

Abstract
Despite curative locoregional treatments for hepatocellular carcinoma (HCC), tumour recurrence rates remain high. The current study was designed to assess the safety and bioactivity of infusion of dendritic cells (DCs) stimulated with OK432, a streptococcus-derived anti-cancer immunotherapeutic agent, into tumour tissues following transcatheter hepatic arterial embolization (TAE) treatment in patients with HCC. DCs were derived from peripheral blood monocytes of patients with hepatitis C virus-related cirrhosis and HCC in the presence of interleukin (IL)-4 and granulocyte-macrophage colony-stimulating factor and stimulated with 0·1 KE/ml OK432 for 2 days. Thirteen patients were administered with 5 × 106 of DCs through arterial catheter during the procedures of TAE treatment on day 7. The immunomodulatory effects and clinical responses were evaluated in comparison with a group of 22 historical controls treated with TAE but without DC transfer. OK432 stimulation of immature DCs promoted their maturation towards cells with activated phenotypes, high expression of a homing receptor, fairly well-preserved phagocytic capacity, greatly enhanced cytokine production and effective tumoricidal activity. Administration of OK432-stimulated DCs to patients was found to be feasible and safe. Kaplan–Meier analysis revealed prolonged recurrence-free survival of patients treated in this manner compared with the historical controls (P = 0·046, log-rank test). The bioactivity of the transferred DCs was reflected in higher serum concentrations of the cytokines IL-9, IL-15 and tumour necrosis factor-α and the chemokines CCL4 and CCL11. Collectively, this study suggests that a DC-based, active immunotherapeutic strategy in combination with locoregional treatments exerts beneficial anti-tumour effects against liver cancer.
Keywords: dendritic cells, hepatocellular carcinoma, immunotherapy, recurrence-free survival, transcatheter hepatic arterial embolization
Introduction
Many locoregional therapeutic approaches including surgical resection, radiofrequency ablation (RFA) and transcatheter hepatic arterial embolization (TAE) have been taken in the search for curative treatments of hepatocellular carcinoma (HCC). Despite these efforts, tumour recurrence rates remain high [1,2], probably because active hepatitis and cirrhosis in the surrounding non-tumour liver tissues causes de novo development of HCC [3,4]. One strategy to reduce tumour recurrence is to enhance anti-tumour immune responses that may induce sufficient inhibitory effects to prevent tumour cell growth and survival [5,6]. Dendritic cells (DCs) are the most potent type of antigen-presenting cells in the human body, and are involved in the regulation of both innate and adaptive immune responses [7]. DC-based immunotherapies are believed to contribute to the eradication of residual and recurrent tumour cells.
To enhance tumour antigen presentation to T lymphocytes, DCs have been transferred with major histocompatibility complex (MHC) class I and class II genes [8] and co-stimulatory molecules, e.g. CD40, CD80 and CD86 [9,10], and loaded with tumour-associated antigens, including tumour lysates, peptides and RNA transfection [11]. To induce natural killer (NK) and natural killer T (NK T) cell activation, DCs have been stimulated and modified to produce larger amounts of cytokines, e.g. interleukin (IL)-12, IL-18 and type I interferons (IFNs)[10,12]. Furthermore, DC migration into secondary lymphoid organs could be induced by expression of chemokine genes, e.g. C-C chemokine receptor-7 (CCR7) [13], and by maturation using inflammatory cytokines [14], matrix metalloproteinases and Toll-like receptor (TLR) ligands [15].
DCs stimulated with OK432, a penicillin-inactivated and lyophilized preparation of Streptococcus pyrogenes, were suggested recently to produce large amounts of T helper type 1 (Th1) cytokines, including IL-12 and IFN-γ and enhance cytotoxic T lymphocyte activity compared to a standard mixture of cytokines [tumour necrosis factor-α (TNF-α), IL-1β, IL-6 and prostaglandin E2 (PGE2)][16]. Furthermore, because OK432 modulates DC maturation through TLR-4 and the β2 integrin system [16,17] and TLR-4-stimulated DCs can abrogate the activity of regulatory T cells [18], OK432-stimulated DCs may contribute to the induction of anti-tumour immune responses partly by reducing the activity of suppressor cells. Recently, in addition to the orchestration of immune responses, OK432-activated DCs have themselves been shown to mediate strong, specific cytotoxicity towards tumour cells via CD40/CD40 ligand interactions [19].
We have reported recently that combination therapy using TAE together with immature DC infusion is safe for patients with cirrhosis and HCC [20]. DCs were infused precisely into tumour tissues and contributed to the recruitment and activation of immune cells in situ. However, this approach by itself yielded limited anti-tumour effects due probably to insufficient stimulation of immature DCs (the preparation of which seems closely related to therapeutic outcome [21,22]). The current study was designed to assess the safety and bioactivity of OK432-stimulated DC infusion into tumour tissues following TAE treatment in patients with cirrhosis and HCC. In addition to documenting the safety of this approach, we found that patients treated with OK432-stimulated DCs displayed unique cytokine and chemokine profiles and, most importantly, experienced prolonged recurrence-free survival.
Patients and methods
Patients
Inclusion criteria were a radiological diagnosis of primary HCC by computed tomography (CT) angiography, hepatitis C virus (HCV)-related HCC, a Karnofsky score of ≥ 70%, an age of ≥ 20 years, informed consent and the following normal baseline haematological parameters (within 1 week before DC administration): haemoglobin ≥ 8·5 g/dl; white cell count ≥ 2000/µl; platelet count ≥ 50 000/µl; creatinine < 1·5 mg/dl and liver damage A or B [23].
Exclusion criteria included severe cardiac, renal, pulmonary, haematological or other systemic disease associated with a discontinuation risk; human immunodeficiency virus (HIV) infection; prior history of other malignancies; history of surgery, chemotherapy or radiation therapy within 4 weeks; immunological disorders including splenectomy and radiation to the spleen; corticosteroid or anti-histamine therapy; current lactation; pregnancy; history of organ transplantation; or difficulty in follow-up.
Thirteen patients (four women and nine men) presenting at Kanazawa University Hospital between March 2004 and June 2006 were enrolled into the study, with an age range from 56 to 83 years (Table 1). Patients with verified radiological diagnoses of HCC stage II or more were eligible and enrolled in this study. In addition, a group of 22 historical controls (nine women and 13 men) treated with TAE without DC administration between July 2000 and September 2007 was included in this study. All patients received RFA therapy to increase the locoregional effects 1 week later [24]. They underwent ultrasound, computed tomography (CT) scan or magnetic resonance imaging (MRI) of the abdomen about 1 month after treatment and at a minimum of once every 3 months thereafter, and tumour recurrences were followed for up to 360 days. The Institutional Review Board reviewed and approved the study protocol. This study complied with ethical standards outlined in the Declaration of Helsinki. Adverse events were monitored for 1 month after the DC infusion in terms of fever, vomiting, abdominal pain, encephalopathy, myalgia, ascites, gastrointestinal disorder, bleeding, hepatic abscess and autoimmune diseases.
Table 1.
Patient characteristics.
| Patient no. | Gender | Age (years) | HLA | TNM stages | No. of tumours | Largest tumour (mm) | Child–Pugh | KPS | Post-TAE Rx |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 60 | A11 A33 | III | 5 | 35 | B | 100 | RFA |
| 2 | M | 57 | A11 A24 | III | 1 | 21 | B | 100 | RFA |
| 3 | M | 57 | A11 A31 | III | 2 | 39 | B | 100 | RFA |
| 4 | M | 77 | A2 A24 | III | 2 | 35 | A | 100 | RFA |
| 5 | F | 83 | A11 A24 | III | 3 | 29 | B | 100 | RFA |
| 6 | F | 74 | A2 A24 | II | 1 | 35 | A | 100 | RFA |
| 7 | F | 72 | A24 A33 | III | 3 | 41 | B | 100 | RFA |
| 8 | F | 65 | A2 A11 | II | 4 | 12 | B | 100 | RFA |
| 9 | M | 71 | A2 A11 | II | 4 | 16 | A | 100 | RFA |
| 10 | M | 79 | A11 A24 | III | 2 | 40 | A | 100 | RFA |
| 11 | M | 71 | A2 A24 | II | 1 | 28 | A | 100 | RFA |
| 12 | M | 56 | A2 A26 | III | 2 | 25 | B | 100 | RFA |
| 13 | M | 64 | A2 A33 | III | 2 | 37 | B | 100 | RFA |
M, male; F, female; TNM, tumour–node–metastasis; Child–Pugh, Child–Pugh classification; KPS, Karnofsky performance scores; TAE, transcatheter arterial embolization; Rx, treatment; HCC, hepatocellular carcinoma; HLA, human leucocyte antigen; RFA, percutaneous radiofrequency ablation.
Preparation and injection of autologous DCs
DCs were generated from blood monocyte precursors, as reported previously [25]. Briefly, peripheral blood mononuclear cells (PBMCs) were isolated by centrifugation in Lymphoprep™ Tubes (Nycomed, Roskilde, Denmark). For generating DCs, PBMCs were plated in six-well tissue culture dishes (Costar, Cambridge, MA, USA) at 1·4 × 107 cells in 2 ml per well and allowed to adhere to plastic for 2 h. Adherent cells were cultured in serum-free media (GMP CellGro® DC Medium; CellGro, Manassas, VA, USA) with 50 ng/ml recombinant human IL-4 (GMP grade; CellGro®) and 100 ng/ml recombinant human granulocyte–macrophage colony-stimulating factor (GM-CSF) (GMP grade; CellGro®) for 5 days to generate immature DC, and matured for a further 2 days in 0·1 KE/ml OK432 (Chugai Pharmaceuticals, Tokyo, Japan) to generate OK-DC. On day 7, the cells were harvested for injection, 5 × 106 cells were suspended in 5 ml normal saline containing 1% autologous plasma, mixed with absorbable gelatin sponge (Gelfoam; Pharmacia & Upjohn, Peapack, NJ, USA) and infused through an arterial catheter following Lipiodol (iodized oil) (Lipiodol Ultrafluide, Laboratoire Guerbet, Aulnay-Sous-Bois, France) injection during selective TAE therapy. Release criteria for DCs were viability > 80%, purity > 30%, negative Gram stain and endotoxin polymerase chain reaction (PCR) and negative in process cultures from samples sent 48 h before release. All products met all release criteria, and the DCs had a typical phenotype of CD14- and human leucocyte antigen (HLA)-DR+.
Flow cytometry analysis
The DC preparation was assessed by staining with the following monoclonal antibodies for 30 min on ice: anti-lineage cocktail 1 (lin-1; CD3, CD14, CD16, CD19, CD20 and CD56)-fluorescein isothiocyanate (FITC), anti-HLA-DR-peridinin chlorophyll protein (PerCP) (L243), anti-CCR7-phycoerythrin (PE) (3D12) (BD PharMingen, San Diego, CA, USA), anti-CD80-PE (MAB104), anti-CD83-PE (HB15a) and anti-CD86-PE (HA5.2B7) (Beckman Coulter, Fullerton, CA, USA). Cells were analysed on a fluorescence activated cell sorter (FACS0Calibur™ flow cytometer. Data analysis was performed with CELLQuest™ software (Becton Dickinson, San Jose, CA, USA).
DC phagocytosis
Immature DCs and OK432-stimulated DCs were incubated with 1 mg/ml FITC dextran (Sigma-Aldrich, St Louis, MO, USA) for 30 min at 37°C and the cells were washed three times in FACS buffer before cell acquisition using a FACSCalibur™ cytometer. Control DCs (not incubated with FITC dextran) were acquired at the same time to allow background levels of fluorescence to be determined.
Enzyme-linked immunosorbent assay (ELISA)
DCs were seeded at 200 000 cells/ml, and supernatant collected after 48 h. IL-12p40 and IFN-γ were detected using matched paired antibodies (BD Pharmingen) following standard protocols.
Cytotoxicity assays
The ability of DCs to exert cytotoxicity was assessed in a standard 51Cr release assay [19]. We used the HCC cell lines Hep3B and PLC/PRF/5 [American Type Culture Collection (ATCC), Manassas, VA, USA] and a lymphoblastoid cell line T2 that expresses HLA-A*0201 (ATCC) as target cells. Target cells were labelled with 51Cr. In a 96-well plate, 2·5 × 103 target cells per well were incubated with DCs for 8 h at different effector/target (E/T) ratios in triplicate. Percentage of specific lysis was calculated as follows: (experimental release − spontaneous release)/(maximum release − spontaneous release) × 100. Spontaneous release was always < 20% of the total.
NK cell activity
NK cell cytotoxicity against K562 erythroleukemia target cells was measured by using 51Cr-release assay, according to previously published methods [26], with PBMCs obtained from the patients. All experiments were performed in triplicate. Percentage of cytotoxicity was calculated as follows: {[experimental counts per minute (cpm) − spontaneous cpm]/[total cpm − spontaneous cpm]} × 100.
Intracellular cytokine expression
Freshly isolated PBMCs were stimulated with 25 ng/ml phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich) and 1 µg/ml ionomycin (Sigma-Aldrich) at 37°C in humidified 7% CO2 for 4 h. To block cytokine secretion, brefeldin A (Sigma) [27] was added to a final concentration of 10 µg/ml. After addition of stimuli, the surface staining was performed with anti-CD4-PC5 (13B8·2), anti-CD8-PerCP (SK1) and anti-CD56-PC5 (N901) (Beckman Coulter). Subsequently, the cells were permeabilized, stained for intracellular IFN-γ and IL-4 using the FastImmune™ system (BD Pharmingen), resuspended in phosphate-buffered saline (PBS) containing 1% paraformaldehyde (PFA), and analysed on a flow cytometer (≈ 10 000 gated events acquired per sample).
IFN-γ enzyme-linked immunospot (ELISPOT) assay
ELISPOT assays were performed as described previously with the following modifications [28–30]. HLA-A24 restricted peptide epitopes, squamous cell carcinoma antigen recognized by T cells 2 (SART2)899 (SYTRLFLIL), SART3109 (VYDYNCHVDL), multi-drug resistance protein 3 (MRP3)765 (VYSDADIFL), MRP3503 (LYAWEPSFL), MRP3692 (AYVPQQAWI), alpha-fetoprotein (AFP)403 (KYIQESQAL), AFP434 (AYTKKAPQL), AFP357 (EYSRRHPQL), human telomerase reverse transcriptase (hTERT)167 (AYQVCGPPL) (unpublished), hTERT461 (VYGFVRACL) and hTERT324 (VYAETKHFL) were used in this study. Negative controls consisted of an HIV envelope-derived peptide (HIVenv584). Positive controls consisted of 10 ng/ml PMA (Sigma) or a CMV pp65-derived peptide (CMVpp65328). The coloured spots were counted with a KS ELISPOT Reader (Zeiss, Tokyo, Japan). The number of specific spots was determined by subtracting the number of spots in the absence of antigen from the number of spots in its presence. Responses were considered positive if more than 10 specific spots were detected and if the number of spots in the presence of antigen was at least twofold greater than the number of spots in the absence of antigen.
Cytokine and chemokine profiling
Serum cytokine and chemokine levels were measured using the Bioplex assay (Bio-Rad, Hercules, CA, USA). Briefly, frozen serum samples were thawed at room temperature, diluted 1:4 in sample diluents, and 50 µl aliquots of diluted sample were added in duplicate to the wells of a 96-well microtitre plate containing the coated beads for a validated panel of 27 human cytokines and chemokines (cytokine 27-plex antibody bead kit) according to the manufacturer's instructions. These included IL-1β, IL-1Ra, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12p70, IL-13, IL-15, IL-17, basic fibroblast growth factor (FGF), eotaxin, G-CSF, GM-CSF, IFN-γ, interferon gamma-induced protein (IP)-10, monocyte chemoattractant protein (MCP)-1, MIP-1α, MIP-1β, platelet-derived growth factor (PDGF)-BB, regulated upon activation normal T cell-expressed and secreted (RANTES), TNF-α and vascular endothelial growth factor (VEGF). Eight standards (ranging from 2 to 32 000 pg/ml) were used to generate calibration curves for each cytokine. Data acquisition and analysis were performed using Bio-Plex Manager software version 4.1.1.
Arginase activity
Serum samples were tested for arginase activity by conversion of l-arginine to l-ornithine [31] using a kit supplied by the manufacturer (BioAssay Systems, Hayward, CA, USA). Briefly, sera were treated with a membrane filter (Millipore, Billerica, MA, USA) to remove urea, combined with the sample buffer in wells of a 96-well plate, and incubated at 37°C for 2 h. Subsequently, the urea reagent was added to stop the arginase reaction. The colour produced was read at 520 nm using a microtitre plate reader.
Statistical analysis
Results are expressed as means ± standard deviation (s.d.). Differences between groups were analysed for statistical significance by the Mann–Whitney U-test. Qualitative variables were compared by means of Fisher's exact test. The estimated probability of tumour recurrence-free survival was determined using the Kaplan–Meier method. The Mantel–Cox log-rank test was used to compare curves between groups. Any P-values less than 0·05 were considered statistically significant. All statistical tests were two-sided.
Results
Preparation of OK432-stimulated DCs
Adherent cells isolated from PBMCs of patients with cirrhosis and HCC (Table 1) were differentiated into DCs in the presence of IL-4 and GM-CSF. The cells were stimulated with 0·1 KE/ml OK432 for 3 days; 54·6 ± 9·5% (mean ± s.d.; n = 13) of OK432-stimulated cells showed high levels of MHC class II (HLA-DR) and the absence of lineage markers including CD3, CD14, CD16, CD19, CD20 and CD56, in which 30·9 ± 14·2% were CD11c-positive (myeloid DC subset) and 14·8 ± 11·2 were CD123-positive (plasmacytoid DC subset), consistent with our previous observations [20]. As reported [32,33], greater proportions of the cells developed high levels of expression of the co-stimulatory molecules B7-1 (CD80) and B7-2 (CD86) and an activation marker (CD83) compared to DCs prepared without OK432 stimulation (Fig. 1a). Furthermore, the chemokine receptor CCR7 which leads to homing to lymph nodes [13,34] was also induced following OK432 stimulation.
Fig. 1.
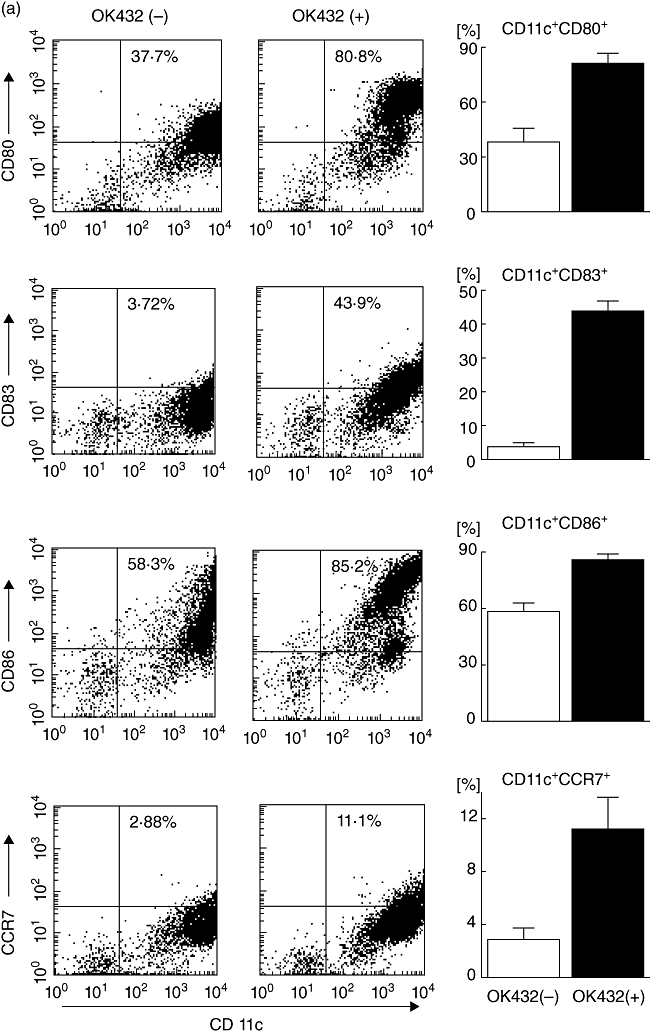
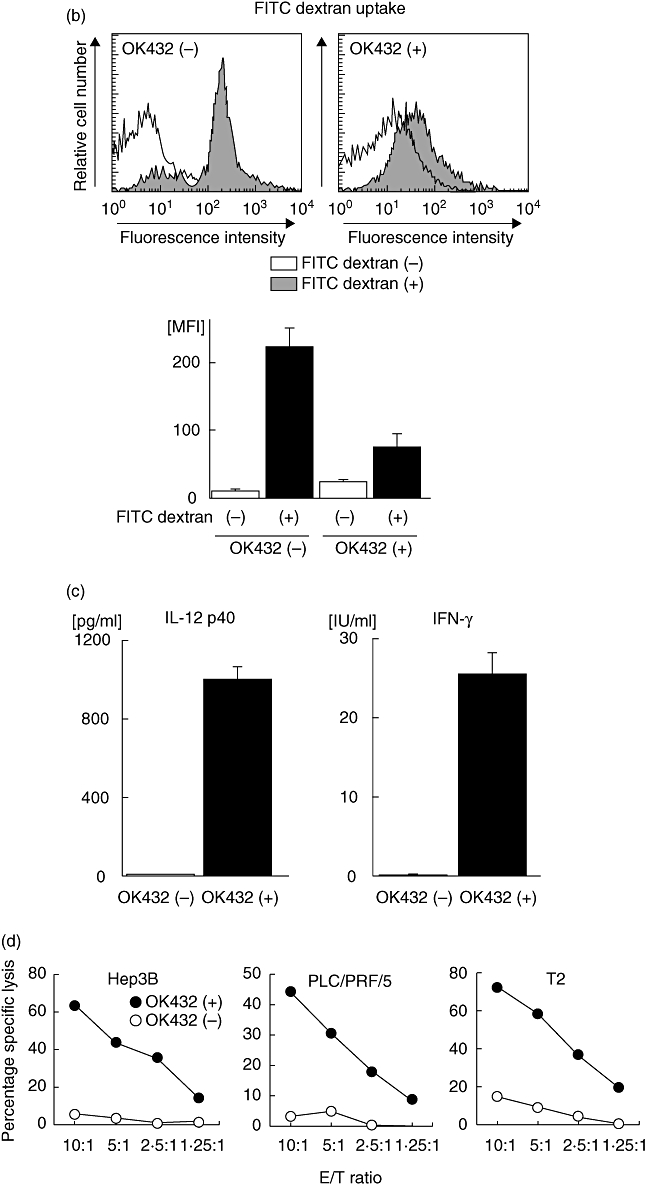
Effects of OK432 stimulation on the properties of dendritic cells (DCs) generated from blood monocyte precursors in patients with cirrhosis and hepatocellular carcinoma (HCC) (n = 13). (a) Lineage cocktail 1 (lin 1-) human leucocyte antigen D-related (HLA-DR-) subsets with [OK432(+)] and without [OK432(-)] stimulation were analysed for surface expression of CD80, CD83, CD86 and CCR7. Dot plots of a representative case are shown in the left-hand panel. Mean percentages [±standard deviation (s.d.)] of positive cells are indicated in the right-hand panel. OK432 stimulation resulted in the expression of high levels of CD80, CD83, CD86 and CCR7 in the lin 1-human leucocyte antigen D-related (HLA-DR-) DC subset. (b) DC subsets with and without OK432 stimulation were incubated with fluorescein isothiocyanate (FITC) dextran for 30 min and the uptake was determined by flow cytometry. A representative analysis is shown in the upper panel. Mean fluorescence intensities (MFIs) (±s.d.) of the positive cells are indicated in the lower panel. OK432-stimulated cells showed lower levels of uptake due to maturation. (c) DC supernatants were harvested and the concentrations of interleukin (IL)-12 and interferon (IFN)-γ measured by enzyme-linked immunosorbent assay (ELISA). OK432-stimulated cells produced large amounts of the cytokines. The data indicate means ± s.d. of the groups with and without the stimulation. All comparisons in (a–c) [OK432(+) versus OK432(-)] were statistically significant by the Mann-Whitney U-test (P < 0·005). (d) Tumoricidal activity of DCs assessed by incubation with 51Cr-labelled Hep3B, PLC/PRF/5 and T2 targets for 8 h at the indicated effector/target (E/T) cell ratios. OK432-stimulated cells displayed high cytotoxic activity against the target cells. The results are representative of the cases studied.
To evaluate the endocytic and phagocytic ability of the OK432-stimulated cells, uptake of FITC-dextran was quantitated by flow cytometry (Fig. 1b). The cells showed lower levels of uptake due to maturation compared to DCs prepared without OK432 stimulation, while the OK432-stimulated cells derived from HCC patients preserved a moderate uptake capacity. As expected, the OK432-stimulated cells produced large amounts of cytokines IL-12 and IFN-γ (Fig. 1c). In addition, they displayed high cytotoxic activity against HCC cell lines (Hep3B and PLC/PRF/5) and a lymphoblastoid cell line (T2) although DCs without OK432 stimulation lysed none of the target cells to any great degree (Fig. 1d). Taken together, these results demonstrate that OK432 stimulation of IL-4 and GM-CSF-induced immature DCs derived from HCC patients promoted their maturation towards cells with activated phenotypes, high expression of a homing receptor, fairly well-preserved phagocytic capacity, greatly enhanced cytokine production and effective tumoricidal activity, consistent with previous observations [16,19].
Safety of OK432-stimulated DC administration
Prior to the administration of OK432-stimulated DCs to patients, the cells were confirmed to be safe in athymic nude mice to which 100-fold cell numbers/weight were injected subcutaneously (data not shown). Subsequently, OK432-stimulated DC administration was performed during TAE therapy in humans, in which DCs were mixed together with absorbable gelatin sponge (Gelfoam) and infused through an arterial catheter following iodized oil (Lipiodol) injection, as reported previously [20]. Adverse events were monitored clinically and biochemically after DC infusion (Table 2). A larger proportion (12 of 13) of the patients were complicated with high fever compared to those treated previously with immature DCs (five of 10) [20], due probably to the proinflammatory responses induced by OK432-stimulated DCs. However, there were no grades III or IV National Cancer Institute Common Toxicity Criteria adverse events, including vomiting, abdominal pain, encephalopathy, myalgia, ascites, gastrointestinal disorders, bleeding, hepatic abscess or autoimmune diseases associated with DC infusion and TAE in this study. There was also no clinical or serological evidence of hepatic failure or autoimmune response in any patients. Thus, concurrent treatment with OK432-stimulated DC infusions can be performed safely at the same time as TAE in patients with cirrhosis and HCC.
Table 2.
Adverse events.
| Patient no. | Fever (days) | Vomiting | Abdominal pain | Encephalopathy | Others† |
|---|---|---|---|---|---|
| 1 | 2 | No | No | No | No |
| 2 | 2 | No | No | No | No |
| 3 | 1 | No | No | No | No |
| 4 | 3 | No | No | No | No |
| 5 | 3 | No | No | No | No |
| 6 | 4 | No | No | No | No |
| 7 | 10 | No | No | No | No |
| 8 | No | No | No | No | No |
| 9 | 2 | No | No | No | No |
| 10 | 1 | No | No | No | No |
| 11 | 2 | No | No | No | No |
| 12 | 2 | No | No | No | No |
| 13 | 1 | No | No | No | No |
Other adverse events include myalgia, ascites, gastrointestinal disorder, bleeding, hepatic abscess and autoimmune diseases.
Recurrence-free survival following DC infusion
A further objective of this study was to determine clinical response following DC infusion. A group of historical controls treated with TAE without DC administration was reviewed for this study (Table 3). The clinical characteristics including tumour burden and hepatic reserve were comparable between patients treated with TAE and OK432-stimulated DC transfer (n = 13) and those historical controls with TAE but without DC administration (n = 22). We compared the recurrence-free survival between these patient groups. Kaplan–Meier analysis indicated that patients treated with TAE and OK432-stimulated DC transfer had prolonged recurrence-free survival compared with the historical controls that had been treated with TAE alone (recurrence rates 360 days after the treatments; two of 13 and 12 of 22, respectively; P = 0·046, log-rank test) (Fig. 2). The results demonstrated that OK432-stimulated DC transfer during TAE therapy reduces tumour recurrence in HCC patients.
Table 3.
Clinical characteristics of patients treated with TAE + OK-DC and TAE alone.
| TAE + OK-DC | TAE | P | |
|---|---|---|---|
| No. of patients | 13 | 22 | |
| Age (years) | 68.2 ± 9.1 | 70.0 ± 7.6 | n.s.† |
| Gender (M/F) | 9/4 | 13/9 | n.s.‡ |
| White cell count (×102/µl) | 34.4 ± 11.6 | 41.4 ± 18.9 | n.s.† |
| Lymphocytes (×102/µl) | 10.4 ± 3.6 | 12.4 ± 4.7 | n.s.† |
| Platelets (×104/µl) | 11.5 ± 10.2 | 10.3 ± 5.8 | n.s.† |
| Hepaplastin test (%) | 64.6 ± 11.6 | 75.5 ± 24.3 | n.s.† |
| ALT (IU/l) | 56.7 ± 38.9 | 67.9 ± 44.6 | n.s.† |
| Total bilirubin (mg/dl) | 1.3 ± 0.7 | 1.1 ± 0.6 | n.s.† |
| Albumin (g/dl) | 3.4 ± 0.6 | 3.6 ± 0.4 | n.s.† |
| Non-cancerous liver parenchyma (no.) | |||
| Chronic hepatitis | 0 | 8 | |
| Cirrhosis (Child–Pugh A/B/C) | 13 (5/8/0) | 14 (6/8/0) | n.s.‡ |
| TNM stages (I/II/III/IV-A/IV-B) | 0/4/9/0/0 | 3/8/11/0/0 | n.s.‡ |
| No. of tumours | 2.5 ± 1.3 | 1.9 ± 1.3 | n.s.† |
| Largest tumour (mm) | 30.2 ± 9.4 | 32.6 ± 15.2 | n.s.† |
| AFP | 204.8 ± 404.1 | 201.8 ± 544.2 | n.s.† |
Results are expressed as means ± standard deviation.
Mann–Whitney U-test.
Fisher's exact test. TAE, transcatheter arterial embolization; OK-DC, OK432-stimulated dendritic cells; ALT, alanine transaminase; TNM, tumour–node–metastasis; AFP, alpha-fetoprotein; Child–Pugh, Child–Pugh classification; n.s., not significant.
Fig. 2.
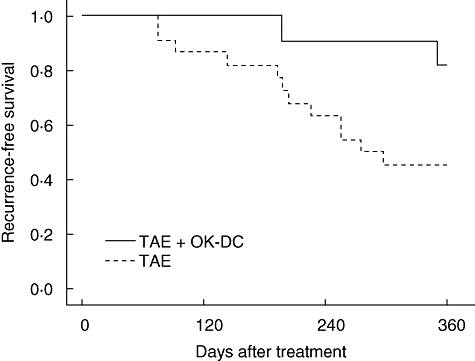
Recurrence-free survival of patients treated with transcatheter hepatic arterial embolization (TAE) with [TAE + OK-stimulated dendritic cells (DC); n = 13] and without (TAE: historical controls; n = 22) OK432-stimulated DC administration. Time zero is the date of TAE. All patients underwent ultrasound, computed tomography (CT) scan or magnetic resonance imaging (MRI) of the abdomen about 1 month after treatment and at a minimum of once every 3 months thereafter. Kaplan–Meier analysis indicated that TAE + OK-DC treatment prolonged recurrence-free survival compared with the TAE-alone group (recurrence rates 360 days after the treatments; two of 13 and 12 of 22, respectively; P = 0·046, log-rank test).
NK cell activity and intracellular cytokine responses in PBMCs
To assess systemic immunomodulatory effects of OK432-stimulated DC transfer, PBMCs were isolated 1 and 3 months after treatment and NK cell cytotoxicity against K562 erythroleukaemia target cells measured using the 51Cr-release assay (Fig. 3). The level of NK cell was unaltered following treatment. In addition, cytokine production capacity of lymphocyte subsets was quantitated by measuring intracellular IFN-γ and IL-4 using flow cytometry. There were also no significant changes in terms of cytokine production capacity in the CD4+, CD8+ and CD56+ subsets in the patients treated with OK432-stimulated DCs.
Fig. 3.
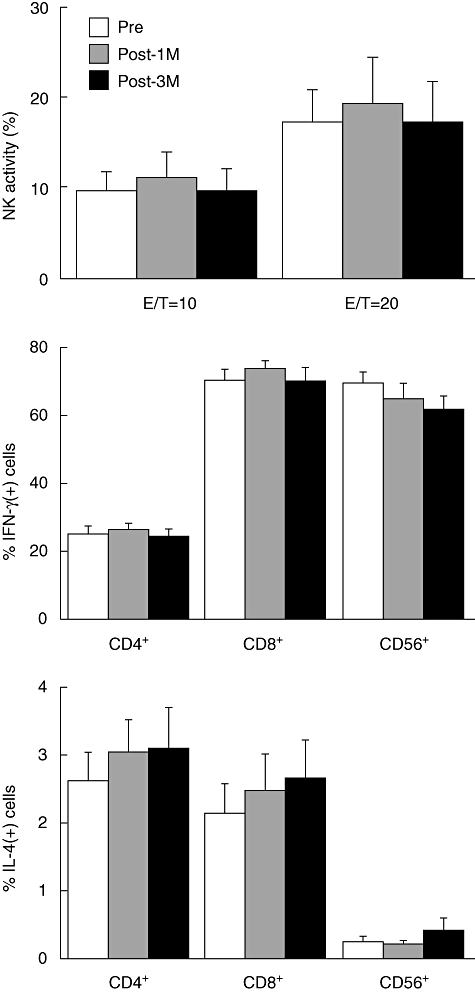
Natural killer (NK) cell activity and intracellular cytokine production in peripheral blood mononuclear cells (PBMCs) of patients treated with OK432-stimulated dendritic cells (DCs) during transcatheter hepatic arterial embolization (TAE) therapy (n = 13). PBMCs were isolated before and 1 and 3 months after treatment and used for the analyses. Upper panel: NK cell cytotoxicity against K562 erythroleukaemia target cells was evaluated at the effector/target (E/T) cell ratios shown. NK cell activities were not changed following treatment. Middle and lower panels: PBMCs were stimulated with phorbol 12-myristate 13-acetate (PMA) and ionomycin, stained for CD4, CD8 and CD56 expression, permeabilized and stained for intracellular interferon (IFN)-γ and interleukin (IL)-4. Percentages of cytokine-positive cells were quantitated by flow cytometry. There were no significant changes in terms of cytokine production capacity in the CD4+, CD8+ and CD56+ subsets following the treatments. The data are given as means ± standard deviation of the groups.
Immune responses to peptide epitopes derived from tumour antigens
To assess the effects on T cell responses to tumour antigens, PBMCs were obtained 4 weeks after DC infusion, pulsed with peptides derived from AFP, MRP3, SART2, SART3 and hTERT. IFN-γ production was then quantitated in an ELISPOT assay. Cells producing IFN-γ in response to stimulation with HLA-A24 [the most common HLA-A antigen (58·1%) in Japanese populations [35]]-restricted peptide epitopes derived from tumour antigens MRP3 and hTERT were induced in three of six HLA-A24-positive patients (numbers 2, 6 and 11) after treatment with TAE and OK432-stimulated DCs (Fig. 4). To understand the immunological and clinical significance of the T lymphocyte responses, PBMCs obtained from the historical control patients who had been treated with TAE without DC administration were also evaluated by ELISPOT. Similarly, positive reactions were observed in four (numbers t8, t19, t20 and t22) of six HLA-A24-positive patients. These data indicate that T lymphocyte responses to HLA-A24 restricted peptide epitopes of tumour antigens were induced following the TAE therapy, but no additional responses were observed as a result of OK432-stimulated DC transfer in the current study.
Fig. 4.
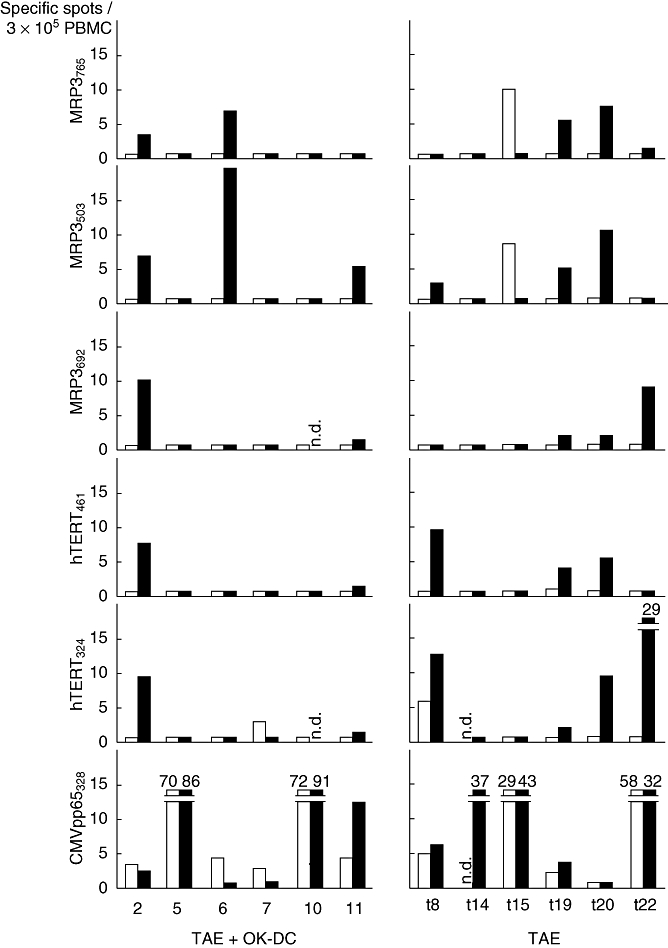
Immune responses to human leucocyte antigen (HLA-DR-)-A24-restricted peptide epitopes derived from tumour antigens in HLA-A24-positive patients treated with OK432-stimulated DCs during transcatheter hepatic arterial embolization (TAE) therapy (numbers 2, 5, 6, 7, 10 and 11) and HLA-A24-positive historical controls treated with TAE without dendritic cell (DC) transfer (numbers t8, t14, t15, t19, t20 and t22). Peripheral blood mononuclear cells (PBMCs) were obtained before (open bars) and 1 month after the infusion (solid bars), pulsed with the peptides derived from squamous cell carcinoma antigen recognized by T cells 2 (SART2), SART3, multi-drug resistance protein 3 (MRP3), alpha-fetoprotein (AFP), human telomerase reverse transcriptase (hTERT) and interferon (IFN)-γ production was quantitated by enzyme-linked immunospot (ELISPOT). Negative controls consisted of a human immunodeficiency virus (HIV) envelope-derived peptide (HIVenv584). Positive controls consisted of 10 ng/ml phorbol 12-myristate 13-acetate (PMA) or a cytomegalovirus (CMV) pp65-derived peptide (CMVpp65328). The number of specific spots was determined by subtracting the number of spots in the absence of antigen from the number of spots in its presence. T lymphocyte responses to the peptide epitopes were induced following TAE therapy, but no additional responses were observed after DC transfer. Numbers denote specific spots beyond the upper limit of y-axis; n.d., not determined.
Serum levels of cytokines, chemokines and arginase activity
To screen for immunobiological responses induced following OK432-stimulated DC transfer, serum levels of cytokines and chemokines were measured simultaneously using the Bio-Plex multiplex suspension array system. The results were compared with the historical control patients treated with TAE without DC administration. Interestingly, serum concentrations of IL-9, IL-15 and TNF-α were greatly increased after OK432-stimulated DC infusion, in contrast to their reduction following TAE treatment alone (Fig. 5a). Furthermore, the chemokines eotaxin (CCL11) and MIP-1β (CCL4) were induced markedly after DC transfer, although they were also decreased after TAE alone. These data indicate that transfer of OK432-stimulated DC during TAE therapy induced unique immune responses that may be mediated by the cytokines IL-9, IL-15 and TNF-α and the chemokines eotaxin and MIP-1β.
Fig. 5.
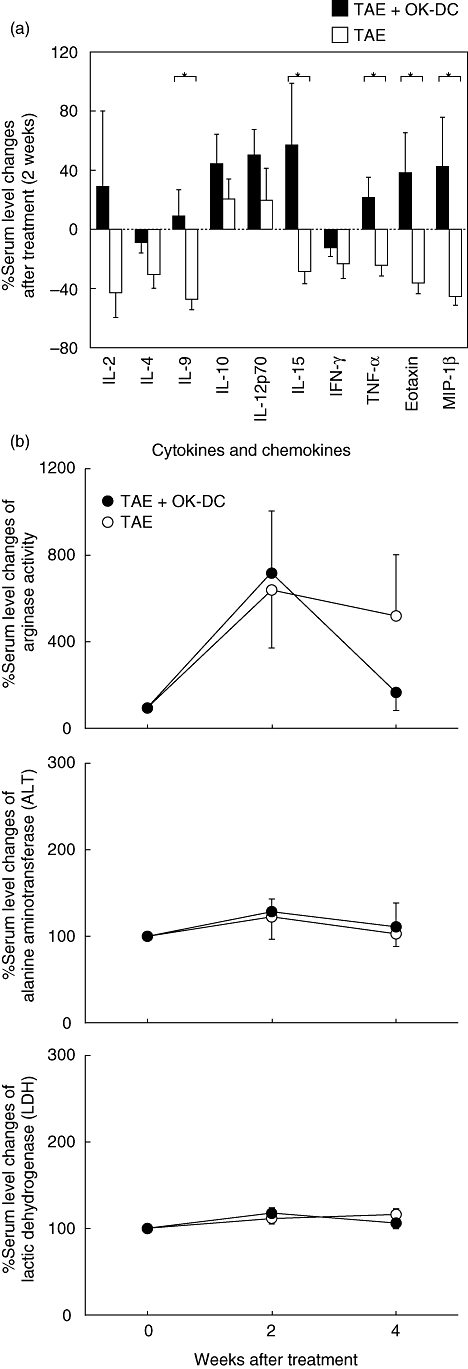
Cytokine and chemokine profiling and arginase activity in sera of patients treated with OK432-stimulated dendritic cells (DCs) during transcatheter hepatic arterial embolization (TAE) therapy (TAE + OK-DC; n = 13) and the historical controls treated with TAE without DC transfer (TAE; n = 22). (a) Serum samples were examined for their content of a validated panel of cytokines and chemokines using the Bioplex assay. Percentage changes in serum levels 2 weeks after the treatments were calculated as follows: [(post-treatment level – pretreatment level)/pretreatment level] × 100. The data are means ± standard error of the mean (s.e.m.) of the groups. *P < 0·05 when compared by the Mann–Whitney U-test. (b) Serum samples were tested for arginase activity by conversion of l-arginine to l-ornithine, and for alanine aminotransferase (ALT) and lactic dehydrogenase (LDH) activities. While there was a trend for the arginase activity in the TAE + OK-DC group to decrease 4 weeks after treatment, the difference did not reach statistical significance (P > 0·05). Percentage changes in serum levels 2 weeks after the treatments were calculated as follows: [(post-treatment level – pretreatment level)/pretreatment level] × 100. The data indicate means ± s.e.m. of the groups.
In addition, serum arginase activity was reported to reflect numbers of myeloid-derived suppressor cells (MDSCs) that may inhibit T lymphocyte responses in cancer patients [36]. Therefore, serum arginase activity was measured after OK432-stimulated DC infusion, and it was found that it was increased six- or sevenfold in patients treated with TAE. However, this increase was independent of the presence or absence of OK432-stimulated DC transfer (Fig. 5b). None the less, serum arginase activity was decreased again 4 weeks after treatment with both TAE and OK432-stimulated DC transfer but tended to be maintained at a high levels in patients treated with TAE without DC transfer. However, these differences did not reach statistical significance (P > 0·05). Because arginase activity is known to be relatively high in liver and HCC cells [37], the influence of tissue injury was assessed biochemically by measuring serum levels of ALT and LDH activities. We did not observe ALT or LDH elevation, indicating that the increase of arginase activity was not due to tissue damage following treatment. Collectively, these results demonstrate that infusion of OK432-stimulated DCs during TAE treatment may reduce the immunosuppressive activities of MDSCs, and assist in developing a favourable environment for the induction of anti-tumour immunity.
Discussion
Although many novel strategies, including immunotherapies, have been developed in an attempt to suppress tumour recurrence after curative treatments for HCC, recurrence rates and survival times have not been improved significantly [38]. In the current study, we first established that OK432-stimulated DC administration during TAE therapy did not cause critical adverse events in patients with cirrhosis and HCC. Most importantly, DC transfer resulted in prolonged recurrence-free survival after combination therapy with TAE and OK432-stimulated DC administration. In terms of the immunomodulatory effects of DC transfer, although NK cell activity, intracellular cytokine production and T lymphocyte-mediated immune responses were not altered in PBMCs from treated patients, serum levels of IL-9, IL-15 and TNF-α and the chemokines eotaxin and MIP-1β were enhanced markedly after DC transfer. In addition, serum levels of arginase activity were decreased following DC transfer. Collectively, this study demonstrated the feasibility, safety and beneficial anti-tumour effects of OK432-stimulated DC infusion into tumour tissues for patients with cirrhosis and HCC, suggesting the ability of an active immunotherapeutic strategy to reduce tumour recurrence after locoregional treatment of HCC.
DCs were stimulated with OK432 prior to infusion into tumour tissues through an arterial catheter. OK432 was reported to activate DCs through its binding to TLR-2 and -4 [16,39] that can be used for cancer therapy [33]. The current results indicate that OK432 stimulation of immature DCs from HCC patients promoted their maturation processes while preserving antigen uptake capacity and enhancing tumoricidal activity, consistent with previous observations [16,19] and supporting the current strategy in which OK432-stimulated DCs were infused directly into tumour tissues. Because the tumoricidal activity of unstimulated DCs was not observed in in vitro experiments, OK432 stimulation obviously altered the cytotoxic properties of DCs. One of the mechanisms of DC killing was reported to be CD40/CD40 ligand interaction [19]. Further studies are needed to determine the killing mechanisms of DCs derived from HCC patients in a direct [TNF, TNF-related apoptosis inducing ligand (TRAIL), Fas ligand, nitric oxide (NO) and perforin/granzyme] and indirect (MHC-restricted) manner [40–43]. Although the main mechanism by which OK432-stimulated DCs prolonged the recurrence-free survival was not elucidated, the tumoricidal activity of mature DCs was implicated in in vivo enhancement of antigen presentation, co-stimulation and inflammatory cytokine production.
Very recent reports document injection of OK432-stimulated DCs into patients with cancer of the gastrointestinal tract or pancreas [44,45], but their anti-tumour effects were not defined clearly. The current study shows for the first time that OK432-stimulated DCs induce beneficial anti-tumour responses when transferred into tumour tissues during TAE therapy. The anti-tumour responses may have been enhanced as a result of optimal activation of the DCs with OK432 or combining infusion of stimulated DCs with TAE therapy. Inappropriately activated DCs may be unable to generate sufficient numbers of properly activated effector T lymphocytes [46]. As shown in Fig. 1, all these alterations could contribute to the further enhancement of anti-tumour effects compared to those in our previous study with immature DCs [20]. Furthermore, the tumour cell death-promoting therapies, e.g. chemotherapy [47] and TAE [48], can be expected to enhance the effects of therapeutic cancer vaccines by redressing the immunosuppressive tumour environment.
NK cell activity and intracellular cytokine responses in CD4+ and CD8+ T lymphocytes and CD56+ NK cell subsets in PBMCs were not changed significantly in patients treated with OK432-stimulated DCs. Furthermore, we did not observe tumour antigen-specific T lymphocyte responses associated clearly with DC administration. The data suggest therefore that the immune responses induced by the therapy applied here were not detectable systemically. Because cytotoxic T lymphocyte responses were enhanced in patients receiving > 3 × 107 cells [49,50], the numbers of transferred OK432-stimulated DCs were apparently not sufficient to induce responses detectable in the peripheral blood, but were enough to exert beneficial anti-tumour effects. In addition, many studies have concluded that cytotoxic T lymphocyte responses rarely predict clinical outcomes of DC-based immunotherapies [51,52] and that in many cases, also including our own studies [28,30], tumour-specific effector T lymphocytes co-exist with the tumours. Consistent with these observations, the current results suggest that cytotoxic T lymphocyte responses in PBMCs are not reliable predictors of beneficial anti-tumour effects in patients treated with the current OK432-stimulated DC strategy.
Serum levels of the cytokines IL-9, IL-15 and TNF-α and the chemokines eotaxin and MIP-1β were increased following OK432-stimulated DC transfer, but decreased after TAE therapy without DC administration. IL-9 and IL-15 belong to the cytokine receptor common gamma chain (γc; CD132) family, a member of the type I cytokine receptor family expressed on most lymphocyte populations [53]. IL-9 exerts pleiotropic activities on T and B lymphocytes, mast cells, monocytes and haematopoietic progenitors [54,55]. IL-15 and TNF-α are known to prime T lymphocytes and NK cells when secreted by DCs [56] and to induce anti-tumour immune responses [57]. Eotaxin is known to selectively recruit eosinophils also contributing to anti-tumour effects [58,59], and MIP-1β is a chemoattractant for NK cells, monocytes and a variety of other immune cells [60]. In addition, serum levels of arginase tended to decrease after DC transfer. Because serum arginase activity reflects the numbers of MDSCs that inhibit T lymphocyte responses in cancer patients [36], the patients treated with OK432-stimulated DCs might have developed lower levels of suppressor cells. Collectively, the results suggest that infusion of OK432-stimulated DCs may orchestrate the immune environment in the whole body that could enhance beneficial anti-tumour effects, although the precise molecular and cellular mechanisms associated with the actions of these cytokines and chemokines were not defined clearly in the current analysis.
Acknowledgments
The authors thank Kazumi Fushimi and Mariko Katsuda for technical assistance. We also thank the patients for participating in this trial. This work was supported in part by research grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, the Ministry of Health, Labour and Welfare of Japan and the Japanese Society of Gastroenterology.
Disclosure
The authors have declared that no conflict of interest exists.
References
- 1.Omata M, Tateishi R, Yoshida H, Shiina S. Treatment of hepatocellular carcinoma by percutaneous tumor ablation methods: ethanol injection therapy and radiofrequency ablation. Gastroenterology. 2004;127:S159–66. doi: 10.1053/j.gastro.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 2.Belghiti J. Resection and liver transplantation for HCC. J Gastroenterol. 2009;44(Suppl. 19):132–5. doi: 10.1007/s00535-008-2250-1. [DOI] [PubMed] [Google Scholar]
- 3.Nakamoto Y, Guidotti LG, Kuhlen CV, Fowler P, Chisari FV. Immune pathogenesis of hepatocellular carcinoma. J Exp Med. 1998;188:341–50. doi: 10.1084/jem.188.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ercolani G, Grazi GL, Ravaioli M, et al. Liver resection for hepatocellular carcinoma on cirrhosis: univariate and multivariate analysis of risk factors for intrahepatic recurrence. Ann Surg. 2003;237:536–43. doi: 10.1097/01.SLA.0000059988.22416.F2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shankaran V, Ikeda H, Bruce AT, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–11. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 6.Vulink A, Radford KJ, Melief C, Hart DN. Dendritic cells in cancer immunotherapy. Adv Cancer Res. 2008;99:363–407. doi: 10.1016/S0065-230X(07)99006-5. [DOI] [PubMed] [Google Scholar]
- 7.Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 8.Lemos MP, Esquivel F, Scott P, Laufer TM. MHC class II expression restricted to CD8alpha+ and CD11b+ dendritic cells is sufficient for control of Leishmania major. J Exp Med. 2004;199:725–30. doi: 10.1084/jem.20030795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ni K, O'Neill HC. The role of dendritic cells in T cell activation. Immunol Cell Biol. 1997;75:223–30. doi: 10.1038/icb.1997.35. [DOI] [PubMed] [Google Scholar]
- 10.Andrews DM, Andoniou CE, Scalzo AA, et al. Cross-talk between dendritic cells and natural killer cells in viral infection. Mol Immunol. 2005;42:547–55. doi: 10.1016/j.molimm.2004.07.040. [DOI] [PubMed] [Google Scholar]
- 11.Heiser A, Coleman D, Dannull J, et al. Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J Clin Invest. 2002;109:409–17. doi: 10.1172/JCI14364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 13.Forster R, Schubel A, Breitfeld D, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 14.MartIn-Fontecha A, Sebastiani S, Hopken UE, et al. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med. 2003;198:615–21. doi: 10.1084/jem.20030448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ratzinger G, Stoitzner P, Ebner S, et al. Matrix metalloproteinases 9 and 2 are necessary for the migration of Langerhans cells and dermal dendritic cells from human and murine skin. J Immunol. 2002;168:4361–71. doi: 10.4049/jimmunol.168.9.4361. [DOI] [PubMed] [Google Scholar]
- 16.Nakahara S, Tsunoda T, Baba T, Asabe S, Tahara H. Dendritic cells stimulated with a bacterial product, OK-432, efficiently induce cytotoxic T lymphocytes specific to tumor rejection peptide. Cancer Res. 2003;63:4112–18. [PubMed] [Google Scholar]
- 17.Okamoto M, Oshikawa T, Tano T, et al. Mechanism of anticancer host response induced by OK-432, a streptococcal preparation, mediated by phagocytosis and Toll-like receptor 4 signaling. J Immunother. 2006;29:78–86. doi: 10.1097/01.cji.0000192106.32206.30. [DOI] [PubMed] [Google Scholar]
- 18.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–6. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 19.Hill KS, Errington F, Steele LP, et al. OK432-activated human dendritic cells kill tumor cells via CD40/CD40 ligand interactions. J Immunol. 2008;181:3108–15. doi: 10.4049/jimmunol.181.5.3108. [DOI] [PubMed] [Google Scholar]
- 20.Nakamoto Y, Mizukoshi E, Tsuji H, et al. Combined therapy of transcatheter hepatic arterial embolization with intratumoral dendritic cell infusion for hepatocellular carcinoma: clinical safety. Clin Exp Immunol. 2007;147:296–305. doi: 10.1111/j.1365-2249.2006.03290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–26. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 22.Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7:790–802. doi: 10.1038/nri2173. [DOI] [PubMed] [Google Scholar]
- 23.Makuuchi M. General rules for the clinical and pathological study of primary liver cancer. 2nd edn. Tokyo: Kanehara & Co., Ltd; 2003. [Google Scholar]
- 24.Veltri A, Moretto P, Doriguzzi A, Pagano E, Carrara G, Gandini G. Radiofrequency thermal ablation (RFA) after transarterial chemoembolization (TACE) as a combined therapy for unresectable non-early hepatocellular carcinoma (HCC) Eur Radiol. 2006;16:661–9. doi: 10.1007/s00330-005-0029-9. [DOI] [PubMed] [Google Scholar]
- 25.Dhodapkar MV, Steinman RM, Sapp M, et al. Rapid generation of broad T-cell immunity in humans after a single injection of mature dendritic cells. J Clin Invest. 1999;104:173–80. doi: 10.1172/JCI6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orange JS, Brodeur SR, Jain A, et al. Deficient natural killer cell cytotoxicity in patients with IKK-gamma/NEMO mutations. J Clin Invest. 2002;109:1501–9. doi: 10.1172/JCI14858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klausner RD, Donaldson JG, Lippincott-Schwartz J. Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–80. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizukoshi E, Nakamoto Y, Marukawa Y, et al. Cytotoxic T cell responses to human telomerase reverse transcriptase in patients with hepatocellular carcinoma. Hepatology. 2006;43:1284–94. doi: 10.1002/hep.21203. [DOI] [PubMed] [Google Scholar]
- 29.Mizukoshi E, Nakamoto Y, Tsuji H, Yamashita T, Kaneko S. Identification of alpha-fetoprotein-derived peptides recognized by cytotoxic T lymphocytes in HLA-A24+ patients with hepatocellular carcinoma. Int J Cancer. 2006;118:1194–204. doi: 10.1002/ijc.21468. [DOI] [PubMed] [Google Scholar]
- 30.Mizukoshi E, Honda M, Arai K, Yamashita T, Nakamoto Y, Kaneko S. Expression of multidrug resistance-associated protein 3 and cytotoxic T cell responses in patients with hepatocellular carcinoma. J Hepatol. 2008;49:946–54. doi: 10.1016/j.jhep.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez PC, Quiceno DG, Zabaleta J, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- 32.Itoh T, Ueda Y, Okugawa K, et al. Streptococcal preparation OK432 promotes functional maturation of human monocyte-derived dendritic cells. Cancer Immunol Immunother. 2003;52:207–14. doi: 10.1007/s00262-002-0337-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuroki H, Morisaki T, Matsumoto K, et al. Streptococcal preparation OK-432: a new maturation factor of monocyte-derived dendritic cells for clinical use. Cancer Immunol Immunother. 2003;52:561–8. doi: 10.1007/s00262-003-0394-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gunn MD, Kyuwa S, Tam C, et al. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J Exp Med. 1999;189:451–60. doi: 10.1084/jem.189.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Imanishi T, Akaza T, Kimura A, Tokunaga K, Gojobori T. HLA 1991, Proceedings of the Eleventh International Histocompatibility Workshop and Conference. Tokyo: Oxford University Press; 1992. [Google Scholar]
- 36.Zea AH, Rodriguez PC, Atkins MB, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005;65:3044–8. doi: 10.1158/0008-5472.CAN-04-4505. [DOI] [PubMed] [Google Scholar]
- 37.Chrzanowska A, Krawczyk M, Baranczyk-Kuzma A. Changes in arginase isoenzymes pattern in human hepatocellular carcinoma. Biochem Biophys Res Commun. 2008;377:337–40. doi: 10.1016/j.bbrc.2008.09.093. [DOI] [PubMed] [Google Scholar]
- 38.Caldwell S, Park SH. The epidemiology of hepatocellular cancer: from the perspectives of public health problem to tumor biology. J Gastroenterol. 2009;44(Suppl. 19):96–101. doi: 10.1007/s00535-008-2258-6. [DOI] [PubMed] [Google Scholar]
- 39.Okamoto M, Oshikawa T, Tano T, et al. Involvement of Toll-like receptor 4 signaling in interferon-gamma production and antitumor effect by streptococcal agent OK-432. J Natl Cancer Inst. 2003;95:316–26. doi: 10.1093/jnci/95.4.316. [DOI] [PubMed] [Google Scholar]
- 40.Liu S, Yu Y, Zhang M, Wang W, Cao X. The involvement of TNF-alpha-related apoptosis-inducing ligand in the enhanced cytotoxicity of IFN-beta-stimulated human dendritic cells to tumor cells. J Immunol. 2001;166:5407–15. doi: 10.4049/jimmunol.166.9.5407. [DOI] [PubMed] [Google Scholar]
- 41.Lu G, Janjic BM, Janjic J, Whiteside TL, Storkus WJ, Vujanovic NL. Innate direct anticancer effector function of human immature dendritic cells. II. Role of TNF, lymphotoxin-alpha(1)beta(2), Fas ligand, and TNF-related apoptosis-inducing ligand. J Immunol. 2002;168:1831–9. doi: 10.4049/jimmunol.168.4.1831. [DOI] [PubMed] [Google Scholar]
- 42.Nicolas A, Cathelin D, Larmonier N, et al. Dendritic cells trigger tumor cell death by a nitric oxide-dependent mechanism. J Immunol. 2007;179:812–18. doi: 10.4049/jimmunol.179.2.812. [DOI] [PubMed] [Google Scholar]
- 43.Stary G, Bangert C, Tauber M, Strohal R, Kopp T, Stingl G. Tumoricidal activity of TLR7/8-activated inflammatory dendritic cells. J Exp Med. 2007;204:1441–51. doi: 10.1084/jem.20070021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.West E, Morgan R, Scott K, et al. Clinical grade OK432-activated dendritic cells: in vitro characterization and tracking during intralymphatic delivery. J Immunother. 2009;32:66–78. doi: 10.1097/CJI.0b013e31818be071. [DOI] [PubMed] [Google Scholar]
- 45.Hirooka Y, Itoh A, Kawashima H, et al. A combination therapy of gemcitabine with immunotherapy for patients with inoperable locally advanced pancreatic cancer. Pancreas. 2009;38:e69–74. doi: 10.1097/MPA.0b013e318197a9e3. [DOI] [PubMed] [Google Scholar]
- 46.Melief CJ. Cancer immunotherapy by dendritic cells. Immunity. 2008;29:372–83. doi: 10.1016/j.immuni.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 47.Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8:59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
- 48.Ayaru L, Pereira SP, Alisa A, et al. Unmasking of alpha-fetoprotein-specific CD4(+) T cell responses in hepatocellular carcinoma patients undergoing embolization. J Immunol. 2007;178:1914–22. doi: 10.4049/jimmunol.178.3.1914. [DOI] [PubMed] [Google Scholar]
- 49.Thurner B, Haendle I, Roder C, et al. Vaccination with mage-3A1 peptide-pulsed mature, monocyte-derived dendritic cells expands specific cytotoxic T cells and induces regression of some metastases in advanced stage IV melanoma. J Exp Med. 1999;190:1669–78. doi: 10.1084/jem.190.11.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Banchereau J, Palucka AK, Dhodapkar M, et al. Immune and clinical responses in patients with metastatic melanoma to CD34(+) progenitor-derived dendritic cell vaccine. Cancer Res. 2001;61:6451–8. [PubMed] [Google Scholar]
- 51.Engell-Noerregaard L, Hansen TH, Andersen MH, Thor Straten P, Svane IM. Review of clinical studies on dendritic cell-based vaccination of patients with malignant melanoma: assessment of correlation between clinical response and vaccine parameters. Cancer Immunol Immunother. 2009;58:1–14. doi: 10.1007/s00262-008-0568-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Itoh K, Yamada A, Mine T, Noguchi M. Recent advances in cancer vaccines: an overview. Jpn J Clin Oncol. 2009;39:73–80. doi: 10.1093/jjco/hyn132. [DOI] [PubMed] [Google Scholar]
- 53.Sugamura K, Asao H, Kondo M, et al. The common gamma-chain for multiple cytokine receptors. Adv Immunol. 1995;59:225–77. doi: 10.1016/s0065-2776(08)60632-x. [DOI] [PubMed] [Google Scholar]
- 54.Temann UA, Geba GP, Rankin JA, Flavell RA. Expression of interleukin 9 in the lungs of transgenic mice causes airway inflammation, mast cell hyperplasia, and bronchial hyperresponsiveness. J Exp Med. 1998;188:1307–20. doi: 10.1084/jem.188.7.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McMillan SJ, Bishop B, Townsend MJ, McKenzie AN, Lloyd CM. The absence of interleukin 9 does not affect the development of allergen-induced pulmonary inflammation nor airway hyperreactivity. J Exp Med. 2002;195:51–7. doi: 10.1084/jem.20011732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Saint-Vis B, Fugier-Vivier I, Massacrier C, et al. The cytokine profile expressed by human dendritic cells is dependent on cell subtype and mode of activation. J Immunol. 1998;160:1666–76. [PubMed] [Google Scholar]
- 57.Shanmugham LN, Petrarca C, Frydas S, et al. IL-15 an immunoregulatory and anti-cancer cytokine. Recent advances. J Exp Clin Cancer Res. 2006;25:529–36. [PubMed] [Google Scholar]
- 58.Kataoka S, Konishi Y, Nishio Y, Fujikawa-Adachi K, Tominaga A. Antitumor activity of eosinophils activated by IL-5 and eotaxin against hepatocellular carcinoma. DNA Cell Biol. 2004;23:549–60. doi: 10.1089/dna.2004.23.549. [DOI] [PubMed] [Google Scholar]
- 59.Simson L, Ellyard JI, Dent LA, et al. Regulation of carcinogenesis by IL-5 and CCL11: a potential role for eosinophils in tumor immune surveillance. J Immunol. 2007;178:4222–9. doi: 10.4049/jimmunol.178.7.4222. [DOI] [PubMed] [Google Scholar]
- 60.Bystry RS, Aluvihare V, Welch KA, Kallikourdis M, Betz AG. B cells and professional APCs recruit regulatory T cells via CCL4. Nat Immunol. 2001;2:1126–32. doi: 10.1038/ni735. [DOI] [PubMed] [Google Scholar]