

Abstract
The aim of this study was to determine the genetic regulation of macrophage migration inhibitory factor (MIF). DNase I hypersensitivity was used to identify potential hypersensitive sites (HS) across the MIF gene locus. Reporter gene assays were performed in different human cell lines with constructs containing the native or mutated HS element. Following phylogenetic and transcription factor binding profiling, electrophoretic mobility shift assay (EMSA) and RNA interference were performed and the effects of incubation with mithramycin, an antibiotic that binds GC boxes, were also studied. An HS centred on the first intron of MIF was identified. The HS acted as an enhancer in human T lymphoblasts (CEMC7A), human embryonic kidney cells (HEK293T) and human monocytic cells (THP-1), but not in a fibroblast-like synoviocyte (FLS) cell line (SW982) or cultured FLS derived from rheumatoid arthritis (RA) patients. Two cis-elements within the first intron were found to be responsible for the enhancer activity. Mutation of the consensus Sp1 GC box on each cis-element abrogated enhancer activity and EMSA indicated Sp1 binding to one of the cis-elements contained in the intron. SiRNA knock-down of Sp1 alone or Sp1 and Sp3 together was incomplete and did not alter the enhancer activity. Mithramycin inhibited expression of MIF in CEMC7A cells. This effect was specific to the intronic enhancer and was not seen on the MIF promoter. These results identify a novel, cell type-specific enhancer of MIF. The enhancer appears to be driven by Sp1 or related Sp family members and is highly sensitive to inhibition via mithramycin.
Keywords: gene regulation, inflammation, MIF, mithramycin
Introduction
Macrophage migration inhibitory factor (MIF) is an early modulator of inflammation and is active in both innate and adaptive immunity [1–3]. Although its role as a direct cytokine is not entirely resolved [4], the case for MIF as an important modulator of inflammation is increasingly strong. Rheumatoid arthritis (RA) is a chronic inflammatory disease of unknown aetiology affecting ∼ 1% of the world's population. It is characterized by progressive destruction of synovial joints. A substantial body of evidence supports a key role for MIF in RA. Multiple different animal models show a role of MIF in the inflammatory joint process [5–9]. Furthermore, elevated levels of MIF have been shown in RA synovial fluid and serum samples, compared to healthy and osteoarthritic controls [10,11]. MIF stimulates macrophage release of tumour necrosis factor (TNF)-α, interleukin (IL)-1, IL-6 and IL-8, all of which occur at increased levels in RA patients. RA fibroblast-like synoviocyte (FLS)-derived MIF induces monocyte TNF-α production in vitro[10], and MIF up-regulates FLS IL-1 mRNA expression [12]. In addition to MIF's role as a potential upstream regulator of synovial cytokine expression, MIF contributes to the processes involved in synovial joint destruction: MIF induces metalloproteinases (MMP)-1 (interstitial collagenase) and MMP-3 (stromelysin) in FLS [12]. Murine osteoblasts express high levels of MIF [13]. MIF stimulates MMP-9 (gelatinase B) and MMP-13 (collagenase 3) from rat calvarial osteoblasts [13] and RA FLS induction of MMP-2 by MIF has also been described [14]. MIF-DNA vaccine protects from ovariectomy-induced bone loss [15] and MIF transgenic mice have high turnover osteoporosis associated with increased MMP-3, 9 and 13 expression [16]. This cumulative work supports the involvement of MIF in the cartilage damage and bone remodelling that is central to the progressive joint destruction that occurs in RA.
MIF has an unusual relationship with glucocorticoids (Gc) acting as a counter-regulator as well as being found, in some studies, to be induced by them [7,17–20]. Despite the use of new biological disease-modifying drugs, more than half of patients with RA are treated with Gc [18]. The metabolic side effects of Gc cause considerable morbidity, especially with long-term use, as occurs in RA. Steroid-sparing therapies, such as those that limit elevation of MIF expression, would therefore be clinically advantageous.
The MIF gene is situated on chromosome 22q11.2 within a cytosine–guanine–dinucleotide (CpG) island, and is highly conserved (90%) between mammalian species [21]. The MIF promoter is GC-rich and has no TATA box, suggesting the presence of other regulatory elements. Despite MIF being a key modulator of RA aetiopathogenesis, little is currently known about MIF gene regulation. Baugh et al. [22], Welford et al. [23] and Elsby [24] have shown MIF to be regulated via the hypoxia inducible transcription factor (HIF-1), and previous work found regulation of the proximal MIF promoter by cyclic AMP response element binding protein [25]. There is also evidence demonstrating the cell type-specific inhibition of MIF gene expression by Gc [20]. Importantly, MIF, unlike other proinflammatory mediators, is not regulated by TNF-α[24]. The aim of this study was therefore to identify functional regulatory elements for the MIF gene, so allowing identification of factors important in the control of MIF expression. Using an unbiased DNase I hypersensitivity approach, we have identified a novel cell type-specific enhancer within the first intron of MIF. Further characterization of the enhancer demonstrated that it was physiologically relevant and that regulation of the enhancer with the antibiotic mithramycin resulted in repression of MIF expression.
Materials and methods
Cell lines
CEMC7A, a human T lymphoblast cell line, THP-1, a monocytic cell line and RA FLS were cultured in RPMI-1640 (Gibco PRL, Paisley, UK) with 10% fetal bovine serum (FBS) (PerBio; Sigma, Poole, UK). RA FLS were isolated and cultured as described previously [10] and used between passages 4 and 8. The human fibroblast-like synoviocytes cell line (SW982) was cultured in l–15 Leibovitch (Gibco) with 10% FBS. Human lung carcinoma cells (A549) and kidney cells (HEK293T) were cultured in Dulbecco's modified Eagle's medium (DMEM) with Glutamax (Gibco) with 10% FBS (PerBio). All cell lines were grown in a 37°C incubator with 5% CO2. THP-1, A549, HEK293T and SW982 were obtained from the American Type Culture Collection (ATCC); CEMC7A was kindly provided by Dr Ged Brady.
Generation of probes
All polymerase chain reactions (PCR) were performed on a PTC-0225 DNA Engine Tetrad Thermal cycler (MJ Research, Waltham, MA, USA). Sequence for the probe was acquired via a search for MIF DNA on the UCSC Genome Bioinformatics website (http://genome.ucsc.edu/). Sequence was verified for specificity using blast on the National Center for Biotechnology Information (NCBI) website (http://www.ncbi.nlm.nih.gov/BLAST/) and a 465 base pairs (bp) sequence was chosen to target the 5′ end of the 10·2 Kb DNA fragment containing the MIF gene. This fragment was generated by BglII restriction digest of human genomic DNA. The probe was generated by PCR forward: 5′-GGAGGTAAGGGGTCAGGAGG-3′, probe 4 reverse 5′-GTGTTCACCTGACATAGAGG-3′ primers. The PCR product was then cloned into pCR®4Blunt-TOPO (Invitrogen, Paisley, UK). Probe sequence was confirmed by sequencing using the same primer as for PCR.
DNase I hypersensitivity assay
Confluent cells (∼108 cells) were washed four times with cold phosphate-buffered saline (PBS) and then pelleted. Cells were lysed in 20 ml 1× reticulocyte standard buffer (RSB) buffer (10 mM Tris pH7·4, 10 mM NaCl, 3 mM MgCl2) with 0·1% NP-40 for 7–10 min and the nuclei pelleted. Nuclei were washed in 20 ml of 1× RSB only, pelleted at 783 g for 5 min and resuspended in a final volume of 1 ml of 1× RSB. Different concentrations of DNase I (Worthington, Lakewood, NJ, USA) were added to each tube (0, 0·3, 1, 3, 10, 30 and 100 µg/ml) and incubated at 37°C for 10 min. DNA samples were stored immediately at −20°C or purified with DNeasy columns (Qiagen, Crawley, UK) as per the manufacturer's instructions. Ten µg of each isolated DNA sample was digested with Bgl II (Roche, Burgess Hill, UK) at 37°C overnight. Samples were then run on agarose or stored at 4°C.
Southern blotting
DNA was resolved on 0·8% 1× Tris–borate–ethylenediamine tetraacetic acid (EDTA) (TBE) agarose and transferred onto Hybond+ membrane (Amersham, Little Chalfont, Buckinghamshire, UK). Probes were labelled with [α-32P]dCTP using Random Primers DNA Labelling System (Invitrogen) according to the manufacturer's instructions. Labelled DNA was cleaned up using a microspin G50 column (Amersham), boiled for 5 min, then added to the prehybridization buffer and left to hybridize overnight at 65°C. Membranes were then washed twice for a minimum of 10 min in 2× saline sodium citrate (SSC), 0·1% sodium dodecyl sulphide (SDS) at 65°C and autoradiography performed.
Generation of reporter constructs
The whole of MIF intron 1 (194 bp) was amplified by PCR using the following primers: forward primer 5′-GGATCCGCGGGTCTCCTGGTCCTTCT-3′ and reverse primer 5′-GGATCCCGCGATGTACTGCGAGGAAAG-3′. The amplicons were cloned initially into pBLUNT vector (Invitrogen), subcloned into pGL3-promoter vector (Promega, Southampton, UK) and then sequenced, and for each construct a clone with the intron in the sense (S) or anti-sense (AS) orientation was isolated. These were digested with XhoI and HindIII to open up the promoter site and replace the SV40 promoter with the MIF promoter region. MIF promoter was obtained by digesting the pGL3-enhancer/MIF CATT promoter construct described previously [20] with XhoI and HindIII. The gel-purified promoter was then ligated into pGL3-promoter/intron wild-type (WT) S, pGL3-promoter/intron WT AS, pGL3-promoter/intron single nucleotide polymorphism (SNP) S and pGL3-promoter/intron SNP AS using the XhoI and HindIII sites.
Tiled oligonucleotide constructs
Overlapping WT (Metabion, Martinsried, Germany) or mutated oligonucleotides (Invitrogen), each ∼50 bp and spanning the whole of MIF intron 1 (oligo-1–oligo-7), were designed and phosphorylated at the 5′ end. The sequences are given in Table 1. Complementary oligonucleotides were annealed and ligated into the pGL3 promoter vector using the BamHI sites. All plasmids were sequenced.
Table 1.
Tiled ∼50 base pairs overlapping oligonucleotide constructs designed across migration inhibitory factor (MIF) intron 1.
| oligo1 | 5′-GATCCGTTTGCCGGGAGGGGACAGGAAGAGGGGGGTGCCCACCGGG-3′ |
| oligo1comp | 5′-TCGACCCGGTGGGCACCCCCCTCTTCCTGTCCCCTCCCGGCAAACG-3′ |
| oligo2 | 5′-GATCCGGGGGTGCCCACCGGACGAGGGGTTCCGCGCTGGGAGCTGG-3′ |
| oligo2comp | 5′-TCGACCAGCTCCCAGCGCGGAACCCCTCGTCCGGTGGGCACCCCCG-3′ |
| oligo3 | 5′-GATCCCCGCGCTGGGAGCTGGGGAGGCGACTCCTGAACGGAGCTGG-3′ |
| oligo3comp | 5′-TCGACCAGCTCCGTTCAGGAGTCGCCTCCCCAGCTCCCAGCGCGGG-3′ |
| oligo4 | 5′-GATCCAACGGAGCTGGGGGGCGGGGCGGGGGGAGGACGGTGGCTCG-3′ |
| oligo4comp | 5′-TCGACGAGCCACCGTCCTCCCCCCGCCCCGCCCCCCAGCTCCGTTG 3-3′ |
| oligo5 | 5′-GATCCACGGTGGCTCGGGCCCGAAGTGGACGTTCGGGGCCCGACGG-3′ |
| oligo5comp | 5′-TCGACCGTCGGGCCCCGAACGTCCACTTCGGGCCCGAGCCACCGTG-3′ |
| oligo6 | 5′-GATCCGTTCGGGGCCCGACGAGGTCGCTGGGGCGGGCTG-3′ |
| oligo6comp | 5′-TCGACAGCCCGCCCCAGCGACCTCGTCGGGCCCCGAACG-3′ |
| oligo7 | 5′-GATCCTCGCTGGGGCGGGCTGACCGCGCCCTTTCCTCGCAGG-3′ |
| oligo7comp | 5′-TCGACCTGCGAGGAAAGGGCGCGGTCAGCCCGCCCCAGCGAG-3′ |
| oligo1mut | 5′-GATCCGTTTGCCTTGAGGTGACAGGAAGAGGGGGGTGCCCACCGGG-3′ |
| oligo1mutcomp | 5′-TCGACCCGGTGGGCACCCCCCTCTTCCTGTCACCTCAAGGCAAACG-3′ |
| oligo6mut | 5′-GATCCGTTCGGGGCCCGACGAGGTCGCTGTTGCGTGCTG-3′ |
| oligo6mutcomp | 5′-TCGACAGCACGCAACAGCGACCTCGTCGGGCCCCGAACG-3′ |
Comp: complementary sequence.
Transfection of plasmid DNA
Cells were transfected transiently in triplicate using either electroporation, Lipofectamine 2000 (Invitrogen) or nucleofection. For electroporation, CEMC7A and THP-1 cells were resuspended in serum-free medium at 107 cells per ml. Plasmid DNA (10 µg) was added with renilla (1 µg) as transfection efficiency control. Lipofectamine 2000 was used for SW982, HEK293T and CEMC7A cells. Plasmid DNA (0·2–1·5 µg) and renilla control (0·2–0·8 µg) were used for each transfection in a 96-well or a 24-well plate. HEK293T and SW982 were seeded at 2 × 105 cells/ml and CEM cells at 5 × 105 cells/ml to 3 × 106 cells/ml and left to grow overnight. Transfections were performed as per the manufacturer's instructions. For nucleofection, CEMC7A were resuspended in medium at 5 × 106 cells/ml and grown overnight. Using the nucleofector Kit V, a total of 1 × 106 cells/ml was used per reaction to co-transfect siRNA with plasmid DNA with the program C16. For human RA FLS, a total of 0·5 × 106 cells were used per reaction to transfect 2·5 µg of plasmid DNA with the program U23 using the nucleofection kit for NHLF (Amaxa, Wokingham, Berkshire, UK). Transfections were performed according to the manufacturer's protocol.
All luciferase/renilla assays were performed 24 h post-transfection as described previously, and the normalized luciferase data presented [26].
SP1 knock-down
A pool of four siRNA sequences directed against Sp1 (M-026959-00 siGENOME SMARTpool reagent; Dharmacon, Lafayette, CO, USA) or lamin A/C control (D-001620-02-05 siGLO lamin A/C; Dharmacon) as a negative control were co-transfected with plasmid DNA (SV40/MIF intron WT S and SV40 promo) in HEK293T and CEMC7A, according to the manufacturer's instructions. Transfections were performed in 24-well plates by nucleofection for CEMC7A or using Lipofectamine 2000 (Invitrogen) for HEK293T. iRNA (100 nM) and plasmid DNA (1–5 µg) were transfected, cells were incubated for 48 h and each reaction was split into three fractions; Sp1 knock-down was assessed by Western blot and enhancer activity was assessed by luciferase assay.
SP1 and SP3 combined knock-down
HEK293T cells were co-transfected with siRNA directed against Sp1 (s8222; Ambion) (15 nM) and Sp3 (s13326, Ambion) (15 nM) or lamin A/C (s58222; Ambion) (30 nM). Transfections were performed in 24-well plates using Lipofectamine™ RNAiMAX Transfection Reagent (Invitrogen) as per the manufacturer's instructions and incubated at 37°C for 24 h before harvesting.
Western blotting
Cell lysates were separated by SDS-polyacrylamide gel electrophoresis (PAGE) using either 4–20% gradient precast gels (Biorad, Hemel Hempstead, UK) in 1× Tris–glycine SDS (TGS) buffer (Biorad) or Nupage® Novex 3–8% Tris–acetate precast gels (Invitrogen) in 1× Tris–acetate running buffer (50 mM Trisbase, 50 mM Tricine, 0·4% SDS). Proteins were transferred onto supported nitocellulose (Biorad) or polyvinylidene difluoride (PVDF) membrane (Biorad), respectively. Membranes were blocked with 2% milk in PBS/0·05% Tween-20 overnight and incubated with primary antibody for 2 h followed by secondary antibodies conjugated to horseradish peroxidase (HPP) (1:1000–1:3000 dilution). Chemiluminescent results were obtained with ECL Plus (Amersham), according to the manufacturer's protocol. Washes were performed with PBS/0·05% Tween-20. Antibodies used were as follows: anti-Sp1 rabbit antibody [1:3000; Upstate 07-645, Upstate (Millipore), Watford, UK], anti-Sp3 D-20 rabbit antibody (1:200; Santa Cruz SC-644, Santa Cruz, CA, USA), rabbit anti-goat IgG HRP conjugate (Upstate), anti-alpha-tubulin clone DM1A mouse antibody (1:10000; Upstate), anti-human MIF goat antibody (1:300–1:500; R&D Systems, Abingdon, UK), goat anti-mouse immunoglobulin (Ig)G, HRP conjugate (Upstate), anti-rabbit IgG HRP (R&D Systems) secondary antibodies.
Sp1 inhibition assay
CEMC7A were treated with mithramycin (Sigma) at 1 nM to 50 nM for 48 h, then analysed by Western blotting for endogenous MIF production. Alternatively, CEMC7A were treated with mithramycin at 200 nM for 24 h and transfected transiently with pGL3-control (which possesses an SV40 promoter and an SV40 enhancer), pGL3 promo/oligo-1 and pGL3. Cell death was analysed using Trypan blue exclusion (Sigma).
Preparation of nuclear extract
CEMC7A cells were grown to a density of 1 × 105 cells/ml in 1 l of RPMI-1640 medium in order to gain a sufficient final volume of nuclear extract. Cells were harvested and washed twice in PBS. Nuclear extract was then prepared as described previously [27], with a minor modification. Cells were disrupted by passing the cell suspensions through a 23-gauge needle five times.
Electrophoretic mobility shift assay (EMSA)
EMSA was performed using a radiolabelled double-stranded DNA oligonucleotide corresponding to the intronic oligonucleotide sequence 1 (oligo-1), as described above.
The annealed oligonucelotide was end-labelled with [γ-32P]ATP (Amersham) using T4 polynucleotide kinase (Promega) and purified using G50 columns (Amersham).
Reactions contained 10 µl buffer C [10 mM Tris pH 7·8, 50 mM NaCl, 1 mM dithiothreitol (DTT), 1 mM EDTA 5% glycerol], 2 µg bovine serum albumin (BSA), 100 ng poly dI : dC, 0·1 ng labelled probe and 5 µg nuclear extract, as indicated in the figure legends. Competition experiments were performed by adding 100× unlabelled oligonucelotide to the reaction mixes prior to addition of radiolabelled probe, as indicated. Supershift experiments were performed by the addition of 2 µg of antibody as indicated prior to the addition of radiolabelled probe. Reactions were then incubated at room temperature for 1 h and loaded onto a 5% 0·5× TBE gel run at 150 V for approximately 3 h. Gels were then dried and autoradiographed.
Bioinformatics
To compare MIF DNA sequences across species, University of California Santa Cruz (UCSC) at http://genome.ucsc.edu and the NCBI at http://www.ncbi.nlm.nih.gov websites were used. Analysis of MIF intron 1 sequence for potential transcription factor sites was performed using the following database: AliBaba 2·1 and P-Match at http://www.gene-regulation.com and MatInspector from Genomatix at http://www.genomatix.de.
Data analysis
All luciferase results were adjusted to the CMV-renilla control and calculated as a fold induction relative to the control promoter. Statistical analysis was performed using spss version 11·5 or Prism 5. Multiple comparison analysis was performed using a one-way analysis of variance (anova) with a post-hoc Bonferroni test and comparison of a group of data to a control group was performed using a one-way anova comparative test with a post-hoc two-sided Dunnet's t-test.
Results
DNase I hypersensitivity assay
Nuclei from a total of 108 CEMC7A cells were isolated and treated with increasing amounts of DNase I. The genomic DNA was then isolated and restriction digested with BglII. This generated a 10·2 Kb fragment containing 3354 bp 5′ to the transcriptional start of the MIF gene and 5981 bp 3′ of the termination codon (Fig. 1b). Hybridization with a 468 bp probe specific to the 5′ end of the 10·2 Kb fragment revealed that it was digested by DNase I, indicating the presence of a hypersensitive site (HS). A shorter fragment of ∼3·6 Kb appeared (Fig. 1a) as the DNase I concentration increased and interpolation of the band height of the ∼3·6 Kb fragment indicated the HS spanned ∼911 bp (Fig. 1b). The HS encompassed the first intron of MIF, which was isolated for further study.
Fig. 1.
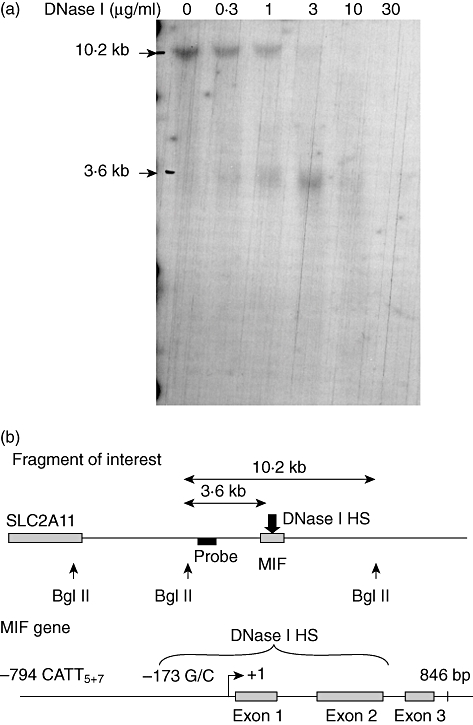
Mapping of the DNase I hypersensitive site. (a) Genomic DNA was migrated overnight in a 0·8% agarose gel then transferred to a nylon membrane and hybridized overnight with a probe specific for the 10·2 Kb fragment of interest. Results are representative of two independent experiments. (b) Interpolation of hypersensitive site (HS) in untreated CEMC7A cells. The hypersensitive site spans ∼911 base pairs but is centred on the first intron.
The HS acts as an enhancer
The first intron of the MIF gene (containing the HS) was cloned into the pGL3 promoter vector, which contains an SV40 promoter upstream of the luciferase gene. MIF intron 1 was inserted at a site distal to the SV40 promoter in either the sense or anti-sense orientation (pGL3 SV40-MIF intron 1 sense/anti-sense). CEMC7A cells were then transfected with either pGL3 SV40-MIF intron 1 sense/anti-sense plasmids, or a pGL3 vector containing the SV40 promoter and SV40 enhancer (pGL3 SV40-SV40) or the SV40 promoter only (pGL3-SV40). Constructs were transfected transiently in CEMC7A cells, and luciferase gene expression was measured. The intron exhibited orientation-independent enhancer activity, showing a fivefold increase over the pGL3 promoter-only vector (P< 0·0001) (Fig. 2a).
Fig. 2.
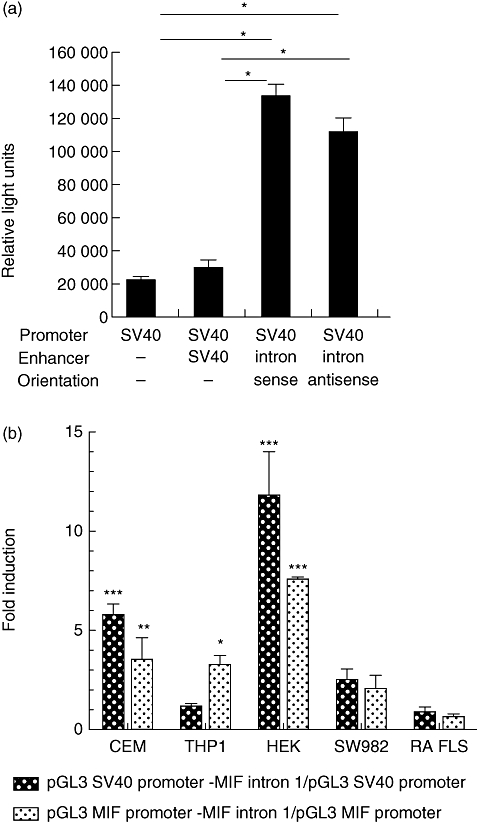
The DNase I hypersensitive site is a cell type-specific enhancer. Reporter assays of the intronic constructs. (a) The first intron of the MIF gene was cloned into the pGL3 promoter vector (SV40) in both the sense and anti-sense orientation. As a positive control of enhancer activity the pGL3 control vector was used, which possesses an SV40 enhancer together with the SV40 promoter. The plasmids were transfected in CEMC7A cells and cells were left to incubate for 24 h before harvest and luciferase assay. *P< 0·0001. (b) The hypersensitive site is a cell type-specific enhancer. The transcriptional activity of the macrophage migration inhibitory factor (MIF) intron 1 enhancer was analysed in four different human cell lines: a T lymphoblast cell line (CEMC7A), an embryonic kidney cell line (HEK293T), a monocytic cell line (THP-1), a fibroblast-like synoviocyte cell line (FLS) and in primary fibroblast isolated from rheumatoid arthritis (RA) patients (RA FLS). Cells were transfected with constructs containing the MIF intron 1 sequence with either the SV40 promoter or the MIF promoter initiating luciferase gene expression. Following transfection, cells were incubated for 24 h before luciferase assays were performed. Results are representative of a minimum of two independent experiments (± standard error of the mean) performed in biological triplicates for all cell lines, and three different patients in biological duplicates for RA FLS. Results are expressed as a fold induction over the pGL3 SV40 or pGL3 MIF promoter vector. *P< 0·05; **P< 0·01; ***P< 0·001.
The enhancer acts in a cell type-specific manner
In order to determine any cell type-specificity of intronic enhancer activity, HEK293T, SW982 (FLS), human RA FLS and THP-1 cells were transfected with constructs containing intron 1 of the MIF gene, in which luciferase gene transcription is initiated at the SV40 promoter. Enhancer activity was observed within the CEMC7A cells, as shown previously (Fig. 2a), with a fivefold induction above the SV40 promoter control (P< 0·01) (Fig. 2b). Enhanced transcriptional activity was also observed in HEK293T cells, with a sevenfold induction over the SV40 promoter (P< 0·0001) (Fig. 2b). However, no enhancer activity was noted in the FLS cell line SW982 and cultured RA FLS (Fig. 2b).
To assess if the intron had enhancer activity on the MIF promoter, the SV40 promoter was substituted for the full-length MIF promoter −1075 to +85 (CATT5) described previously [20]. The MIF promoter construct showed similar levels of transcriptional activation as the SV40 promoter control and this was consistent with all cell lines (Fig. 2b), with the exception of the THP-1 cell line, where enhanced transcriptional activity was detected when the MIF enhancer was coupled the MIF promoter (P< 0·05) but not with the SV40 promoter (Fig. 2b). This demonstrated that the intron could enhance the transcriptional activity of both a heterologous promoter or the MIF promoter in a cell type-specific manner, with a preference for the MIF promoter in THP-1 s, where no enhancer activity was detected with the SV40 promoter (Fig. 2b).
Mutation of Sp1 binding site abrogated enhancer activity
Comparative genomics combined with TF database analysis were used in order to identify potential TF for the enhancer activity of the first intron of MIF. Search results suggested a high density of Sp1 binding sites in the 5′ region of the MIF intron 1 (Fig. 3a). In order to determine a role for Sp1 in the enhancer activity of the HS, we cloned seven oligonucleotides of ∼50 bp each, spanning the full length of the HS, at the enhancer site of the luciferase reporter vector pGL3, under the SV40 promoter, to determine the cis-acting elements in the enhancer (Fig. 3a). The reporter constructs were assayed in CEMC7A cells and oligo-1 and oligo-6 were shown to carry the cis elements responsible for the enhancer activity, as fold induction of luciferase activity over SV40-only control was increased significantly (oligo-1 P< 0·001, oligo-6 P< 0·05) (Fig. 3b). Oligos-2, -3, -4, -5 and -7 had no luciferase activity greater than that of the control (data not shown). Consensus binding site for Sp1 in oligo-1 and oligo-6 were mutated in order to disrupt Sp1 binding and the enhancer activity was assayed as per the WT oligonucleotides in CEMC7A cells (Fig. 3c). Disruption of the Sp1 consensus binding site in both oligo-1 and oligo-6 abrogated enhancer activity (oligo-1mut P< 0·001, oligo-6mut P< 0·01) (Fig. 3b), further suggesting a role for Sp1, or its family members, in driving the HS enhancer activity.
Fig. 3.
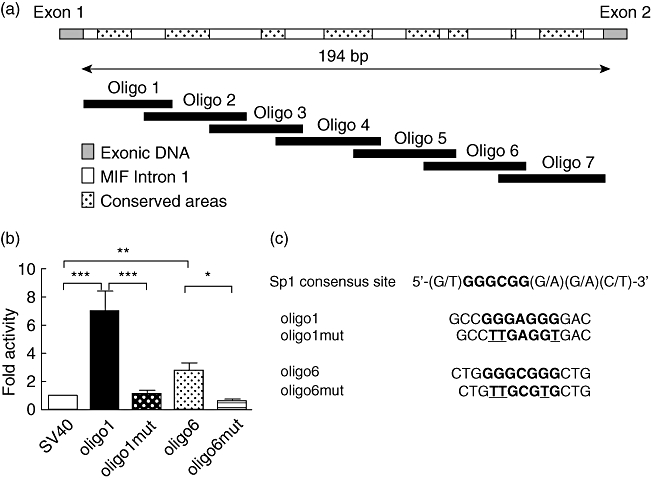
Mutation of Sp1 binding sites on cis-acting elements abrogates enhancer activity. (a) Schematic representation of the migration inhibitory factor (MIF) intronic enhancer and putative Sp1 binding sites. Seven oligonucleotides spanning the length of the intronic hypersensitive sites (HS) were cloned into pGL3 promoter. The hashed areas represent highly conserved Sp1 transcription factor binding sites. (b) Oligo-1 and oligo-6 are responsible for MIF enhancer activity. Cells were transfected with constructs containing wild-type (WT) or mutated oligo-1 or oligo-6 sequences. The pGL3 SV40 promoter-only construct was used as control and results are expressed as fold induction over the control. Following transfection, cells were incubated for 24 h before luciferase assays were performed. Results are representative of four to 10 independent experiments (±standard error of the mean) performed in single replicates. Results are expressed as a fold induction over the pGL3 SV40 or pGL3 MIF promoter vector. *P< 0·05; **P< 0·01; ***P< 0·001. (c) Sp1 binding site mutation. Three base pairs in each Sp1 consensus binding site were mutated from G to T, disrupting the core element for Sp1 binding.
Sp1 binds the MIF enhancer
In order to determine whether Sp1 is recruited to the MIF intron 1 enhancer, EMSA was performed using CEMC7A nuclear extract and a radiolabelled DNA probe of the 5′ region of the intronic sequence (oligo-1). We were able to demonstrate that Sp1 bound this sequence, as competition with excess cold Sp1 consensus sequence and addition of Sp1-specific antibody (αSP1) resulted in the abrogation of a shifted complex (Fig. 4). The 3′ region of the intron also possessed putative Sp1 binding sites (oligo-6); however, no transcription factor binding to this sequence was observed using the same method (data not shown).
Fig. 4.
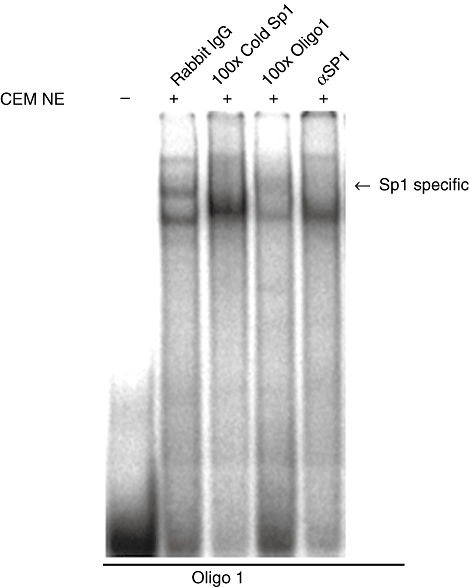
Sp1 binds MIF intron 1. Electrophoretic mobility shift assay (EMSA) analysis demonstrating the binding of Sp1 to oligo-1. Nuclear extracts prepared from CEMC7A cells were incubated with radiolabelled oligo-1. Competition experiments were performed by the addition of 100× unlabelled oligonucleotide (cold Sp1) as indicated; 2 µg of rabbit IgG or Sp1 antibody added as indicated. Complexes were then resolved by native gel electrophoresis. Sp1 sequence specific complex marked by an arrow.
Knock-down of Sp1/Sp3 has no effect on enhancer activity
The presence of Sp1 in the different cell types in which the enhancer activity and Sp1 EMSA were studied was determined using immunoblots of whole cell lysates in untreated cells. In HEK293T and CEMC7A Sp1 was highly expressed, whereas THP-1 and FLS cells showed less expression (Fig. 5a). To analyse further the potential regulatory role of Sp1 in the enhancer function, RNA interference was used to deplete Sp1 in order to observe whether the enhancer activity was diminished. Western blotting showed that the endogenous Sp1 expression was knocked down in HEK293T cells (Fig. 5b). However, Sp1 knock-down had no effect on endogenous MIF expression (Fig. 5b).
Fig. 5.
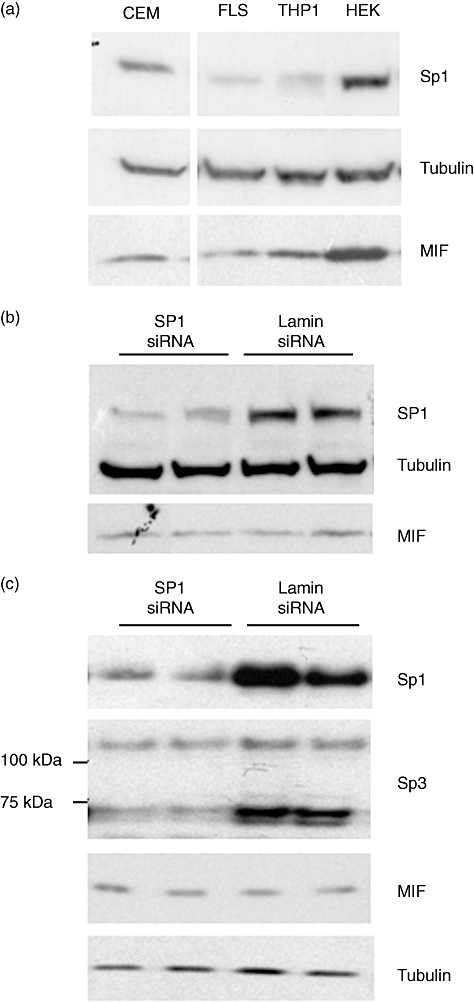
Neither Sp1 alone nor Sp1/Sp3 combined knock-down effects the enhancer activity. (a) Endogenous levels of Sp1 protein in different cell lines. Cell lysates of CEMC7A, FLS, THP-1 and HEK293T cells were immunoblotted for Sp1, alpha-tubulin and migration inhibitory factor (MIF). Results are representative of a minimum of two independent experiments performed in biological triplicates. (b) A pool of four siRNA sequences directed against Sp1 or Lamin A/C (negative control), 100 nM siRNA and 1–5 µg plasmid DNA were used per reaction. Transfected cells were incubated for 48 h Sp1 knock-down and endogenous MIF production was assessed by Western blotting. No down-regulation of the MIF protein was seen on endogenous MIF expression in HEK293T. (c) SiRNA directed against Sp1 (15 nM) and Sp3 (15 nM) or lamin A/C (negative control) (30 nM) were co-transfected into HEK293T cells. Cells were harvested 48 h post-transfection and subject to Western blot analysis with anti-Sp1, anti-Sp3, anti-MIF and anti-α-tubulin antibodies as indicated.
Sp1 is part of a family of transcription factors (Sp1–Sp4) which share highly conserved DNA binding domains and can function co-operatively to regulate transcription of a multitude of genes [28]. Other members of the Sp1 family may be important in the control of the enhancer activity. Both Sp3 and Sp4 share high affinity for Sp1 consensus binding sites; however, Sp4 is expressed predominantly in the brain and shows limited expression in other tissues. Sp3 is expressed ubiquitously, therefore HEK293T cells were co-transfected with siRNA directed against Sp3 and Sp1 or lamin A/C as a negative control. Cells were harvested 48 h post-transfection and cell lysates analysed by Western blotting with anti-Sp1, anti-Sp3, anti-MIF and anti-tubulin antibodies. Both Sp1 and Sp3 showed a clear knock-down in HEK293T cells treated with Sp1 and Sp3 siRNA compared with cells treated with lamin siRNA (Fig. 5b,c). The prominent doublet at ∼75 kDa (Sp3) was down-regulated markedly by siRNA; a higher molecular weight band (∼100 kDa) is also Sp3-related and is knocked down by the siRNA (Fig. 5c). However, the knock-down of Sp1 and Sp3 in combination had no effect on endogenous MIF expression (Fig. 5c).
Mithramycin specifically inhibits the enhancer activity
In parallel to the Sp1/Sp3 knock-down assay and mutation analysis of the Sp1 binding sites in the cis-elements, the Sp1 inhibitor mithramycin was used to analyse further the role of Sp1 on the MIF intronic enhancer. CEMC7A cells were transfected transiently with luciferase reporter constructs containing oligo-1 and oligo-6. Following transfection, cells were incubated with mithramycin at 200 nM for 24 h. The enhancer activity was significantly higher than that of the control vector (oligo-1 P< 0·01, oligo-6 P< 0·01) and mithramycin completely inhibited the enhancer activity of both oligo-1 and oligo-6, compared to the control vector (both P< 0·05) (Fig. 6a).
Fig. 6.
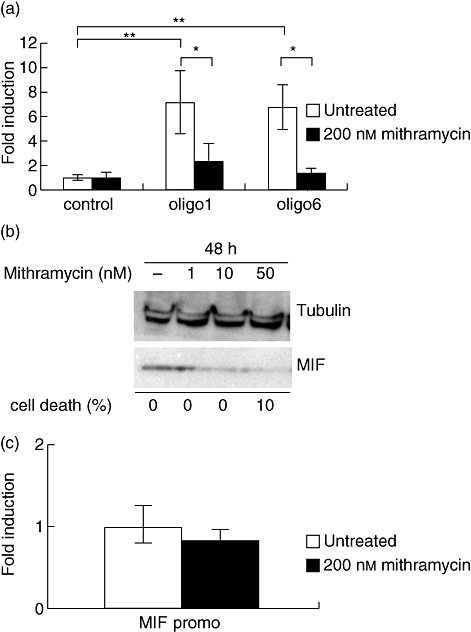
Mithramycin specifically inhibits the enhancer activity and endogenous migration inhibitory factor (MIF) expression. (a) Mithramycin inhibits the enhancer activity in CEMC7A cells. CEMC7A cells were transfected transiently either with a control vector possessing the SV40 promoter (control) or a vector containing oligo-1 or oligo-6. The cells were then treated with 200 nM mithramycin and left to incubate for 24 h. In untreated cells (white columns), the oligo-1 and oligo-6 reporter assays show enhancer activity. This activity was abrogated by the addition of mithramycin to the cells (black columns). Results are representative of a minimum of two independent experiments (±standard error of the mean) performed in biological triplicates. Results are expressed in fold induction over the untreated control vector. *P< 0·05; **P< 0·01. (b) Mithramycin inhibits endogenous expression of MIF in CEMC7A cells. CEMC7A cells were treated with increasing amounts of mithramycin (0, 1, 10 and 50 nM) for 24 h. Cell lysates were then collected and immunoblotted with the relevant primary anti-sera: anti-MIF goat antibody for MIF and anti-alpha-tubulin clone DM1A mouse antibody for tubulin. Cell death was counted by trypan blue exclusion. Results are representative of three independent experiments peformed in biological triplicates. (c) Mithramycin has no effect on the MIF promoter. CEMC7A cell were transfected with the MIF promoter only plasmid (MIF promo) and were left untreated (white column) or were treated with 200 nM mithramycin (black column) and left to incubate 24 h before luciferase assay was performed. Results are representative of two independent experiments performed in biological triplicates. Results are expressed in fold induction over untreated cells.
To investigate whether mithramycin inhibition of the enhancer activity in CEMC7A cells affected endogenous MIF production, CEMC7A cells were incubated with increasing concentrations of mithramycin for 48 h. Mithramycin inhibited MIF production from very low concentrations of 1 nM up to 50 nM, as analysed by Western blotting. No significant cell death, as measured by Trypan blue exclusion, was found to occur over these time-points (Fig. 6b). Finally, a reporter assay was carried out in CEMC7A cells to assess specificity of the action of mithramycin to the intronic enhancer element (Fig. 6c). No inhibition of the MIF promoter was detected after treating cells with 200 nM mithramycin for 24 h, indicating that the inhibitory effect of mithramycin on MIF endogenous production is specific to its capacity to block the enhancer activity in this cell line.
Discussion
This study aimed to expand the understanding of MIF gene regulation in cell types of relevance to inflammatory pathologies such as RA.
DNase I treatment revealed a strong HS centred on the first intron of MIF. The intronic constructs examined showed strong enhancer activity in the CEMC7A cells, regardless of orientation, suggesting classical enhancer activity [29,30]. This enhancer was cell type-specific, with enhancer activity observed in immune cells, namely a T lymphoblast and a monocytic cell line, but not in cells from mesenchymal origin (SW982 and cultured RA FLS), potentially allowing for a ‘fine tuning’ of the regulation of MIF expression in immune cells.
Database searches for transcription factor binding sites (TFBS) showed highly conserved Sp1 sites within oligo-1 and oligo-6 sequences. Sp1 is part of a large family of transcription factors which bind similar consensus sequences, although they have highly diverse functions (reviewed in [31,32]). Sp1 drives the expression of several constitutive genes and the Sp1 null mouse dies at day 11 of gestation [33]. We therefore focused initially on Sp1 as the candidate TF mediating the enhancer activity. First, we used mutation analysis of the Sp1 binding site in oligo-1 and oligo-6. Disruption of the Sp1 consensus binding sites completely abrogated the enhancer activity in both oligonucleotides, demonstrating the crucial function of these sequences and suggesting Sp1 is the TF driving the enhancer activity. Secondly, we used EMSA to determine Sp1 binding to cis-elements present in the HS site that were shown to have enhancer activity. We determined that oligo-1 was able to bind Sp1 present in CEMC7A nuclear extracts by competition with unlabelled Sp1 consensus sequence and incubation with Sp1-specific antibody. This further supported Sp1 as the TF responsible the enhancer activity of the HS site.
Sp1 is expressed ubiquitously in most cell types, but levels of expression may differ from one cell type to another and influence the activity of the TF. We looked at the endogenous expression of Sp1 in the several cell types where we had observed the HS site to have an enhancer activity. Sp1 was expressed at higher levels in both CEMC7A and HEK293T cells, which was concordant with the highest level of enhancer activity of the HS site in these cells.
We used reverse genetics and depleted HEK293T cells, which had the highest level of Sp1 expression, of Sp1 and of Sp3 using small interfering RNA (siRNA). The enhancer activity was not changed when Sp1 was knocked down alone or in combination with Sp3, and there was no effect on MIF protein production. Multiple isoforms are known to exist for Sp3 (UniProtKB/Swiss-Prot Q0227), and two were observed in the HEK293T cells. Furthermore, due to the high endogenous levels of Sp1 in these cells, it is possible that a sufficient concentration of Sp1 remained for it to bind effectively to the MIF intronic enhancer.
Mithramycin (or plicamycin) is an aureolic acid which has been used in the treatment of cancer and hypercalcaemia [34,35], and has been used in many studies of the Sp family [36–40]. Mithramycin specifically abrogated the enhancer activity for both cis-acting elements of the first intron of MIF, without having any effect on the MIF promoter vector in CEMC7A cells. This suggests that the GC-rich elements within oligo-1 and oligo-6 are the core enhancer elements.
Mithramycin binds GC boxes as opposed to specifically inhibiting Sp1 binding to DNA [37,41]. The Sp family possesses eight members; Sp1–Sp4 have similar structures and Sp5–Sp8 also have similar structures, but appear to be truncated forms of Sp1– Sp4 (reviewed in [32]). Co-operation between Sp1, Sp3 and other members of the Sp family has been described previously [42,43], and as such these remain possible interaction partners for Sp1 and could influence MIF enhancer function.
To determine the physiological relevance of the enhancer, CEMC7A cells were treated with increasing concentrations of mithramycin, and comparison of MIF protein expression between treated and untreated cells was undertaken. Clearly, endogenous MIF production was inhibited after treating CEMC7A cells with 10 nM of mithramycin for 48 h (Fig. 5). Importantly, the inhibitory effect of mithramycin on MIF production was specific to the enhancer activity, as mithramycin had no effect on the MIF promoter itself, despite its high GC content, and the presence of a number of putative Sp1 sites.
Roger et al. found that mithramycin inhibited the transcriptional activity of a MIF promoter reporter assay and MIF mRNA expression in monocytic THP-1 cells [44]. We have looked at the effects of mithramycin in a different cell type, a T lymphoblastic cell line, CEMC7A, and found no effect of mithramycin on the MIF promoter but inhibition of the MIF intronic enhancer. These observations show that mithramycin, albeit potentially through different mechanisms, plays an immunosuppressive role on immune cells via the down-regulation of MIF.
As an anti-neoplastic drug, the role of mithramycin in apoptosis [45] and in inflammation has been investigated previously [46]. Mithramycin has also been shown to inhibit bone resorption [47]. Analogues of mithramycin have been developed recently which have enhanced efficacy and specificity [48,49]. Use of such drugs for the selective reduction of MIF in circulating immune cells could enhance the therapeutic advantage of glucocorticoids, which has important implications in the long-term management of chronic inflammatory conditions such as RA.
Acknowledgments
We are grateful to Dr May Tassabehji for expert help with Southern blotting. The authors' laboratories are supported by the Manchester Academic Health Sciences Centre (MAHSC), the NIHR Biomedical Research Centre and by the National Health and Medical Research Council of Australia. This work was supported by grants from the Arthritis Research Campaign, UK and the NHMRC, Australia. E. B. is a recipient of a Frederick Craven Fellowship Award.
Disclosure
The authors have nothing to declare.
References
- 1.Calandra T, Bernhagen J, Mitchell RA, Bucala R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J Exp Med. 1994;179:1895–902. doi: 10.1084/jem.179.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernhagen J, Calandra T, Mitchell RA, et al. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365:756–9. doi: 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- 3.Bernhagen J, Bacher M, Calandra T, et al. An essential role for macrophage migration inhibitory factor in the tuberculin delayed-type hypersensitivity reaction. J Exp Med. 1996;183:277–82. doi: 10.1084/jem.183.1.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kudrin A, Scott M, Martin S, et al. Human macrophage migration inhibitory factor: a proven immunomodulatory cytokine. J Biol Chem. 2006;281:29641–51. doi: 10.1074/jbc.M601103200. [DOI] [PubMed] [Google Scholar]
- 5.Mikulowska A, Metz CN, Bucala R, Holmdahl R. Macrophage migration inhibitory factor is involved in the pathogenesis of collagen type II-induced arthritis in mice. J Immunol. 1997;158:5514–17. [PubMed] [Google Scholar]
- 6.Leech M, Metz C, Santos L, et al. Involvement of macrophage migration inhibitory factor in the evolution of rat adjuvant arthritis. Arthritis Rheum. 1998;41:910–17. doi: 10.1002/1529-0131(199805)41:5<910::AID-ART19>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 7.Santos L, Hall P, Metz C, Bucala R, Morand EF. Role of macrophage migration inhibitory factor (MIF) in murine antigen-induced arthritis: interaction with glucocorticoids. Clin Exp Immunol. 2001;123:309–14. doi: 10.1046/j.1365-2249.2001.01423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leech M, Lacey D, Xue JR, et al. Regulation of p53 by macrophage migration inhibitory factor in inflammatory arthritis. Arthritis Rheum. 2003;48:1881–9. doi: 10.1002/art.11165. [DOI] [PubMed] [Google Scholar]
- 9.Santos LL, Dacumos A, Yamana J, Sharma L, Morand EF. Reduced arthritis in MIF deficient mice is associated with reduced T cell activation: down-regulation of ERK MAP kinase phosphorylation. Clin Exp Immunol. 2008;152:372–80. doi: 10.1111/j.1365-2249.2008.03639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leech M, Metz C, Hall P, et al. Macrophage migration inhibitory factor in rheumatoid arthritis: evidence of proinflammatory function and regulation by glucocorticoids. Arthritis Rheum. 1999;42:1601–8. doi: 10.1002/1529-0131(199908)42:8<1601::AID-ANR6>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 11.Onodera S, Tanji H, Suzuki K, et al. High expression of macrophage migration inhibitory factor in the synovial tissues of rheumatoid joints. Cytokine. 1999;11:163–7. doi: 10.1006/cyto.1998.0402. [DOI] [PubMed] [Google Scholar]
- 12.Onodera S, Kaneda K, Mizue Y, Koyama Y, Fujinaga M, Nishihira J. Macrophage migration inhibitory factor up-regulates expression of matrix metalloproteinases in synovial fibroblasts of rheumatoid arthritis. J Biol Chem. 2000;275:444–50. doi: 10.1074/jbc.275.1.444. [DOI] [PubMed] [Google Scholar]
- 13.Onodera S, Suzuki K, Matsuno T, Kaneda K, Kuriyama T, Nishihira J. Identification of macrophage migration inhibitory factor in murine neonatal calvariae and osteoblasts. Immunology. 1996;89:430–5. doi: 10.1046/j.1365-2567.1996.d01-751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pakozdi A, Amin MA, Haas CS, et al. Macrophage migration inhibitory factor: a mediator of matrix metalloproteinase-2 production in rheumatoid arthritis. Arthritis Res Ther. 2006;8:R132. doi: 10.1186/ar2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Onodera S, Oshima S, Nishihira J, et al. Active immunization against macrophage migration inhibitory factor using a novel DNA vaccine prevents ovariectomy-induced bone loss in mice. Vaccine. 2008;26:829–36. doi: 10.1016/j.vaccine.2007.11.066. [DOI] [PubMed] [Google Scholar]
- 16.Onodera S, Sasaki S, Ohshima S, et al. Transgenic mice overexpressing macrophage migration inhibitory factor (MIF) exhibit high-turnover osteoporosis. J Bone Miner Res. 2006;21:876–85. doi: 10.1359/jbmr.060310. [DOI] [PubMed] [Google Scholar]
- 17.Roger T, Chanson AL, Knaup-Reymond M, Calandra T. Macrophage migration inhibitory factor promotes innate immune responses by suppressing glucocorticoid-induced expression of mitogen-activated protein kinase phosphatase-1. Eur J Immunol. 2005;35:3405–13. doi: 10.1002/eji.200535413. [DOI] [PubMed] [Google Scholar]
- 18.Aeberli D, Leech M, Morand EF. Macrophage migration inhibitory factor and glucocorticoid sensitivity. Rheumatology (Oxf) 2006;45:937–43. doi: 10.1093/rheumatology/kel142. [DOI] [PubMed] [Google Scholar]
- 19.Calandra T, Bernhagen J, Metz CN, et al. MIF as a glucocorticoid-induced modulator of cytokine production. Nature. 1995;377:68–71. doi: 10.1038/377068a0. [DOI] [PubMed] [Google Scholar]
- 20.Alourfi Z, Donn RP, Stevens A, Berry A, McMaster A, Ray DW. Glucocorticoids suppress macrophage migration inhibitory factor (MIF) expression in a cell-type specific manner. J Mol Endocrinol. 2005;34:583–95. doi: 10.1677/jme.1.01647. [DOI] [PubMed] [Google Scholar]
- 21.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3:791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baugh JA, Gantier M, Li L, Byrne A, Buckley A, Donnelly SC. Dual regulation of macrophage migration inhibitory factor (MIF) expression in hypoxia by CREB and HIF-1. Biochem Biophys Res Commun. 2006;347:895–903. doi: 10.1016/j.bbrc.2006.06.148. [DOI] [PubMed] [Google Scholar]
- 23.Welford SM, Bedogni B, Gradin K, Poellinger L, Broome PM, Giaccia AJ. HIF1{alpha} delays premature senescence through the activation of MIF. Genes Dev. 2006;20:3366–71. doi: 10.1101/gad.1471106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elsby LM, Donn RP, Alourfi Z, Green LM, Beaulieu E, Ray DW. Hypoxia and glucocorticoid signalling converge to regulate MIF gene expression. Arthritis Rheum. 2009;60:2220–31. doi: 10.1002/art.24659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waeber G, Thompson N, Chautard T, et al. Transcriptional activation of the macrophage migration-inhibitory factor gene by the corticotropin-releasing factor is mediated by the cyclic adenosine 3′,5′-monophosphate responsive element-binding protein CREB in pituitary cells. Mol Endocrinol. 1998;12:698–705. doi: 10.1210/mend.12.5.0109. [DOI] [PubMed] [Google Scholar]
- 26.Donn R, Berry A, Stevens A, et al. Use of gene expression profiling to identify a novel glucocorticoid sensitivity determining gene, BMPRII. FASEB J. 2007;21:402–14. doi: 10.1096/fj.06-7236com. [DOI] [PubMed] [Google Scholar]
- 27.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucl Acids Res. 1983;11:1475–89. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wierstra I. Sp1: emerging roles – beyond constitutive activation of TATA-less housekeeping genes. Biochem Biophys Res Commun. 2008;372:1–13. doi: 10.1016/j.bbrc.2008.03.074. [DOI] [PubMed] [Google Scholar]
- 29.Blackwood EM, Kadonaga JT. Going the distance: a current view of enhancer action. Science. 1998;281:61–3. doi: 10.1126/science.281.5373.60. [DOI] [PubMed] [Google Scholar]
- 30.Lee TI, Young RA. Transcription of eukaryotic protein-coding genes. Annu Rev Genet. 2000;34:77–137. doi: 10.1146/annurev.genet.34.1.77. [DOI] [PubMed] [Google Scholar]
- 31.Safe S, Kim K. Nuclear receptor-mediated transactivation through interaction with Sp proteins. Prog Nucleic Acid Res Mol Biol. 2004;77:1–36. doi: 10.1016/S0079-6603(04)77001-4. [DOI] [PubMed] [Google Scholar]
- 32.Safe S, Abdelrahim M. Sp transcription factor family and its role in cancer. Eur J Cancer. 2005;41:2438–48. doi: 10.1016/j.ejca.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 33.Marin M, Karis A, Visser P, Grosveld F, Philipsen S. Transcription factor Sp1 is essential for early embryonic development but dispensable for cell growth and differentiation. Cell. 1997;89:619–28. doi: 10.1016/s0092-8674(00)80243-3. [DOI] [PubMed] [Google Scholar]
- 34.Koller CA, Miller DM. Preliminary-observations on the therapy of the myeloid blast phase of chronic granulocytic-leukemia with plicamycin and hydroxyurea. N Engl J Med. 1986;315:1433–8. doi: 10.1056/NEJM198612043152301. [DOI] [PubMed] [Google Scholar]
- 35.Dutcher JP, Coletti D, Paietta E, Wiernik PH. A pilot study of alpha-interferon and plicamycin for accelerated phase of chronic myeloid leukemia. Leuk Res. 1997;21:375–80. doi: 10.1016/s0145-2126(96)00108-7. [DOI] [PubMed] [Google Scholar]
- 36.Blume SW, Snyder RC, Ray R, Thomas S, Koller CA, Miller DM. Mithramycin inhibits Sp1 binding and selectively inhibits transcriptional activity of the dihydrofolate-reductase gene in vitro and in vivo. J Clin Invest. 1991;88:1613–21. doi: 10.1172/JCI115474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ray R, Snyder RC, Thomas S, Koller CA, Miller DM. Mithramycin blocks protein-binding and function of the Sv40 early promoter. J Clin Invest. 1989;83:2003–7. doi: 10.1172/JCI114110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qiu ZH, Norflus F, Singh B, et al. Sp1 is up-regulated in cellular and transgenic models of Huntington disease, and its reduction is neuroprotective. J Biol Chem. 2006;281:16672–80. doi: 10.1074/jbc.M511648200. [DOI] [PubMed] [Google Scholar]
- 39.Remsing LL, Bahadori HR, Carbone GM, McGuffie EM, Catapano CV, Rohr J. Inhibition of c-src transcription by mithramycin: structure–activity relationships of biosynthetically produced mithramycin analogues using the c-src promoter as target. Biochemistry (Mosc) 2003;42:8313–24. doi: 10.1021/bi034091z. [DOI] [PubMed] [Google Scholar]
- 40.Cheng YH, Imir A, Suzuki T, et al. SP1 and SP3 mediate progesterone-dependent induction of the 17beta hydroxysteroid dehydrogenase type 2 gene in human endometrium. Biol Reprod. 2006;75:605–14. doi: 10.1095/biolreprod.106.051912. [DOI] [PubMed] [Google Scholar]
- 41.Miller DM, Polansky DA, Thomas SD, et al. Mithramycin selectively inhibits transcription of G-C containing Dna. Am J Med Sci. 1987;294:388–94. doi: 10.1097/00000441-198711000-00015. [DOI] [PubMed] [Google Scholar]
- 42.Saito T, Takahashi Y, Hashimoto H, Kamataki T. Novel transcriptional regulation of the human CYP3A7 gene by Sp1 and Sp3 through nuclear factor kappa B-like element. J Biol Chem. 2001;276:38010–22. doi: 10.1074/jbc.M106130200. [DOI] [PubMed] [Google Scholar]
- 43.Miki N, Ikuta M, Matsui T. Hypoxia-induced activation of the retinoic acid receptor-related orphan receptor alpha 4 gene by an interaction between hypoxia-inducible factor-1 and Sp1. J Biol Chem. 2004;279:15025–31. doi: 10.1074/jbc.M313186200. [DOI] [PubMed] [Google Scholar]
- 44.Roger T, Ding X, Chanson AL, Renner P, Calandra T. Regulation of constitutive and microbial pathogen-induced human macrophage migration inhibitory factor (MIF) gene expression. Eur J Immunol. 2007;37:3509–21. doi: 10.1002/eji.200737357. [DOI] [PubMed] [Google Scholar]
- 45.Leroy I, Laurent G, Quillet-Mary A. Mithramycin A activates Fas death pathway in leukemic cell lines. Apoptosis. 2006;11:113–19. doi: 10.1007/s10495-005-3089-z. [DOI] [PubMed] [Google Scholar]
- 46.Liacini A, Sylvester J, Li WQ, Zafarullah M. Mithramycin downregulates proinflammatory cytokine-induced matrix metalloproteinase gene expression in articular chondrocytes. Arthritis Res Ther. 2005;7:R777–83. doi: 10.1186/ar1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kiang DT. Effect of mithramycin on bone beta-glucuronidase and resorption. Calcif Tissue Res. 1978;26:209–13. doi: 10.1007/BF02013260. [DOI] [PubMed] [Google Scholar]
- 48.Lombo F, Menendez N, Salas JA, Mendez C. The aureolic acid family of antitumor compounds: structure, mode of action, biosynthesis, and novel derivatives. Appl Microbiol Biotechnol. 2006;73:1–14. doi: 10.1007/s00253-006-0511-6. [DOI] [PubMed] [Google Scholar]
- 49.Albertini V, Jain A, Vignati S, et al. Novel GC-rich DNA-binding compound produced by a genetically engineered mutant of the mithramycin producer Streptomyces argillaceus exhibits improved transcriptional repressor activity: implications for cancer therapy. Nucl Acids Res. 2006;34:1721–34. doi: 10.1093/nar/gkl063. [DOI] [PMC free article] [PubMed] [Google Scholar]