

Abstract
We are increasingly aware that cellular metabolism plays a vital role in diseases such as cancer, and that p53 is an important regulator of metabolic pathways. By transcriptional activation and other means, p53 is able to contribute to the regulation of glycolysis, oxidative phosphorylation, glutaminolysis, insulin sensitivity, nucleotide biosynthesis, mitochondrial integrity, fatty acid oxidation, antioxidant response, autophagy and mTOR signalling. The ability to positively and negatively regulate many of these pathways, combined with feedback signalling from these pathways to p53, demonstrates the reciprocal and flexible nature of the regulation, facilitating a diverse range of responses to metabolic stress. Intriguingly, metabolic stress triggers primarily an adaptive (rather than pro-apoptotic) p53 response, and p53 is emerging as an important regulator of metabolic homeostasis. A better understanding of how p53 coordinates metabolic adaptation will facilitate the identification of novel therapeutic targets and will also illuminate the wider role of p53 in human biology.
Keywords: p53, Homeostasis, Metabolism, Glycolysis, Oxidative phosphorylation, Autophagy
The emerging role of p53 in cellular metabolism
Control of cellular metabolism is a key requirement of normal cell behaviour, and the role that aberrant cellular metabolism plays in disease—particularly cancer—is becoming increasingly apparent. It is, therefore, not surprising that the tumour suppressor p53—a key player in the cellular response to stress—is emerging as an important regulator of cellular metabolism. p53 is a transcription factor that responds to numerous extrinsic and intrinsic challenges to the cell, including DNA damage, oncogene activation and hypoxia, to promote a variety of responses depending on the type, severity and persistence of the stress [1]. By facilitating DNA repair and the activation of apoptosis or senescence, p53 activation represents an efficient mechanism to prevent the accumulation of abnormal cells, particularly those with heritable DNA damage and so protect from tumour formation. Indeed, p53 function is lost in most cancers, underscoring the importance of p53 as a tumour suppressor. However, the diversity of cellular processes influenced by p53 is becoming more evident and the traditional view of p53 as simply a tumour suppressor is being challenged, as roles for p53 in normal cellular homeostasis and cancer cell homeostasis are revealed.
The ability of p53 to respond to nutrient deficiencies is consistent with the established function of p53 as a mediator of stress. However, it is likely that the requirements for the p53-dependent response to metabolic stress are quite different compared to other p53-activating signals. While damage that is likely to promote heritable genetic changes signals to p53 to eliminate the affected cell, metabolic stress more likely requires an adaptive response. DNA damage is unlikely to spontaneously resolve if left unchecked, whereas the simple restoration of nutrients can rapidly restore a starved cell to full health, permitting a more subtle response to nutrient deficiency. p53 is emerging as an important component in a cell's ability to deal with metabolic fluctuations, both by helping to balance proliferation and growth with nutrient availability, and by limiting the accumulation of further damage—ultimately promoting cell survival.
Control of metabolic pathways by p53
The pathways underlying control of metabolism are well established, but the complexities of how they are regulated are still being revealed. p53 has been shown to contribute to the control of many of these pathways, with an emerging theme that p53 can promote the use of catabolic pathways that would maintain energy production under periods of limiting nutrients, thereby maintaining cell viability. These activities of p53 interdigitate well with p53 functions that ameliorate oxidative stress while inhibiting cell growth and cell cycle progression—thereby establishing a coordinated and multi-faceted response to transient periods of starvation.
Glucose metabolism
Glucose is the major source of energy for most cells, providing for both the generation of energy (ATP) and the metabolites for various anabolic pathways [2, 3]. Glycolysis produces two molecules of ATP per glucose, along with pyruvate that can be transferred to the tricarboxylic acid (TCA) cycle for further energy production. Alternatively, glycolytic intermediates can be diverted away from energy production into anabolic pathways. The oxidative arm of the pentose phosphate pathway (PPP) is an important source of NADPH for antioxidant functions and lipid synthesis, as well as providing precursors for de novo nucleotide biosynthesis, while the hexosamine pathway provides substrates to allow protein and lipid glycosylation. An elevated rate of glycolysis under aerobic conditions drives anabolism and increased cell proliferation, and is a characteristic of stem cells [4] and many cancers [5]. p53 plays an important role in regulating glucose metabolism.
Several studies have found that p53 can limit glycolytic flux through a number of mechanisms. p53 lowers the expression of glucose transporters through direct repression of gene expression [6] or more indirectly, through the inhibition of NF-κB [7, 8]. The ability of p53 to repress the insulin receptor promoter provides another mechanism by which p53 can limit the transport of glucose into cells [9]. The negative regulation of phosphoglycerate mutase (PGM) by p53 (through the control of protein stability, rather than a direct effect on transcription) also reduces glycolytic rate and suppresses transformation [4]. TP53-induced glycolysis and apoptosis regulator (TIGAR) functions as a fructose-2,6-bisphosphatase, limiting the activity of phosphofructokinase 1 (PFK1) and so lowering the rate of glycolysis, and promoting the diversion of glycolytic intermediates into the PPP [10] (Fig. 1). Basal expression of carbohydrate responsive element-binding protein (ChREBP) promotes aerobic glycolysis, supporting cell proliferation by stimulating lipid and nucleotide biogenesis. ChREBP levels are elevated in p53-deficient cells, demonstrating that basal p53 levels suppress ChREBP [11].
Fig. 1.
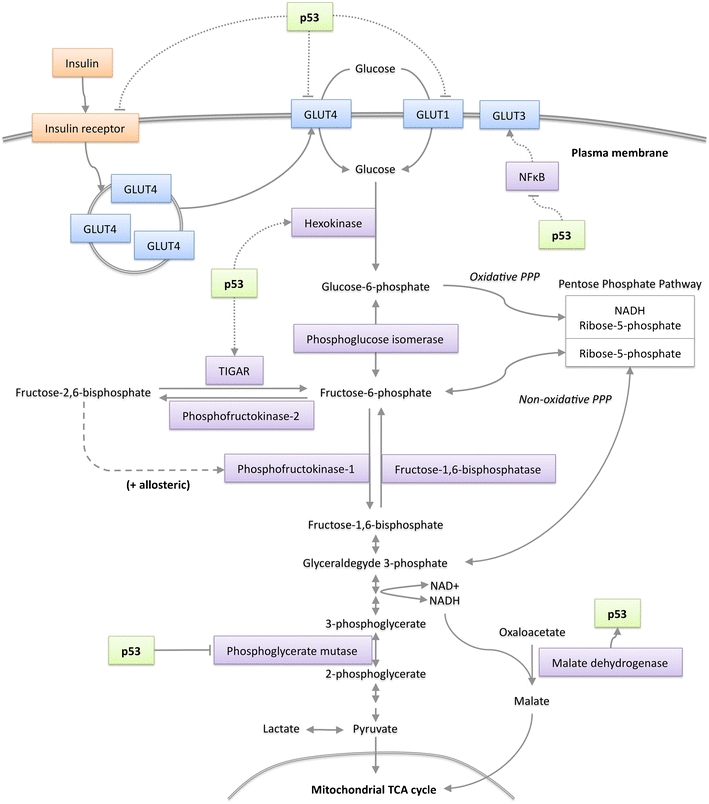
p53 signalling in glucose metabolism. p53 can suppress the transcription of glucose transporters GLUT1 and GLUT4 (and via NFκB inhibits GLUT3) along with the insulin receptor to inhibit cellular glucose uptake. By transcriptional activation of TIGAR, p53 can suppress the rate of glycolysis and increase diversion of glycolytic intermediates into the PPP. p53 can also suppress glycolysis by promoting the degradation of phosphoglycerate mutase (PGM). By activating the transcription of hexokinase II (HK II) p53 can stimulate glycolysis. Malate dehydrogenase (MDH1) forms part of the malate/aspartate shuttle that links glycolysis to mitochondrial respiration; MDH1 binds to and modulates the activity of p53
Overall, there is convincing evidence that p53 can be a negative regulator of glycolysis, although there is also evidence to suggest that p53 can enhance some steps in this pathway. p53 activates the transcription of the muscle isoform of PGM (PGM-M) in cardiac myocytes [12] and hexokinase II (HK2), which catalyses the first step in glycolysis, is under the control of a p53-responsive promoter [13]. The effects of p53 on glycolytic pathways are likely to be very cell and context dependent, but it is of interest to note that the combined effect of TIGAR and HK2 activity could further enhance the supply of glycolytic intermediates to the PPP—the consequences of which are discussed below. Cytoplasmic malate dehydrogenase 1 (MDH1) links glycolysis to OXPHOS by facilitating the transport of NADH equivalents (generated from glycolysis) into mitochondria where they can be used to generate ATP [14, 15]. During glucose starvation MDH1 physically interacts with p53 to modulate its transcriptional response [16]. This interaction provides a direct signal from glucose metabolism to p53 and completes a reciprocal signalling loop between energy metabolism and p53.
Mitochondrial respiration
Under normal, aerobic conditions, the pyruvate generated by glycolysis can be fed into the mitochondrial TCA cycle as an efficient mechanism of ATP generation via oxidative phosphorylation (OXPHOS). However, as with glycolysis, intermediates of the TCA cycle can also be diverted into anabolic pathways to facilitate cell growth and proliferation, a process enhanced in many cancers [3, 17]. In coordination with the inhibition of glycolysis, p53 has been shown to promote OXPHOS through mechanisms that include the transcriptional activation of synthesis of cytochrome c oxidase 2 (SCO2), a regulator of complex IV [18], subunit 1 of complex IV itself [19] and the mitochondrial apoptosis-inducing factor protein (AIF), which is essential for mitochondrial complex I function [20, 21] (Fig. 2). p53, therefore, seems to favour the use of the TCA cycle for energy production, while limiting glycolytic flux. These functions would maximise energy production under conditions of nutrient deprivation while opposing the metabolic switch to aerobic glycolysis (the Warburg effect) that underpins the growth of most cancer cells. In addition, p53 plays a role in mitochondrial homeostasis by protecting mitochondrial DNA integrity and maintaining mitochondrial mass [22, 23], through mechanisms that include the activation of ribonucleotide reductase p53R2 [22, 24] and direct effects of p53 localised to the mitochondria [25].
Fig. 2.
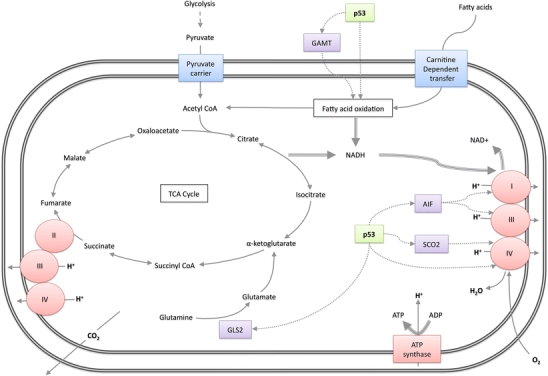
p53 and mitochondrial respiration. Basal p53 levels transcriptionally activate synthesis of cytochrome oxidase 2 (SCO2) and apoptosis-inducing factor (AIF), which support the function of mitochondrial respiratory chain complexes I, III & IV and acts directly on complex IV subunit 1. p53 transcriptionally activates glutaminase 2 (GLS2), which catalyses the conversion of glutamine to glutamate. p53 regulates FAO via transcriptional activation of guanidinoacetate aminotransferase (GAMT), and possibly by other mechanisms
Glutaminolysis
An alternative to glucose as the fuel for bioenergetic pathways is glutamine, which feeds the TCA cycle by providing α-ketoglutarate from glutamate (Fig. 2). This pathway has recently been shown to be important in cancer cells, with one isoform of the enzyme glutaminase (GLS1/KGA)—which converts glutamine to glutamate—showing the characteristics of an oncogene. Indeed, compounds inhibiting GLS1/KGA suppresses tumour growth and oncogenic transformation [26, 27]. It is intriguing, then, that p53 plays a role in the regulation of glutaminolysis by activating the expression of another isoform of glutaminsase (GLS2/LGA) [28, 29]. This activity of p53 may be important to help cells deal with periods of glucose deprivation, and GLS2 has been shown to function as a tumour suppressor, found to be down-regulated in hepatic tumours and malignant gliomas [28–30]. Why the expression of these two isoforms of GLS should have such different consequences is not quite clear, but may be related to the observation that the activation of glutaminolysis by GLS1 drives use of TCA cycle intermediates for anabolic pathways, while activation of GLS2 in response to p53 promotes ATP production and antioxidant functions [31]. This is perhaps because in addition to driving GLS2 expression p53 promotes OXPHOS, thus facilitating the conversion of TCA cycle activity into energy.
Fatty acid oxidation
When available glucose levels are low, mitochondrial fatty acid oxidation (FAO) is used to drive the TCA cycle and generate the ATP needed to meet cellular energy demands [32]. Consistent with a role for p53 in promoting the use of alternative energy sources when glucose is lacking, p53 can promote FAO in cultured cells during glucose starvation [33]. Evidence for the general role of p53 in facilitating FAO as an alternative energy source is provided by in vivo studies where starvation of mice expressing wild-type p53 led to increased FAO, whereas p53-null animals had lower basal FAO that was not elevated in response to starvation [34]. The p53-dependent regulation of FAO occurs in part through the activation of guanidinoacetate methyltransferase (GAMT) activity, although in this context, p53-dependent activation of GAMT provides energy to facilitate apoptosis (an energy-dependent process) rather than cell survival.
Autophagy
In the absence of an adequate exogenous nutrient supply, autophagy (i.e. macroautophagy)—which is a process of controlled lysosomal degradation of organelles and proteins—can be engaged to replenish metabolic reserves and promote cell survival [35]. Interestingly, p53 has been shown to both promote and inhibit autophagy [33, 36–38]. Recent evidence suggests that the ability to enhance or suppress autophagy (by regulating autophagy protein LC3) allows p53 signalling to provide the most appropriate cell survival strategy during nutrient starvation [39]. Thus, cells with low autophagic rates show a p53-dependent increase in autophagy in response to starvation, whereas cells with high autophagic rate show a p53-dependent decrease in autophagy in response to the same conditions—the end result in both cases is the promotion of cell survival [39].
Oxidative stress & antioxidant response
Reactive oxygen species (ROS) are constantly produced during normal metabolism (especially OXPHOS), and while low levels of ROS can be pro-proliferative, excess ROS can damage DNA and proteins with potential to contribute to aging, cardiac disease, cancer and other pathologies [40]. Cells engage a range of pathways to eliminate ROS, or modulate ROS levels to facilitate essential cellular tasks such as apoptosis. While the apoptotic response to p53 is linked to p53-dependent elevation of ROS, basal p53 expression drives a number of antioxidant responses that can limit oxidative stress. p53 induces a range of antioxidant targets such as GPX1, MnSOD, ALDH4 and TPP53INP1 [41, 42]. The diversion of glucose-6-phosphate into the oxidative PPP by TIGAR produces NADPH, which acts as a co-factor in the production of the antioxidant, reduced glutathione (GSH) [10]. p53-dependent induction of GLS2 expression upregulates the production of the GSH precursor glutamate, again contributing to antioxidant activity [28, 29]. The Sestrins, a family of stress-responsive proteins involved in the regulation of ROS [43], are also induced by p53.
It is likely that under normal conditions (which can be considered to provide basal levels oxidative stress), or mild stress that p53 augments the antioxidant response, protecting cells from potential oxidative damage, including that generated by p53-dependent activation of OXPHOS. However, under severe stress, p53 utilises its ability to promote ROS to facilitate apoptosis [44].
Regulation of cell growth
The ability of p53 to inhibit cell cycle progression was one of the first p53-driven responses to be identified [45, 46], and the activation of this response has been shown to play an important role in allowing cell survival under conditions of glucose deprivation [47]. Recent evidence indicates that p53 can also prevent cell growth, illustrating another facet of the ability of p53 to coordinate an orderly response to nutrient stress. The functions of p53 in the regulation of cell growth reflect a substantial interaction with the mTOR pathway, which coordinates cell growth by sensing nutrient availability and growth factor signalling and balancing anabolic and catabolic processes [48].
AMP-activated protein kinase (AMPK) is a key cellular fuel sensor. Detection of low energy through the ability of falling ATP:AMP ratios to activate AMPK is a pivotal step in mounting a metabolic stress response, which includes inhibition of mTOR and activation of p53. AMPK can induce p53 by promoting phosphorylation on serine-15, a site known to be important for the activation of p53 (although p53 is not necessarily a direct target of AMPK) [47, 49, 50]. Mice expressing p53 with a serine-phosphorylation site mutation display increased metabolic stress and severely defective glucose metabolism [51]. Prolonged culture of p53-proficient cells in the complete absence of glucose leads to p53 serine-46 phosphorylation and p53-dependent apoptosis [52]. This suggests that p53 establishes a cut-off point at which metabolic stress becomes too extreme to warrant pro-survival responses, perhaps associated with serine-46 phosphorylation. Further signals downstream of mTOR can also modulate p53 through the signalling of ribosomal S6K to MDM2, the major ubiquitin ligase that controls p53 stability [53] (Fig. 3). The ability of other ribosomal proteins to control p53 through this route [54] is also likely to play a key role in the response to metabolic stress. Other mechanisms by which p53 is activated in response to metabolic stress have been described, including nucleotide deficiency triggered by inhibition of dihydroorotate dehydrogenase, which catalyses mitochondrial respiratory chain-linked pyrimidine synthesis [55].
Fig. 3.
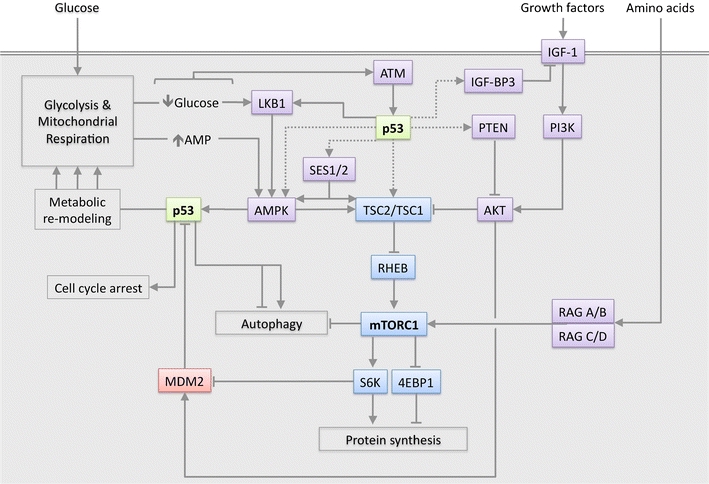
Signalling between p53 and the IGF-AKT-mTORC1 pathway. p53 is activated by metabolic stress signals via AMPK, ATM and mTORC1-S6K-MDM2 signalling, and modulates energy metabolism, cell cycle and autophagy. p53 can inhibit mTORC1 signalling via transcriptional activation of PTEN, IGF-BP3, Sestrin1 & Sestrin2 (SES1/2), TSC2 and AMPKβ. Direct interaction of p53 with LKB1 may directly lead to AMPK activation. Dotted lines indicate transcriptional regulation, unbroken lines indicate a direct protein level effect, arrowheads indicate an activating signal, blunt-ended lines indicate an inhibitory signal
Importantly, the relationship between p53 and AMPK is reciprocal. Genotoxic stress causes p53-dependent suppression of mTOR activity via AMPK and TSC1/TSC2, leading to the suggestion that AMPK is phosphorylated in response to p53 activation, potentially via the interaction of p53 with the AMPK-activating protein LKB1 [50]. A more recent study demonstrates that p53 transcriptionally activates the mTOR suppressing proteins AMPKβ, TSC2, IGF-BP3 and PTEN in response to genotoxic stress [56]. The antioxidant response of p53 also inhibits the mTOR pathway via the Sestrins, which are induced by p53 and can interact with mTOR pathway suppressors AMPK, TSC1 and TSC2 [43]. However, p53 does not always inhibit mTOR in response to metabolic stress [47], suggesting that protein synthesis may aid in metabolic remodelling. Overall, the interaction between p53 signalling and the mTOR pathway is substantial [57], indicating the capacity of these pathways to cooperate (Fig. 3).
Cancer
While the Warburg effect, defined as a high rate of aerobic glycolysis, is a frequent characteristic of cancer cells, it is becomingly increasingly apparent that cancers have a diverse range of metabolic profiles with many relying primarily on OXPHOS for ATP production [58]. Despite this diversity, it is fair to say that rapidly proliferating cancer cells are likely to utilise large amounts of glucose to fuel anabolism and are more likely to display the Warburg effect than not [58, 59]. Inactivation of p53 occurs in over half of all cancers and from a metabolic perspective, loss of p53 signalling represents a double-edged sword. As p53 suppresses glycolysis and promotes OXPHOS, its inactivation serves to promote the Warburg effect. On the other hand, cancer cells must also undergo metabolic adaptation, and loss of p53 would impede this process. Therefore, loss of p53 may initially serve to drive metabolism in support of tumourigenesis via the Warburg effect, but once a tumour is established, p53-deficiency may sensitise tumour cells to metabolic stress. Although studies on cell lines and animal models support these conclusions, more direct evidence from p53-deficient tumours will be needed to qualify these predictions. We anticipate that while loss of p53 would generally favour aerobic glycolysis, the overall metabolic profile of tumours is likely to reflect the combination of many factors, including the contribution of other mutations, tissue of origin and microenvironment.
Clinically, the inability of p53-deficient cells to survive metabolic stress may have already been inadvertently exploited by the use of anti-diabetic drugs such as metformin. Diabetics treated with metformin have reduced risk of cancer compared to those who have not received the drug [60, 61]. In mice, metformin suppresses the growth of p53-deficient xenografts, but does not inhibit the growth of p53-proficent tumours [33]. In the quest for new anti-cancer agents, anti-diabetic drugs are clearly a good starting point for generating compounds active against p53-deficient tumours. It also seems prudent to perform additional retrospective studies of cancer rates in patients treated with anti-diabetic drugs and other established drugs that modulate cellular metabolism.
Induction of wild-type p53 by compounds such as the Nutlins has been used to inhibit tumour growth in cancer models [62]. However, this approach may run the risk of enhancing the pro-survival metabolic adaptation functions of p53 in some tumours. Indeed, the presence of wild-type p53 in breast cancers has been associated with reduced therapeutic response and poor prognosis in at least one study [63]. Elucidating the pathways by which p53 coordinates metabolic adaptation could establish new therapeutic targets in cancers that express wild-type p53. In simple terms, understanding what p53-proficent cells can do (in terms of metabolic adaptation) and what p53-deficient cells cannot do, could provide therapeutic opportunities in each type of tumour. It is also possible that p53 isoforms have specific metabolic functions that promote metabolic adaptation or tumour suppression, a possibility that warrants further investigation. Further complexity is added by the observation that loss of wild-type p53 function in many cancers is the result of a point mutation within the TP53 gene, leading to the expression of a mutant p53 protein. While these tumour-derived p53 mutants fail to exhibit wild-type p53 activity, they have also been shown to gain functions that can contribute to tumour invasion and metastasis [64]. Although not fully explored, it seems possible that these mutant p53s may also show new activities in the control of metabolism, or the response to metabolic stress.
Aging
p53 is thought to have an important but ill defined role in the aging process, with evidence that p53 expression can oppose aging and promote longevity [65]. The ability to promote the anti-oxidant response and inhibit the mTOR pathway are clear mechanisms through which p53 could suppress processes thought to promote aging [40, 41]. A more responsive p53 pathway has been shown to extend the lifespan of mice [66]. Therefore, it has been suggested that enhancing p53 expression via MDM2 modulation could combat aging [65]. Although this must be balanced with clear evidence showing slightly enhanced levels of p53 in normal tissues strongly promotes aging [67]. The ability of p53 to control autophagy and senescence may also play a role in the regulation of aging, and interestingly, mTOR activity has been shown to cooperate with p53 to switch the response to p53-activation from quiescence to senescence [68–70]. While still in speculation, it is interesting to consider how p53 expression and mTOR inhibition (a result of caloric restriction) could cooperate to promote longevity.
Other diseases
A number of recent studies link p53 to a diverse range of physiological processes and diseases, including diabetes, central nervous system disorders, obesity and alcoholic liver disease. A role for p53 in the central nervous system is demonstrated by its ability to transcriptionally activate brain-expressed ring finger protein (BERP), which interacts with GABA receptors to modulate seizure susceptibility [71]. With the ability to regulate apoB and apobec1, p53 has the potential to modulate lipid transport from the intestine to the liver [72]. Mice expressing a p53 mutant that cannot be activated by serine-15 phosphorylation have increased metabolic stress and severe defects in glucose homeostasis. Animals develop glucose intolerance and insulin resistance that correlates with reduced antioxidant gene expression and reduced insulin signalling [51]. p53 also has a role in adipogenesis and protection of adipocytes from lipotoxicity, leading to the description of p53 as ‘guardian of corpulence’ [73]. In a rat model of chronic alcohol consumption, modulation of TIGAR expression and apoptosis by p53 contributes to the metabolic abnormalities associated with hepatic steatosis [74].
Conclusions
Reviewing the role of p53 in cellular metabolism provides crucial insights into p53 biology. p53 is emerging as a key regulator of metabolic homeostasis, an observation that throws up a wider conceptual issue: Which is the primary function of p53? We now know that p53 has many activities, including tumour suppression, metabolic control, maintenance of fecundity and the regulation of stem cells. It seems likely that each of the roles of p53 is intertwined in a way that will make it difficult to untangle them—and indeed, part of the confusion may be caused by a semantic rather than a biological conflict. However, the fact that p53 is mutated in roughly 50% of cancers, but is retained as wild type in the other 50%, makes understanding how the presence or absence of p53 might affect tumour development and therapeutic response an enticing and ever-more achievable goal. Furthermore, the role of p53 in cellular homeostasis explains its involvement in such a wide range of physiological processes and diseases.
Acknowledgments
We thank Dan Tennant and Arnaud Vigneron for critically reading the manuscript and Cancer Research UK for funding.
Conflict of interests
None.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
An erratum to this article can be found at http://dx.doi.org/10.1007/s00109-011-0745-3
References
- 1.Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 2.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tong X, Zhao F, Thompson CB. The molecular determinants of de novo nucleotide biosynthesis in cancer cells. Curr Opin Genet Dev. 2009;19:32–37. doi: 10.1016/j.gde.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, Martinez D, Carnero A, Beach D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65:177–185. [PubMed] [Google Scholar]
- 5.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- 6.Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004;64:2627–2633. doi: 10.1158/0008-5472.CAN-03-0846. [DOI] [PubMed] [Google Scholar]
- 7.Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008;10:611–618. doi: 10.1038/ncb1724. [DOI] [PubMed] [Google Scholar]
- 8.Kawauchi K, Araki K, Tobiume K, Tanaka N. Activated p53 induces NF-kappaB DNA binding but suppresses its transcriptional activation. Biochem Biophys Res Commun. 2008;372:137–141. doi: 10.1016/j.bbrc.2008.05.021. [DOI] [PubMed] [Google Scholar]
- 9.Webster NJ, Resnik JL, Reichart DB, Strauss B, Haas M, Seely BL. Repression of the insulin receptor promoter by the tumor suppressor gene product p53: a possible mechanism for receptor overexpression in breast cancer. Cancer Res. 1996;56:2781–2788. [PubMed] [Google Scholar]
- 10.Bensaad K, Tsuruta A, Selak MA, Vidal MNC, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 11.Tong X, Zhao F, Mancuso A, Gruber JJ, Thompson CB. The glucose-responsive transcription factor ChREBP contributes to glucose-dependent anabolic synthesis and cell proliferation. Proc Natl Acad Sci USA. 2009;106:21660–21665. doi: 10.1073/pnas.0911316106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruiz-Lozano P, Hixon ML, Wagner MW, Flores AI, Ikawa S, Baldwin AS, Chien KR, Gualberto A. p53 is a transcriptional activator of the muscle-specific phosphoglycerate mutase gene and contributes in vivo to the control of its cardiac expression. Cell Growth Differ. 1999;10:295–306. [PubMed] [Google Scholar]
- 13.Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer's double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777–4786. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wade M, Tsernoglou D, Hill E, Webb L, Banaszak L. Chemical and structural relationships of NAD + and platinum binding to malate dehydrogenase. Biochim Biophys Acta. 1973;322:124–132. doi: 10.1016/0005-2795(73)90182-7. [DOI] [PubMed] [Google Scholar]
- 15.Grant PM, Roderick SL, Grant GA, Banaszak LJ, Strauss AW. Comparison of the precursor and mature forms of rat heart mitochondrial malate dehydrogenase. Biochemistry. 1987;26:128–134. doi: 10.1021/bi00375a019. [DOI] [PubMed] [Google Scholar]
- 16.Lee SM, Kim JH, Cho EJ, Youn HD. A nucleocytoplasmic malate dehydrogenase regulates p53 transcriptional activity in response to metabolic stress. Cell Death Differ. 2009;16:738–748. doi: 10.1038/cdd.2009.5. [DOI] [PubMed] [Google Scholar]
- 17.Dang CV. PKM2 tyrosine phosphorylation and glutamine metabolism signal a different view of the Warburg effect. Sci Signal. 2009;2:pe75. doi: 10.1126/scisignal.297pe75. [DOI] [PubMed] [Google Scholar]
- 18.Matoba S, Kang J-G, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 19.Okamura S, Ng CC, Koyama K, Takei Y, Arakawa H, Monden M, Nakamura Y. Identification of seven genes regulated by wild-type p53 in a colon cancer cell line carrying a well-controlled wild-type p53 expression system. Oncol Res. 1999;11:281–285. [PubMed] [Google Scholar]
- 20.Vahsen N, Cande C, Briere J-J, Benit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, et al. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stambolsky P, Weisz L, Shats I, Klein Y, Goldfinger N, Oren M, Rotter V. Regulation of AIF expression by p53. Cell Death Differ. 2006;13:2140–2149. doi: 10.1038/sj.cdd.4401965. [DOI] [PubMed] [Google Scholar]
- 22.Kulawiec M, Ayyasamy V, Singh KK. p53 regulates mtDNA copy number and mitocheckpoint pathway. J Carcinog. 2009;8:8. doi: 10.4103/1477-3163.50893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lebedeva MA, Eaton JS, Shadel GS. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochim Biophys Acta. 2009;1787:328–334. doi: 10.1016/j.bbabio.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bourdon A, Minai L, Serre V, Jais J-P, Sarzi E, Aubert S, Chretien D, de Lonlay P, Paquis-Flucklinger V, Arakawa H, et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39:776–780. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- 25.Nantajit D, Fan M, Duru N, Wen Y, Reed JC, Li JJ. Cyclin B1/Cdk1 phosphorylation of mitochondrial p53 induces anti-apoptotic response. PLoS ONE. 2010;5:e12341. doi: 10.1371/journal.pone.0012341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J-B, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio ALB, Dias SMG, Dang CV, Cerione RA. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18:207–219. doi: 10.1016/j.ccr.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS, Rabinowitz JD, Dang CV, Riggins GJ. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70:8981–8987. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA. 2010;107:7455–7460. doi: 10.1073/pnas.1001006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA. 2010;107:7461–7466. doi: 10.1073/pnas.1002459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szeliga M, Obara-Michlewska M, Matyja E, Lazarczyk M, Lobo C, Hilgier W, Alonso FJ, Marquez J, Albrecht J. Transfection with liver-type glutaminase cDNA alters gene expression and reduces survival, migration and proliferation of T98G glioma cells. Glia. 2009;57:1014–1023. doi: 10.1002/glia.20825. [DOI] [PubMed] [Google Scholar]
- 31.Vousden KH. Alternative fuel–another role for p53 in the regulation of metabolism. Proc Natl Acad Sci USA. 2010;107:7117–7118. doi: 10.1073/pnas.1002656107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jensen MD. Fate of fatty acids at rest and during exercise: regulatory mechanisms. Acta Physiol Scand. 2003;178:385–390. doi: 10.1046/j.1365-201X.2003.01167.x. [DOI] [PubMed] [Google Scholar]
- 33.Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–6752. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- 34.Ide T, Brown-Endres L, Chu K, Ongusaha PP, Ohtsuka T, El-Deiry WS, Aaronson SA, Lee SW. GAMT, a p53-inducible modulator of apoptosis, is critical for the adaptive response to nutrient stress. Mol Cell. 2009;36:379–392. doi: 10.1016/j.molcel.2009.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 37.Yee KS, Wilkinson S, James J, Ryan KM, Vousden KH. PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ. 2009;16:1135–1145. doi: 10.1038/cdd.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C, Harper F, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scherz-Shouval R, Weidberg H, Gonen C, Wilder S, Elazar Z, Oren M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci USA. 2010;107:18511–18516. doi: 10.1073/pnas.1006124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Essick EE, Sam F. Oxidative stress and autophagy in cardiac disease, neurological disorders, aging and cancer. Oxid Med Cell Longev. 2010;3:168–177. doi: 10.4161/oxim.3.3.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Budanov AV, Lee JH, Karin M. Stressin' Sestrins take an aging fight. EMBO Mol Med. 2010;2:388–400. doi: 10.1002/emmm.201000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pani G, Galeotti T. Role of MnSOD and p66shc in mitochondrial response to p53. Antioxid Redox Signal. 2011 doi: 10.1089/ars.2010.3499. [DOI] [PubMed] [Google Scholar]
- 43.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bensaad K, Vousden KH. Savior and slayer: the two faces of p53. Nat Med. 2005;11:1278–1279. doi: 10.1038/nm1205-1278. [DOI] [PubMed] [Google Scholar]
- 45.Kastan MB, Zhan Q, El-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 46.El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-P. [DOI] [PubMed] [Google Scholar]
- 47.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 48.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 49.Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. Cell cycle regulation via p53 phosphorylation by a 5′-AMP activated protein kinase activator, 5-aminoimidazole- 4-carboxamide-1-beta-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem Biophys Res Commun. 2001;287:562–567. doi: 10.1006/bbrc.2001.5627. [DOI] [PubMed] [Google Scholar]
- 50.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Armata HL, Golebiowski D, Jung DY, Ko HJ, Kim JK, Sluss HK. Requirement of the ATM/p53 tumor suppressor pathway for glucose homeostasis. Mol Cell Biol. 2010;30:5787–5794. doi: 10.1128/MCB.00347-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okoshi R, Ozaki T, Yamamoto H, Ando K, Koida N, Ono S, Koda T, Kamijo T, Nakagawara A, Kizaki H. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J Biol Chem. 2008;283:3979–3987. doi: 10.1074/jbc.M705232200. [DOI] [PubMed] [Google Scholar]
- 53.Lai KP, Leong WF, Chau JFL, Jia D, Zeng L, Liu H, He L, Hao A, Zhang H, Meek D, et al. S6K1 is a multifaceted regulator of Mdm2 that connects nutrient status and DNA damage response. EMBO J. 2010;29:2994–3006. doi: 10.1038/emboj.2010.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell. 2009;16:369–377. doi: 10.1016/j.ccr.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khutornenko AA, Roudko VV, Chernyak BV, Vartapetian AB, Chumakov PM, Evstafieva AG. Pyrimidine biosynthesis links mitochondrial respiration to the p53 pathway. Proc Natl Acad Sci USA. 2010;107:12828–12833. doi: 10.1073/pnas.0910885107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67:3043–3053. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- 57.Feng Z, Levine AJ. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol. 2010;20:427–434. doi: 10.1016/j.tcb.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jose C, Bellance N, Rossignol R. Choosing between glycolysis and oxidative phosphorylation: a tumor's dilemma. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbabio.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 59.Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134:703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 60.Evans JMM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29:254–258. doi: 10.2337/diacare.29.02.06.dc05-1558. [DOI] [PubMed] [Google Scholar]
- 62.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 63.Bertheau P, Espie M, Turpin E, Lehmann J, Plassa L-F, Varna M, Janin A, de The H. TP53 status and response to chemotherapy in breast cancer. Pathobiology. 2008;75:132–139. doi: 10.1159/000123851. [DOI] [PubMed] [Google Scholar]
- 64.Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139:1327–1341. doi: 10.1016/j.cell.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 65.Poyurovsky MV, Prives C. P53 and aging: a fresh look at an old paradigm. Aging. 2010;2:380–382. doi: 10.18632/aging.100179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C, Flores JM, Vina J, Blasco MA, Serrano M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007;448:375–379. doi: 10.1038/nature05949. [DOI] [PubMed] [Google Scholar]
- 67.Donehower LA. Using mice to examine p53 functions in cancer, aging, and longevity. Cold Spring Harb Perspect Biol. 2009;1:a001081. doi: 10.1101/cshperspect.a001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci USA. 2010;107:9660–9664. doi: 10.1073/pnas.1002298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging. 2010;2:344–352. doi: 10.18632/aging.100160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8:1888–1895. doi: 10.4161/cc.8.12.8606. [DOI] [PubMed] [Google Scholar]
- 71.Cheung CC, Yang C, Berger T, Zaugg K, Reilly P, Elia AJ, Wakeham A, You-Ten A, Chang N, Li L, et al. Identification of BERP (brain-expressed RING finger protein) as a p53 target gene that modulates seizure susceptibility through interacting with GABA(A) receptors. Proc Natl Acad Sci USA. 2010;107:11883–11888. doi: 10.1073/pnas.1006529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ashur-Fabian O, Har-Zahav A, Shaish A, Amram HW, Margalit O, Weizer-Stern O, Dominissini D, Harats D, Amariglio N, Rechavi G. apoB and apobec1, two genes key to lipid metabolism, are transcriptionally regulated by p53. Cell Cycle. 2010;9:3761–3770. doi: 10.4161/cc.9.18.12993. [DOI] [PubMed] [Google Scholar]
- 73.Buzuine M, Stenkula KG, Cam M, Arroyo M, Cushman SW. Guardian of Corpulence: a hypothesis on p53 signaling in the fat cell. Clin Lipidol. 2009;4:231–243. doi: 10.2217/clp.09.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Derdak Z, Lang CH, Villegas KA, Tong M, Mark NM, de la Monte SM, Wands JR. Activation of p53 enhances apoptosis and insulin resistance in a rat model of alcoholic liver disease. J Hepatol. 2011;54:164–172. doi: 10.1016/j.jhep.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]