.
Initially, ion mobility spectrometers were used only in the field of physical chemistry and for plasma physics research. The development of the matrix-assisted laser desorption and ionization (MALDI) [13–15] and electrospray ionization (ESI) [16, 17] methods in the late 1980s enabled the measurement of large biomolecules by MS. This also resulted in a major breakthrough in the field of ion mobility MS (IM-MS) in the 1990s. The first work where the mobility of biomolecules was measured was undertaken by Clemmer et al. [18]. In this publication, the possibility of measuring the conformation of proteins with IM-MS was demonstrated. The results led to the maturation of the technique and since then IM-MS has been used for structural studies of several different biomolecules, such as proteins [18–20], lipids [21–26], carbohydrates [27–30] and even intact viruses [31–33]. It was also shown in several works that the separation of optical isomers is possible using chiral modifiers in the collision gas [34–36]. The potential of using ion mobility mass spectrometers in biological and biochemical research accelerated the development of the instrumentation and moved the field from custom-built instruments to commercially available instruments. Nowadays, several vendors sell ion mobility mass spectrometers and these instruments are becoming more and more popular.
In parallel to the development of IM-MS, another bioanalytical technology has emerged: imaging MS [37, 38]. Imaging MS is an MS technique where the spatial distribution of different classes of molecules can be determined on a surface. It is perceived as one of the emerging tools in proteomics, lipidomics and metabolomics. Recent developments in imaging MS allow the direct detection of biomolecules as proteins, lipids and drugs on tissue. Label-free imaging with MS uses an intrinsic molecular parameter, the molecular mass, to identify and visualize the distribution of a wide variety of biomolecules at surfaces. This allows the combined determination of molecular weight and localization. An imaging MS experiment requires surface molecules to be desorbed and ionized. Two general approaches can be distinguished in imaging MS. The first and oldest approach uses energetic charged particles to generate surface ions for mass analysis. This approach is common to secondary ion MS (SIMS). The second approach uses photons to generate surface ions for mass analysis. This is the domain of laser-based MS technologies such as MALDI. There is a continuous stream of new desorption and ionization technologies that extend the capabilities of imaging MS. Desorption ESI and laser spray are examples of recently conceived ambient desorption and ionization methods that further the application areas of imaging MS. The interested reader is referred to recent imaging MS reviews [37, 38] for an overview of all relevant innovation in desorption and ionization for imaging MS. Here we will briefly introduce the key concepts of SIMS and MALDI for imaging MS.
SIMS involves the analysis of secondary ions generated from a surface when this surface is bombarded with primary particles. These primary particles are typically elemental ions generated from a liquid metal ion source. The ‘sputtered’ secondary particles will be electrons, neutrals and ions. These ions are amenable to analysis with a mass spectrometer and provide insight into the chemical composition of the surface. As a rule of thumb, with a primary ion dose below 1013 ions/cm2, each individual primary ion samples a fresh spot on the surface. This is referred to as the ‘static limit’. An ion-induced modification of the surface is sampled with a primary ion dose above this static limit. Under these conditions the surface changes rapidly and is sputtered with a fixed sputter rate. The regime where the primary ion dose exceeds the static limit is hence referred to as ‘dynamic SIMS’. Traditionally, SIMS has been applied in the domain of surface physics and solid-state physics. Over recent decades, SIMS has developed more and more as an analytical tool for biological materials such as cells and tissue. Abundant lipids and small peptides can be studied from these surfaces through the use of sample modification strategies [39] and new elemental cluster ion sources (Aum+n and Bim+n) [40]. SIMS is traditionally implemented on a TOF mass spectrometer that ensures a sensitive and high-throughput analysis.
MALDI is a versatile and effective method to desorb and ionize polymers, small organic molecules, lipids and amino acids but also especially biomolecules such as peptides, proteins and oligonucleotides [14]. The basic experiment involves irradiating the sample with a pulsed laser to generate analyte ions from the surface. A matrix solution is applied to the tissue sample to facilitate effective MALDI analysis. These solutions are used for the extraction of the analyte molecules from the tissue and to create analyte-doped matrix crystals. During the analysis, the surface is irradiated by a pulsed laser, thus inducing ablation of these analyte-rich crystals, which results in cooperative motion of the analyte and matrix ions into the vacuum. MALDI can be applied directly for the analysis of biological surfaces and tissue sections. This makes it a key technology for imaging MS.
The added separation power of IMS has its advantages for imaging MS. Several research groups are combining these technologies to enable ion-mobility-based imaging MS. This review describes the evolution of ion-mobility-based imaging MS from a historical perspective to the current state of the art. The developments described have resulted in an innovative (bio-)analytical technology for the analysis of complex surfaces. Ion-mobility-based imaging MS enables the direct determination of three of the most important parameters of molecules on the surface of biological systems: size, weight and position.
Size: the basics of IMS
The main physical phenomenon behind all of the ion mobility methods is the collision of the ions with neutral gas molecules or atoms. The change in the speed of the ions due to the collisions is influenced by the collision cross-section of the ions. Thus, IMS separates the ions on the basis of their size, conformation and charge state instead of their mass. This way it provides complementary shape information when it is coupled with MS. The time frame of an ion mobility separation is in the millisecond range, which is significantly slower than the average time it takes to acquire a mass spectrum. This makes the coupling of the two methods easy because for every ion-mobility-separated peak several mass spectra can be measured. A given molecular class usually follows a trend line when the ion mobility is plotted as the function of the m/z of the ions. This is illustrated in Fig. 1 with the different trend lines for peptides and lipids. The various ion mobility methods can be grouped on the basis of whether their ion separation selectivity is based on spatial dispersion such as in drift tubes or travelling-wave instruments or on temporal dispersion such as in high-field asymmetric IMS (FAIMS) instruments.
Fig. 1.

Matrix-assisted laser desorption and ionization (MALDI)–ion mobility mass spectrometry (MS) conformation space obtained for a mixture of model species representing each molecular class (ranging from seven to 17 model species for each class, spanning a range of masses up to 1,500 Da). Dashed lines are for visualization purposes, where each molecular class occurs in conformation space. Signals in the vicinity of the asterisk arise from limited post-ion-mobility fragmentation of the parent ion species. (Reproduced with permission from [41])
Drift-time IMS
The first and simplest design ion mobility spectrometers were drift-tube instruments. In this type of spectrometer the ion mobility cell consists of series of stacked-ring electrodes (Fig. 2). With use of these electrodes a near-uniform electric field is created along the axis of the cell. An important part of the drift tube is the collision gas, which is mostly nitrogen, helium or argon. During the measurement ion packets are injected into the cell and the ions drift from one end of the cell to the other in the electric field. During their drift they collide with the particles of the collision gas. This way the time that it takes the ions to travel through the cell is affected by their average collision cross-section. This is the basic principle behind ion mobility separation. In drift-time spectrometers the field strength is usually chosen such that the diffusion processes will be dominant. To achieve this, the electric field should be low enough so the thermal energy the ions obtain from the collisions with the buffer gas is larger than the energy they obtain from the electric field. This is called the ‘low-field limit’. In this case the velocity of the ions is directly proportional to the electric field.
Fig. 2.

A high-resolution drift-time ion mobility mass spectrometer. ESI electrospray ionization, TOF time of flight analyser. (Reproduced with permission from [42])
This is one of the main advantages of the drift-time ion mobility methods because the proportionality constant, called the ‘ion mobility constant’ is simply related to the collision cross-section of the ions. This way the collision cross-sections can be determined directly from the drift time using only the experimental parameters with the following formula:
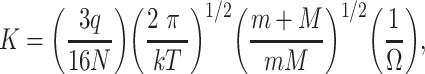 |
where K is the ion mobility constant, which is directly proportional to the electric field and the velocity of the ions, q is the charge on the ion, N is the number density of the buffer gas, k is Boltzmann’s constant, T is the absolute temperature, m is the mass of the buffer gas, M is the mass of the ion and Ω is the collision cross-section of the ion. Drift-time ion mobility spectrometers can be either ambient-pressure or reduced-pressure devices. The ambient-pressure configuration provides better resolution and greater separation selectivity, but because of the inefficient transfer of the ions from the ion mobility cell to the mass spectrometer such instruments suffer from reduced sensitivity. Because of this, ambient-pressure instruments are mostly used as standalone ion mobility spectrometers for field analytical applications such as the detection of explosives or dangerous chemicals at airports. In addition to the ion transfer issues with the ambient-pressure devices, both types of instruments suffer from the need for narrow ion packets for the separation. This seriously compromises their sensitivity, especially when they are coupled to continuous ion sources. This problem can be reduced using a trap cell preceding the drift tube that traps the ions and releases them as narrow ion packets. Still, with this ion accumulation the duty cycle of these instruments is low. Thus, their sensitivity is lower than that of the other type of instruments. Several attempts were made to improve the ion transmission to the drift tube. These include the use of various ion funnel designs [43] or the use multiplex methods such as Fourier transform [44] and Hadamard transform [45, 46] to deconvolve the signals after the continuous introduction of ions into the ion mobility cell instead of using ion packets and thus improving the duty cycle of the instrument.
The other direction in which ion mobility spectrometers have been improved is the resolution of the separation. Several developments have recently been reported in this field as well, such as the so-called ion cyclotron mobility spectrometer developed by Clemmer and co-workers [47]. This device consists of four curved drift tubes and four ion funnels in cyclotron geometry. In this configuration the flight path of the ions in the drift tube can be increased significantly since it depends on the number of cycles in the spectrometer. There have been several developments to overcome the sensitivity issues of the drift-time instruments, such as overtone ion mobility spectrometers, another recent development by Clemmer and co-workers [48, 49]. Their goal was to build a drift-tube instrument which operates as a continuous ion mobility filter rather than using the above-mentioned gating of ion packets that results in decreased sensitivity. To achieve this they divided the linear drift tube into multiple segmented regions and they created two sawtooth voltage gradients by applying the electric field to pairs of segments. The sawtooth voltage is offset by one segment across the whole drift tube and the voltages are alternated during the separation. This way the ion transmission will be a function of the frequency, so an intensity–frequency plot is used to visualize the measured data. The interesting feature of the method is that a given ion gives a signal not only at the fundamental frequency but also at multiples of the fundamental frequency and usually the resolution of these so-called overtone peaks is better than that at the fundamental frequency.
It is possible to couple two drift cells and to do IMS2 experiments [50–52]. In this case the ions are excited between the two ion mobility cells, so their conformation will be different. These types of experiments can provide valuable information about the energetics of conformational changes of gas-phase ions. This way the peak capacity of the method can be further increased as well as making possible the high-throughput analysis of proteins in biological samples such as human plasma.
Travelling-wave IMS
Travelling-wave IMS is the youngest of the ion mobility separation methods. The idea is based on a patent from 1993 by Kirchner [53] but the first commercial instruments were produced only in 2004 [54]. Travelling-wave IMS is based on the use of stacked-ring ion guides (SRIGs) similar to the configuration in drift-time instruments. In these devices an opposite phase of RF is applied to consecutive ring electrodes. The potential generated in one of these ion guides can be seen in Fig. 3.
Fig. 3.

a Calculated effective potential for a stacked-ring ion guide (SRIG). b Series of SIMION plots showing an ion surfing on the front of a travelling wave as it passes along the SRIG. c Mobility separation in the travelling-wave ion guide. (Reproduced with permission from [54])
The main flaw of this design is that there are undulations in the potential surface in the z direction where ions can be trapped. This can significantly slow down the progress of the ions through the ion guide or can even prevent their exiting the cell. As a result, the ions need to be propelled through the ion guide. In travelling-wave instruments a DC potential is superimposed on the RF of an electrode which is subsequently swept over successive electrodes. This creates a moving electric field called a ‘travelling wave’ [54]. The simulation of the ion trajectories indicates that the ions roll down the potential gradient, then the potential pulse moves forward and the process is repeated (Fig. 3b). This way the ions are propelled through the cell by the wave. For ion mobility separations one of these ion guides is filled with a neutral gas, usually nitrogen or argon. In this case the ions not only ‘surf’ on the waves as described above but they also roll over the waves owing to collisions with the gas particles. Low-mobility ions roll over the waves more often than high-mobility ions; thus, they leave the cell later, as can be seen in Fig. 3.
There is one commercially available instrument equipped with a travelling-wave cell as shown in Fig. 4. This is essentially a quadrupole TOF instrument where the travelling-wave ion mobility cell is located between the quadrupole and the TOF analyser. The ion mobility cell consists of three consecutive SRIGs. The first is called ‘the trap cell’, intended to trap the ions and to release them in packets in the mobility cell. In the second one the actual ion mobility separation takes place. The third one is called ‘the transfer cell’ and its role is to transfer mobility-separated ion packets to the TOF analyser (Fig. 4). The advantages of this design are the relatively high duty cycle because of the trap SRIG and the quadrupole analyser before the ion mobility cell, and the possibility to enhance the detection of certain mobility-separated ions by synchronizing the pusher of the TOF and the release of the ions by the transfer cell. Also, these instruments are capable of performing collision-induced-dissociation fragmentation before the ion mobility separation in the trap cell or after the mobility separation in the transfer region, which has the advantage that the fragments will have the same drift time as the corresponding parent ions. It is also possible to fragment the ions in both cells, which leads to MS3-like experiments.
Fig. 4.

The Waters SYNAPT ESI HDMS mass spectrometer equipped with a travelling-wave ion mobility cell. The abbreviation IMS refers to the ion mobility separation device. (Reproduced with permission from [55])
The main issue with this type of instrument is the relatively low resolution, although with recent developments it was increased to around 40 Ω/ΔΩ. This was achieved by increasing the length of the mobility region and adding an additional region filled with helium before the mobility ion guide.
Another drawback of the method is the lack of a simple relation between the drift times and the collision cross-sections of the analytes. There have been recent attempts to find the relation between these two parameters. Shvartsburg and Smith [56] have proposed a physical model to describe the properties of a travelling-wave cell that addresses this issue. Despite these attempts, the experimental collision cross-sections are still determined using different compounds with known collision cross-sections as calibration standards [57, 58]. Since this is the youngest of the three ion mobility separation methods, it is expected that the above-mentioned issues will be dramatically reduced in the future.
Differential mobility analysers
The idea of the method first appeared in a Soviet patent in the 1980s [59] and was published in referenced literature in 1993 [60]. The mobility of ions depends on the ratio of the electric field intensity and the gas number density. The electric field density between two electrodes oscillates between high and low field and the ions are separated on the basis of their mobility difference in high and low field. This is achieved by passing the ions between two parallel electrodes through a carrier gas while on one electrode a voltage waveform is applied. This causes an oscillation of the ions. Their mobility difference between the high and low field will force them to drift in the direction of an electrode, where they will be neutralized. For each species a given compensation voltage has to be applied to one electrode to allow those ions to pass between the electrodes. It is possible to scan with the compensation voltage over a wide range of species with their respective mobility differences, which results in mobility spectrometry. Alternatively, one compensation voltage can be set that enables the exclusive transmission of ions with a specific mobility difference, i.e. mobility filtering. This is why this type of ion mobility analyser is often considered to be analogous to quadrupole mass analysers.
Mainly two electrode geometries are used: plate electrodes (Fig. 5a) or electrodes with cylindrical geometry (Fig. 5b). The latter design not only separates the ions on the basis of their mobility but also focuses the ions and reduces ion loss due to diffusion and Coulomb repulsion [63]. One concern regarding this design is the limited resolution. This is a direct result from the wider compensation voltage range needed to keep the ions stable and allowing them to pass the analyser. Although simulations indicate that the optimal resolution/sensitivity balance can be achieved with planar geometry [64], it is important to improve the sensitivity of these instruments. One solution is to use a slit aperture instead of the common circular orifice between the ion mobility analyser and the mass spectrometer, which will improve the ion transmission and refocus the ions in the mass spectrometer in an electrodynamic ion funnel [65].
Fig. 5.

Planar geometry (a) and cylindrical geometry (b) high-field asymmetric ion mobility spectrometry (FAIMS) analysers and the ion motion in a planar FAIMS device (c). (a Reproduced with permission from [61]; b and c reproduced with permission from [62])
One of the main advantages of the method is the size of the instrument. Field asymmetric ion mobility spectrometers can be produced with dimensions under a few centimetres without seriously compromising the resolution of the instruments. This makes these devices well suited for the production of portable ion mobility spectrometers. One of the potential applications of such instruments is the detection of explosives [66, 67].
Owing to their slightly differing principles, FAIMS and the other ion mobility methods provide slightly different conformational information about the ions. Tandem ion mobility separation becomes possible through the coupling of FAIMS with another ion mobility method [68]. This is especially advantageous as there are currently no means to extract structural information from FAIMS results. A 2D-FAIMS/IMS measurement will reveal the ion geometry separated by FAIMS. Despite their low resolution, isomers or isobars are often distinguished by FAIMS better than by conventional IMS. This is because the differential mobility is much more weakly correlated with the ion mass than the absolute mobility.
Two directions of development were made possible by the miniaturization of the FAIMS analyser. One is increasing the speed of the method by applying higher fields on the two electrodes [69, 70]. The separation speed scales with the fourth to sixth power of the electric field, so this way the separation speed can be increased extensively (about 100–10,000 times higher when the electric field intensity is increased to over 60 kV/cm) although the resolution of these devices is lower than that of the other designs. The other method was proposed to increase the resolution of FAIMS instruments [71, 72]. It has been known for a relatively long time that using He/N2 mixtures in these instruments with as high He content as possible can increase the resolution. A typical planar high-field asymmetric IM spectrometer design with a 2-mm gap between the two electrodes precludes the use of gas mixtures with He content higher than 65%. Electrical breakdown has been observed between the two electrodes at higher He contents. Shvartsburg et al. [71, 72] proved with a slightly modified instrument that He/N2 gas mixtures up to 75% He content can be used without observing electrical breakdown. This way the resolution of the instruments can be increased to around 100, in line with the resolution of common drift tubes.
Weight: IM-MS coupled with a MALDI ion source
Since its development in the late 1980s by Karas et al. [13–15], MALDI-MS has become one of the main ionization techniques used in the field of proteomics. The method has several advantages over ESI-MS, such as its speed and tolerance of salts. It mostly produces singly charged ions, whereas during the electrospray process several multiply charged species arise, making the mass spectra and also the ion mobility spectra more complex. Because of the lack of separation, usually ion suppression and significant chemical noise such as matrix-related peaks can decrease the sensitivity of the method. Combined with low-resolution MS it is difficult to separate ions that are nominally isobaric. Although it is possible to use off-line high-performance liquid chromatography (HPLC) separation before MALDI-MS measurement, this solution has some disadvantages, mainly the significantly increased measurement time needed to analyse a sample. Most of these limitations can be overcome or at least reduced when IM-MS is used with a MALDI ion source. MALDI ion sources operate in pulsed mode, so they are more easily coupled with ion mobility devices than are electrospray ion sources. One of the first examples where a MALDI ion source and IM-MS were coupled was the instrument developed by the group of Bowers [73] in 1995. They used a magnetic sector instrument equipped with a MALDI ion source and they coupled a drift-time ion mobility spectrometer after it. They used this instrument to study the conformation of several polymer ions, namely sodiated poly(ethylene glycol) ions. Since then several papers have been published wherein it was reported that ion mobility mass spectrometers were used to examine even biomolecules and biological mixtures.
Koomen et al. [74] developed a method for rapid screening of oligonucleotides. This work in 2002 was the first where MALDI-IM-MS was used for the study of different oligonucleotides. It was demonstrated that an ion mobility cell with a mobility resolution of 20–30 is sufficient to separate oligonucleotides of different length and to remove the chemical noise caused by matrix signals. However, they did not have enough resolution to separate different structural isomers. The resolution of the ion mobility cells alone is also not enough to separate oligonucleotides with the same length but different mass; in this case the MS data have to be used to differentiate between the species. Also, they studied the separation of different DNA fragment ions and determined chemical modifications of DNA using MALDI-IM-MS. Their results suggest that MALDI-IM-MS can be used to study chemically modified DNA oligonucleotides and DNA–drug and DNA–metal complexes.
McLean and Russell [75] proved in a publication from 2003 that sub-femtomole amounts of peptides can be detected using MALDI-IM-MS . They used a high-repetition-rate laser to increase the duty cycle of the instrument. Sub-femtomole amounts of angiotensin I and angiotensin II and also a tryptic digest of bovine haemoglobin were measured. This proved that using IM-MS with a MALDI ion source will not decrease the detection sensitivity significantly owing to the pulsed nature of the ion source.
In 2004 Woods et al. [21] demonstrated that it is possible to use MALDI-IM-MS for the separation of complex biological mixtures. It was shown in this work that peptides, lipids and oligonucleotides form distinct trend lines which make possible the rapid separation of these different compound classes. Also, MALDI-IM-MS was used for the rapid screening of drugs in saliva.
One of the fields that can benefit tremendously from the use of MALDI-IM-MS is proteomics [76]. One of the main advantages of the method compared with classic HPLC-MS workflows is its speed. The liquid chromatography separation of complex biological mixtures can take from several minutes to hours, whereas ion mobility separation takes only milliseconds. There are several proteomics experiments where IM-MS can be effectively used, such as increasing the confidence of protein identification by separating isobaric tryptic peptides. In conventional experiments to determine post-translational modifications the interpretation of the mass spectra can be difficult because of neutral losses and the small concentration of the modified protein. Some simulations suggest that, for example, phosphorylation changes the protein conformation, leading to a more compact structure, which can thus be beneficial for the study of such modifications by IM-MS; thus, IM-MS is potentially a powerful tool to study post-translational modifications and protein–small molecule complexes. IM-MS also provides a means to study the conformation of gas-phase ions and with variable-temperature ion mobility separation protein folding and unfolding processes, which can be important to understand diseases that are related to protein misfolding such as Parkinson’s or Alzheimer’s disease. Another technique that can benefit from IM-MS is imaging MS. This technique will be discussed in detail in the next section.
In a recent paper McLean’s group [77] reported a custom-built drift-time instrument with interchangeable MALDI and nano-ESI sources and evaluated the instrument for comparison of the mobility of ions produced by ESI and MALDI processes. These experiments can be beneficial to better understand how the conformation of the ions will change during the ionization process in different ion sources. This information is crucial for biochemical studies where the conformation of biomolecules is investigated by IM-MS.
Position: IM-MS in imaging experiments
Imaging MS emerged in recent years as a powerful discovery tool for biochemical studies. However, the performance of the technique is compromised by the complexity of the sample. This can cause ion suppression and also several nominally isobaric species can be present that cannot be resolved using TOF analysers. Recently, it was demonstrated that this can be improved using a Fourier transform ion cyclotron resonance mass spectrometer, an instrument which has mass resolution far superior to that of TOF instruments [78]. Another solution is the use of a separation technique, but because of the nature of the samples, chromatographic techniques cannot be used. An effective way to solve this problem is to add a separation step after the ionization; thus, the use of IM-MS can address some of the aforementioned limitations. With IMS it is possible to separate isobaric species, although it will not prevent ion suppression because the separation takes place after the ionization process. Another useful property of the method is the possibility to remove chemical noise such as matrix-related peaks, this way simplifying the interpretation of the spectra and also improving the contrast of the ion images.
Lipids are one of the most abundant biomolecules in tissue sections. They also ionize easily; thus, they can be easily studied by imaging MS. They can also be associated with several diseases, so they are interesting also from a medical point of view. The imaging of lipids can benefit from ion mobility separations since different classes and structures of possible isobaric species exist. It is not surprising that there are several published reports of the study of the distribution of lipids, especially phospholipids, using ion mobility separation before the mass-spectrometric detection. One work was by Jackson et al. [23] and another was by McLean’s group [79], both from 2007, where the possibility and usefulness of using ion mobility separation for the imaging of different lipid classes, mainly phospholipids from brain samples, were demonstrated.
One of the main advantages of imaging MS is the capability to measure the distribution of drugs and their metabolites selectively and simultaneously in whole-body sections without using labelling [80, 81], thus giving information complementary to that obtained from conventional whole-body autoradiography measurements. Trim et al. [82] investigated whether the coupling of ion mobility separation with imaging MS has additional benefits for pharmaceutical studies. The distribution of vinblastine and its metabolites was studied in mouse whole-body sections using ion mobility imaging MS and MS/MS. An endogenous lipid isobaric to vinblastine and also one isobaric to one of its metabolites were observed. Removing these isobaric backgrounds by ion mobility separation alters the spatial distribution of the mass related to vinblastine or its metabolite and increases the confidence in the results (Fig. 6).
Fig. 6.

Comparison of a m/z 811 and b m/z 811-751 images obtained using MALDI–ion mobility spectrometry (IMS)–MS/MS and conventional MALDI-MS/MS. These images clearly demonstrate the advantages of ion mobility separation within MALDI xenobiotic imaging. The main difference observed is indicated: the distribution of the ions of interest within the renal pelvis. c whole-body autoradiograph showing the distribution of 3H in a 1-h postdose rat dosed with 6 mg/kg 3H-vinblastine intravenously, confirming the distribution observed with MALDI-IMS-MS/MS. (Reproduced with permission from [82])
In 2010 Ridenour et al. [24] described a method to determine the collision cross-section of phospholipids and tryptic peptides directly from tissue using MALDI-T-wave-IM-MS data. It was indicated that it is important to calibrate the instrument with the same class of compounds. They observed that if the instrument was calibrated with the drift-time collision cross-section values of peptides for phospholipid measurement, then the average difference between the drift-time and the T-wave collision cross-section values was 7%, whereas with phospholipid drift-time data this value can be decreased below 0.5%.
Recently, the use of MALDI-IM-MS was reported for on-tissue bottom-up proteomics experiments using formalin-fixed, paraffin-embedded (FFPE) tissue samples in several papers. FFPE samples are amongst the most challenging samples. However, formalin fixing and parafin embedding is the standard protocol that pathologists use to fix tissue sections for storage and these samples are available in great numbers for the study of different diseases. One of the problems is that FFPE samples require extensive washing with different solvents to remove the paraffin, but this can easily damage the sample. The formalin fixing cross-links the proteins, so enzymatic digestion is necessary to study these tissue sections.
In one report, a 78-kDa protein called glucose-regulated protein 78 kDa (Grp78) was identified from pancreatic adenocarcinoma tissue after tryptic digestion. The spatial distribution of the tryptic peptides related to Grp78 suggested high expression of the protein in the adenocarcinoma cells [83]. This result demonstrates that the use of MALDI-IM-MS imaging allows the localization of a tumour biomarker in on-tissue-digested FFPE samples.
In other work, FFPE rat brain and frozen human brain samples were analysed after on-tissue digestion of the proteins [84]. It was shown that with the additional ion mobility separation the number of detected peaks increased owing to the separation of isobaric species. Also the improved confidence of protein identification was proved when ion mobility separation is used. Tubulin and ubiquitin have isobaric tryptic peptides at 1,039 m/z that have different spatial distributions in the brain (Fig. 7). If an MS/MS experiment without ion mobility separation is performed, the resulting spectrum will contain a mixture of fragment peaks from the fragmentation of both species. MASCOT identification of the proteins is severely hampered by this phenomenon. Imaging experiments without ion mobility result in an average image of the two proteins. With drift-time separation the fragmentation products of the two peptides are separated, thus providing higher scores for a MASCOT search and increasing the confidence of the identification of the proteins. It reveals different spatial organization of the proteins.
Fig. 7.

Images of separated and identified isobaric ions of tubulin and ubiquitin peptide fragments (1,039 m/z) are presented directly on tissue by ion mobility MS imaging (BioMap). For these images, two different drift times were selected: 100–127 and 133–148 bins to reconstruct the respective images. Without any ion mobility, one image is obtained corresponding to the superposition of these two images. (Reproduced with permission from [84])
Also, recent work by Djidja et al. [85] used imaging combined with ion mobility separation for tumour classification. The samples studied were pancreatic cancer tissue microarray (TMA) samples and human FFPE tissue sections. After tryptic digestion of the TMA samples, MALDI-IM-MS, MALDI-IM-MS/MS and principal component analysis–discriminant analysis (PCA-DA) were employed to generate tumour classification models. It was found that the use of IM-MS is beneficial for similar studies as it provides the exact distribution of proteins as a result of minimization of peak interferences. This also results in better protein identification from database searches. The classification model they built up was verified after statistical analysis of the TMA results by measuring the digested FFPE tissue sections with MALDI-IM-MS. The distribution of characteristic peptides of the tumour region highlighted by the PCA-DA was in good agreement with the histological data, thus confirming the PCA-DA statistical method.
Summary and outlook
The importance of IM-MS has increased during the past decade and most likely this trend will continue. The separation speed obtainable, the possibility to acquire additional information about the structure of different molecular classes and the easy coupling with MS contributes to the maturation of this technique. We have described some of the technological developments that have contributed to the growing importance of IM-MS. Several research areas have been described that can benefit from the use of IM-MS. These examples also demonstrate the utility of IM-MS to address the need for additional separation steps in imaging and MALDI proteomics applications. Another possible utilization of imaging IM-MS is in the field of quantitative imaging. Quantitation of molecules on tissue is seriously hampered by ion suppression and nominally isobaric species present. With use of ion mobility separation the precision of the quantitation can be increased through the removal of the nominally isobaric ions. This approach has great potential in future quantitative molecular imaging experiments using MS.
As we showed IM-MS has several limitations, such as the significantly decreased sensitivity that needs to be overcome to study low-abundance species with IM-MS. There is a strong need to further increase the resolution of the existing IM-MS methods to obtain more structural separation. Most of the current instrumental developments aim at improving these areas.
Besides the instrumental developments, we expect better understanding of the connection between ion drift times and collision cross-sections, especially in the case of the T-wave [56] and FAIMS [86] devices. There is growing interest in predicting the collision cross-sections of peptides on the basis of their sequence [87]. In the future, developments in bioinformatics may also help to handle the increased datasets resulting from the use of ion mobility separation as an additional dimension in the data.
It was shown that the combination of IMS and imaging MS is capable of determining the size, weight and position of biomolecules simultaneously from complex samples, rendering it an essential tool in the field of analytical biochemistry.
Acknowledgements
This work is part of the research programme of the Stichting voor Fundamenteel Onderzoek der Materie (FOM), which is financially supported by the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO). The authors also gratefully acknowledge funding from the Netherlands Proteomics Centre (NPC).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Published in the special issue Advances in Analytical Mass Spectrometry with Guest Editor Maria Careri.
References
- 1.Rutherford E. Philos Mag. 1897;44:422–440. [Google Scholar]
- 2.McDaniel EW, Martin DW, Barnes WS (1962) Rev Sci Instrum 33
- 3.McAfee KBJ, Sipler DP, Edelson D (1967) Phys Rev 160
- 4.McKnight LG, McAfee KB, Sipler DP (1967) Phys Rev 164
- 5.Karasek FW, Cohen MJ, Carroll DI (1971) J Chromatogr Sci 9
- 6.Wu C, Siems WF, Asbury GR, Hill HH. Anal Chem. 1998;70(23):4929–4938. doi: 10.1021/ac980414z. [DOI] [PubMed] [Google Scholar]
- 7.Hoaglund CS, Valentine SJ, Clemmer DE. Anal Chem. 1997;69(20):4156–4161. doi: 10.1021/ac970526a. [DOI] [Google Scholar]
- 8.Creaser CS, Benyezzar M, Griffiths JR, Stygall JW. Anal Chem. 2000;72(13):2724–2729. doi: 10.1021/ac991409d. [DOI] [PubMed] [Google Scholar]
- 9.Clowers BH, Hill HH. J Mass Spectrom. 2006;41(3):339–351. doi: 10.1002/jms.994. [DOI] [PubMed] [Google Scholar]
- 10.Parent DC, Bowers MT (1981) Chem Phys 60
- 11.Bluhm BK, Gillig KJ, Russell DH. Rev Sci Instrum. 2000;71(11):4078–4086. doi: 10.1063/1.1288235. [DOI] [Google Scholar]
- 12.Tang XT, Bruce JE, Hill HH. Rapid Commun Mass Spectrom. 2007;21(7):1115–1122. doi: 10.1002/rcm.2928. [DOI] [PubMed] [Google Scholar]
- 13.Karas M, Hillenkamp F. Anal Chem. 1988;60(20):2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 14.Karas M, Bahr U, Hillenkamp F. Int J Mass Spectrom Ion Process. 1989;92:231–242. doi: 10.1016/0168-1176(89)83030-7. [DOI] [Google Scholar]
- 15.Karas M, Bahr U, Ingendoh A, Hillenkamp F. Angew Chem Int Ed Engl. 1989;28(6):760–761. doi: 10.1002/anie.198907601. [DOI] [Google Scholar]
- 16.Wong SF, Meng CK, Fenn JB. J Phys Chem. 1988;92(2):546–550. doi: 10.1021/j100313a058. [DOI] [Google Scholar]
- 17.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Science. 1989;246(4926):64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 18.Clemmer DE, Hudgins RR, Jarrold MF. J Am Chem Soc. 1995;117(40):10141–10142. doi: 10.1021/ja00145a037. [DOI] [Google Scholar]
- 19.Ruotolo BT, Giles K, Campuzano I, Sandercock AM, Bateman RH, Robinson CV. Science. 2005;310(5754):1658–1661. doi: 10.1126/science.1120177. [DOI] [PubMed] [Google Scholar]
- 20.Uetrecht C, Rose RJ, van Duijn E, Lorenzen K, Heck AJR. Chem Soc Rev. 2010;39(5):1633–1655. doi: 10.1039/b914002f. [DOI] [PubMed] [Google Scholar]
- 21.Woods AS, Ugarov M, Egan T, Koomen J, Gillig KJ, Fuhrer K, Gonin M, Schultz JA. Anal Chem. 2004;76(8):2187–2195. doi: 10.1021/ac035376k. [DOI] [PubMed] [Google Scholar]
- 22.Jackson SN, Wang HYJ, Woods AS. J Am Soc Mass Spectrom. 2005;16(2):133–138. doi: 10.1016/j.jasms.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 23.Jackson SN, Ugarov M, Egan T, Post JD, Langlais D, Schultz JA, Woods AS. J Mass Spectrom. 2007;42(8):1093–1098. doi: 10.1002/jms.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ridenour WB, Kliman M, McLean JA, Caprioli RM. Anal Chem. 2010;82(5):1881–1889. doi: 10.1021/ac9026115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson SN, Ugarov M, Post JD, Egan T, Langlais D, Schultz JA, Woods AS. J Am Soc Mass Spectrom. 2008;19(11):1655–1662. doi: 10.1016/j.jasms.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trimpin S, Tan B, Bohrer BC, O'Dell DK, Merenbloom SI, Pazos MX, Clemmer DE, Walker JM. Int J Mass Spectrom. 2009;287(1–3):58–69. [Google Scholar]
- 27.Dwivedi P, Bendiak B, Clowers BH, Hill HH. J Am Soc Mass Spectrom. 2007;18(7):1163–1175. doi: 10.1016/j.jasms.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 28.Fenn LS, McLean JA (2008) Chem Commun 5505-5507. doi:10.1039/B810421b [DOI] [PubMed]
- 29.Plasencia MD, Isailovic D, Merenbloom SI, Mechref Y, Clemmer DE. J Am Soc Mass Spectrom. 2008;19(11):1706–1715. doi: 10.1016/j.jasms.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vakhrushev SY, Langridge J, Campuzano I, Hughes C, Peter-Katlinic J. Anal Chem. 2008;80(7):2506–2513. doi: 10.1021/ac7023443. [DOI] [PubMed] [Google Scholar]
- 31.Bothner B, Siuzdak G. Chembiochem. 2004;5(3):258–260. doi: 10.1002/cbic.200300754. [DOI] [PubMed] [Google Scholar]
- 32.Thomas JJ, Bothner B, Traina J, Benner WH, Siuzdak G. Spectrosc Int J. 2004;18(1):31–36. [Google Scholar]
- 33.Uetrecht C, Versluis C, Watts NR, Wingfield PT, Steven AC, Heck AJR. Angew Chem Int Ed. 2008;47(33):6247–6251. doi: 10.1002/anie.200802410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dwivedi P, Wu C, Matz LM, Clowers BH, Siems WF, Hill HH. Anal Chem. 2006;78(24):8200–8206. doi: 10.1021/ac0608772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holliday AE (2008) Abstr Pap Am Chem Soc 236
- 36.Enders JR, McLean JA. Chirality. 2010;21(1E):E253–E264. doi: 10.1002/chir.20806. [DOI] [PubMed] [Google Scholar]
- 37.McDonnell LA, Heeren RMA. Mass Spectrom Rev. 2007;26(4):606–643. doi: 10.1002/mas.20124. [DOI] [PubMed] [Google Scholar]
- 38.Chughtai K, Heeren RMA. Chem Rev. 2010;110(5):3237–3277. doi: 10.1021/cr100012c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Altelaar AFM, van Minnen J, Jimenez CR, Heeren RMA, Piersma SR. Anal Chem. 2005;77(3):735–741. doi: 10.1021/ac048329g. [DOI] [PubMed] [Google Scholar]
- 40.Winograd N. Anal Chem. 2005;77(7):142A–149A. doi: 10.1021/ac053355f. [DOI] [Google Scholar]
- 41.Fenn LS, Kliman M, Mahsut A, Zhao SR, McLean JA. Anal Bioanal Chem. 2009;394(1):235–244. doi: 10.1007/s00216-009-2666-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kemper PR, Dupuis NF, Bowers MT. Int J Mass Spectrom. 2009;287(1–3):46–57. [Google Scholar]
- 43.Tang K, Shvartsburg AA, Lee HN, Prior DC, Buschbach MA, Li FM, Tolmachev AV, Anderson GA, Smith RD. Anal Chem. 2005;77(10):3330–3339. doi: 10.1021/ac048315a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knorr FJ, Eatherton RL, Siems WF, Hill HH., Jr Anal Chem. 1985;57(2):402–406. doi: 10.1021/ac50001a018. [DOI] [PubMed] [Google Scholar]
- 45.Clowers BH, Siems WF, Hill HH, Massick SM. Anal Chem. 2006;78(1):44–51. doi: 10.1021/ac050615k. [DOI] [PubMed] [Google Scholar]
- 46.Szumlas AW, Ray SJ, Hieftje GM. Anal Chem. 2006;78(13):4474–4481. doi: 10.1021/ac051743b. [DOI] [PubMed] [Google Scholar]
- 47.Merenbloom SI, Glaskin RS, Henson ZB, Clemmer DE. Anal Chem. 2009;81(4):1482–1487. doi: 10.1021/ac801880a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kurulugama RT, Nachtigall FM, Lee S, Valentine SJ, Clemmer DE. J Am Soc Mass Spectrom. 2009;20(5):729–737. doi: 10.1016/j.jasms.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Valentine SJ, Stokes ST, Kurulugama RT, Nachtigall FM, Clemmer DE. J Am Soc Mass Spectrom. 2009;20(5):738–750. doi: 10.1016/j.jasms.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koeniger SL, Merenbloom SI, Clemmer DE. J Phys Chem B. 2006;110(13):7017–7021. doi: 10.1021/jp056165h. [DOI] [PubMed] [Google Scholar]
- 51.Merenbloom SI, Koeniger SL, Valentine SJ, Plasencia MD, Clemmer DE. Anal Chem. 2006;78(8):2802–2809. doi: 10.1021/ac052208e. [DOI] [PubMed] [Google Scholar]
- 52.Valentine SJ, Kurulugama RT, Bohrer BC, Merenbloom SI, Sowell RA, Mechref Y, Clemmer DE. Int J Mass Spectrom. 2009;283(1–3):149–160. [Google Scholar]
- 53.Kirchner NJ. US Patent. 1993;5(206):506. [Google Scholar]
- 54.Giles K, Pringle SD, Worthington KR, Little D, Wildgoose JL, Bateman RH. Rapid Commun Mass Spectrom. 2004;18(20):2401–2414. doi: 10.1002/rcm.1641. [DOI] [PubMed] [Google Scholar]
- 55.Pringle SD, Giles K, Wildgoose JL, Williams JP, Slade SE, Thalassinos K, Bateman RH, Bowers MT, Scrivens JH. Int J Mass Spectrom. 2007;261(1):1–12. doi: 10.1016/j.ijms.2006.07.021. [DOI] [Google Scholar]
- 56.Shvartsburg AA, Smith RD. Anal Chem. 2008;80(24):9689–9699. doi: 10.1021/ac8016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Williams JP, Scrivens JH. Rapid Commun Mass Spectrom. 2008;22(2):187–196. doi: 10.1002/rcm.3346. [DOI] [PubMed] [Google Scholar]
- 58.Thalassinos K, Grabenauer M, Slade SE, Hilton GR, Bowers MT, Scrivens JH. Anal Chem. 2009;81(1):248–254. doi: 10.1021/ac801916h. [DOI] [PubMed] [Google Scholar]
- 59.Gorshkov MP (1982) USSR inventors certificate no 966583
- 60.Buryakov IA, Krylov EV, Nazarov EG, Rasulev UK. Int J Mass Spectrom Ion Process. 1993;128(3):143–148. doi: 10.1016/0168-1176(93)87062-W. [DOI] [Google Scholar]
- 61.Miller RA, Eiceman GA, Nazarov EG, King AT. Sens Actuators B Chem. 2000;67(3):300–306. doi: 10.1016/S0925-4005(00)00535-9. [DOI] [Google Scholar]
- 62.Guevremont R. J Chromatogr A. 2004;1058(1–2):3–19. [PubMed] [Google Scholar]
- 63.Guevremont R, Purves RW. Rev Sci Instrum. 1999;70(2):1370–1383. doi: 10.1063/1.1149599. [DOI] [Google Scholar]
- 64.Shvartsburg AA, Li FM, Tang KQ, Smith RD. Anal Chem. 2006;78(11):3706–3714. doi: 10.1021/ac052020v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mabrouki R, Kelly RT, Prior DC, Shvartsburg AA, Tang KQ, Smith RD. J Am Soc Mass Spectrom. 2009;20(9):1768–1774. doi: 10.1016/j.jasms.2009.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eiceman GA, Krylov EV, Nazarov EG, Miller RA. Anal Chem. 2004;76(17):4937–4944. doi: 10.1021/ac035502k. [DOI] [PubMed] [Google Scholar]
- 67.Eiceman GA, Stone JA. Anal Chem. 2004;76(21):390a–397a. doi: 10.1021/ac041665c. [DOI] [PubMed] [Google Scholar]
- 68.Tang KQ, Li FM, Shvartsburg AA, Strittmatter EF, Smith RD. Anal Chem. 2005;77(19):6381–6388. doi: 10.1021/ac050871x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shvartsburg AA, Smith RD, Wilks A, Koehl A, Ruiz-Alonso D, Boyle B. Anal Chem. 2009;81(15):6489–6495. doi: 10.1021/ac900892u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shvartsburg AA, Tang KQ, Smith RD, Holden M, Rush M, Thompson A, Toutoungi D. Anal Chem. 2009;81(19):8048–8053. doi: 10.1021/ac901479e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shvartsburg AA, Danielson WF, Smith RD. Anal Chem. 2010;82(6):2456–2462. doi: 10.1021/ac902852a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shvartsburg AA, Tang KQ, Smith RD. Anal Chem. 2010;82(1):32–35. doi: 10.1021/ac902133n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vonhelden G, Wyttenbach T, Bowers MT. Int J Mass Spectrom Ion Process. 1995;146:349–364. doi: 10.1016/0168-1176(95)04211-3. [DOI] [Google Scholar]
- 74.Koomen JM, Ruotolo BT, Gillig KJ, McLean JA, Russell DH, Kang MJ, Dunbar KR, Fuhrer K, Gonin M, Schultz JA. Anal Bioanal Chem. 2002;373(7):612–617. doi: 10.1007/s00216-002-1363-2. [DOI] [PubMed] [Google Scholar]
- 75.McLean JA, Russell DH. J Proteome Res. 2003;2(4):427–430. doi: 10.1021/pr034004p. [DOI] [PubMed] [Google Scholar]
- 76.McLean JA, Ruotolo BT, Gillig KJ, Russell DH. Int J Mass Spectrom. 2005;240(3):301–315. doi: 10.1016/j.ijms.2004.10.003. [DOI] [Google Scholar]
- 77.Sundarapandian S, May JC, McLean JA. Anal Chem. 2010;82(8):3247–3254. doi: 10.1021/ac902980r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Taban IM, Altelaar AFM, Van der Burgt YEM, McDonnell LA, Heeren RMA, Fuchser J, Baykut G. J Am Soc Mass Spectrom. 2007;18(1):145–151. doi: 10.1016/j.jasms.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 79.McLean JA, Ridenour WB, Caprioli RM. J Mass Spectrom. 2007;42(8):1099–1105. doi: 10.1002/jms.1254. [DOI] [PubMed] [Google Scholar]
- 80.Rohner TC, Staab D, Stoeckli M. Mech Ageing Dev. 2005;126(1):177–185. doi: 10.1016/j.mad.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 81.Stoeckli M, Staab D, Schweitzer A. Int J Mass Spectrom. 2007;260(2–3):195–202. [Google Scholar]
- 82.Trim PJ, Henson CM, Avery JL, McEwen A, Snel MF, Claude E, Marshall PS, West A, Princivalle AP, Clench MR. Anal Chem. 2008;80(22):8628–8634. doi: 10.1021/ac8015467. [DOI] [PubMed] [Google Scholar]
- 83.Djidja MC, Claude E, Snel MF, Scriven P, Francese S, Carolan V, Clench MR. J Proteome Res. 2009;8(10):4876–4884. doi: 10.1021/pr900522m. [DOI] [PubMed] [Google Scholar]
- 84.Stauber J, MacAleese L, Franck J, Claude E, Snel M, Kaletas BK, Wiel IMVD, Wisztorski M, Fournier I, Heeren RMA. J Am Soc Mass Spectrom. 2010;21(3):338–347. doi: 10.1016/j.jasms.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 85.Djidja MC, Claude E, Snel MF, Francese S, Scriven P, Carolan V, Clench MR. Anal Bioanal Chem. 2010;397(2):587–601. doi: 10.1007/s00216-010-3554-6. [DOI] [PubMed] [Google Scholar]
- 86.Prasad S, Tang KQ, Manura D, Papanastasiou D, Smith RD. Anal Chem. 2009;81(21):8749–8757. doi: 10.1021/ac900880v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang B, Valentine S, Plasencia M, Raghuraman S, Zhang XA. BMC Bioinform. 2010;11:182. doi: 10.1186/1471-2105-11-182. [DOI] [PMC free article] [PubMed] [Google Scholar]