

Abstract
Systemic sclerosis (SSc) is an autoimmune disease characterized by fibrotic changes in skin and other organs involving excessive collagen deposition. Here we investigated the effect of intravenous immunoglobulin (IVIG) on fibrosis in a murine model of bleomycin (BLM)-induced scleroderma. Scleroderma was induced in C3H/He J mice by subcutaneous BLM injections daily for 35 days. The collagen content in skin samples from the BLM-injected group (6·30 ± 0·11 mg/g tissue) was significantly higher than the PBS group (5·80 ± 0·10 mg/g tissue), and corresponded with dermal thickening at the injection site. In contrast, mice treated with IVIG for 5 consecutive days after initiating BLM injection showed lesser collagen content significantly (IVIG group, 5·61 ± 0·09 mg/g tissue; BLM vs. IVIG). In order to investigate the cellular and protein characteristics in the early stage of the model, the skin samples were obtained 7 days after the onset of experiment. Macrophage infiltration to the dermis, monocyte chemoattractant protein (MCP-1)-positive cells, and increased TGF-β1 mRNA expression were also observed in the BLM group. IVIG inhibited these early fibrogenic changes; MCP-1 expression was significantly lesser for the IVIG group (1·52 ± 0·19 pg/mg tissue) than for the BLM group (2·49 ± 0·26 pg/mg tissue). In contrast, TGF-β1 mRNA expression was significantly inhibited by IVIG. These results suggest that IVIG treatment may inhibit macrophage recruitment to fibrotic sites by down regulating MCP-1 and TGF-β production, and thus could be a potential drug for managing fibrotic disorders such as SSc.
Keywords: bleomycin-induced fibrosis, immunomodulatory action, intravenous immunoglobulin
Introduction
Systemic sclerosis (SSc) is an autoimmune disease characterized by severe alterations in the microvasculature, prominent inflammatory and immunological changes, and excessive accumulation of extracellular matrix in the skin and internal organs. Despite research efforts, no standard protocol for treating SSc has been established to date. Controlled trials have demonstrated that some immunosuppressive therapies interrupt the immune-mediated portion of its pathogenic cycle, but such findings have been rather inconsistent.
For over 30 years, human intravenous immunoglobulin (IVIG) has been an important treatment option for a number of clinical indications, such as primary immunodeficiency disease, autoimmune disease, and acute inflammatory conditions [1]. IVIG has been also reported to reduce skin stiffness in SSc patients in several open trials [2,3]. Although the precise mechanisms of IVIG are unclear, animal models are crucial for investigating SSc pathogenesis and its various therapeutic approaches. We previously reported several immunomodulatory activities in IVIG using various experimental models of autoimmunity [4–6]. IVIG is classically manufactured from pooled plasma of healthy donors, and contains various antibodies specific to bacteria and toxins. However, this may not be the only mechanism for managing autoimmune diseases. Since pathology of SSc is complicated, IVIG might show some immunological effects on activated fibroblast to produce excessive collagen.
The therapeutic effects of IVIG most likely reflect the functions of natural antibodies in maintaining immune homeostasis in healthy individuals [7]. In this study, we confirm that infiltration of macrophage in the dermis is suppressed by IVIG.
Animal models that exhibit all aspects of SSc are currently unavailable, but a few experimental systems replicating its pathogenic aspects have been reported by us and others [8–10]. In addition, we previously established a murine SSc model by injecting animals with bleomycin (BLM) daily for 35 days. Histological examination on skin samples of BLM-treated mice revealed thickened dermal areas with collagen bundles, and deposition of homogeneous material with cellular infiltrates mimicking human scleroderma at injection sites thus confirming SSc onset.
We investigated the influence of IVIG administration on inducing dermal sclerosis using the BLM-based murine model, and identified that IVIG attenuated excessive collagen accumulation by inhibiting monocyte chemoattractant protein (MCP-1) and transforming growth factor beta (TGF-β) in this model.
Materials and methods
Mice
Specific, pathogen-free, female C3H/HeJ mice (age: 6 weeks, weight 17–22 g) were purchased from Japan SLC, Inc. (Shizuoka, Japan). Mice were kept in our animal facility with free access to food and water.
IVIG
Human immunoglobulin preparation (Venoglobulin-IH™, Benesis Corporation, Osaka, Japan) was administrated intravenously at 400 mg/kg/day into tail veins of the IVIG group mice 5 consecutive days after initiating BLM treatment. The same volume of saline was administered to the BLM group. Amount of the drug used are correspond to that used in clinical.
BLM-induced murine fibrosis model
The BLM-induced murine fibrosis model was performed using a previously described method [11]. Briefly, 100 µL of 600 µg/ml BLM (Nippon Kayaku Co., Japan) in PBS was injected subcutaneously into the shaved back of mice using 27-gauge needles daily for 35 days.
Collagen content in the skin
Punched skin samples (diameter: 14 mm) were obtained from shaved backs of mice 35 days after treatment. Each sample was treated with 0·5 mol/l acetic acid containing pepsin (0·3 mg/10 mg tissue) at 4°C overnight. Then the collagen content in skin was determined using the Sircol™ Soluble Collagen Assay kit (Biocolor Ltd, Northern Ireland).
Quantification of MCP-1
Punched skin samples (diameter: 8 mm) were obtained from shaved backs of mice 7 days after BLM treatment. Each sample homogenized in Dulbecco's modified Eagle's medium, centrifuged for 5 min at 200g, and the supernatants were collected. Immunoreactive MCP-1 levels were determined using a commercial ELISA kit (Biosciences, San Diego, CA) following the manufacturer's instructions.
Histological examination
Skin samples were obtained from shaved backs of mice 7 days after BLM treatment. Skin sections were routinely stained with hematoxylin and eosin. In addition, collagen production was identified by Masson's trichrome stain. The dermal thickness of BLM- (BLM and IVIG groups) and PBS- (PBS group) injected skin were calculated using an image processor Win Roof (Mitani Corp., Tokyo, Japan). To avoid bias, the histologist was single blinded to the treatment groups of mice.
Immunohistochemical examination
For immunohistochemical experiments, skin samples were cut into halves to divide the BLM-injected region equally, fixed in 10% formalin solution, embedded in paraffin, and sections were stained. Rabbit anti-Iba-1 polyclonal antibody (Wako, Tokyo Japan) with final concentration 500 ng/ml was used for staining macrophages after 7 days of initiating BLM treatment. Iba-1 mainly infiltrate macrophage-like and fibroblastic cells in BLM-treated skin. Primary antibodies against MCP-1 (Novus Biologicals, Littleton, CO) and collagen I (abcam, cambridge, MA) were used according to manufacturers guidelines. Samples were subsequently incubated with a secondary antibody labeled with peroxidase (Nichirei Bioscience Inc., Tokyo, Japan). The skin sections were developed with diaminobenzidine solution as chromogen, counterstained with hematoxylin, dehydrated, cleared, and mounted.
RNA isolation and RT-PCR
Total RNA was isolated from skin samples of the model mice (7 days after initiating BLM treatment) using the Ribo Pure™ extraction kit (Ambion Inc., Austin, TX). All samples were treated with RNA Stabilization Reagent (RNAlater, Qiagen, Valencia, CA) at 37°C overnight and stored at −80°C until use. Total RNA was reverse-transcribed into cDNA according to the manufacturer's protocol for the Reverse Transcription System (Promega, Madison, WI). TGF-β1 mRNA expression was analyzed by quantitative RT-PCR according to the manufacturer's instructions (Applied Biosystems, Foster City, CA). Sequence-specific primers and probes were designed using pre-developed TaqMan® assay reagents (Applied Biosystems). RT-PCR (1 cycle at 48°C for 30 min, 1 cycle at 95°C for 10 min, and 40 cycles at 95°C for 15 s and 60°C for 60 s) was performed using a real-time PCR System 7500 (Applied Biosystems). GAPDH was used to normalize mRNA. To compare TGF-β1 mRNA and housekeeping GAPDH mRNA expression, the relative expression of PCR products was determined using the ΔΔCt method [12]. Fold induction is equal to 2 − [ΔΔCt], where Ct = threshold cycle, and ΔΔCt = [Ct gene interest (unknown) − Ct GAPDH (unknown)] − [Ct gene interest (calibrator) − Ct GAPDH (calibrator)]. One control was chosen as the calibrator. Each was examined in duplicate and the mean Ct was used in the equation.
Statistical analysis
All data are expressed as means ± SEM Statistical significance was determined using Student's t-test. P-values < 0·05 were considered significant.
Results
IVIG reduced collagen production in BLM-treatment skin
Representative histological skin sections from mice used in the BLM-induced mouse fibrosis model are shown in Fig. 1a. Mice treated with BLM for 35 days exhibited a thicker dermal layer compared to those treated with PBS. However, mice administered IVIG (400 mg/kg/day) for 5 consecutive days after initiating BLM treatment did not exhibit these fibrotic changes. Thickness of dermal layer of each mouse was calculated using an image processor and subsequent statistical analysis (Fig. 1b). The BLM group had significantly increased skin thickness (335·3 ± 14·9 µm) compared to PBS group (159·4 ± 4·9 µm; PBS vs. BLM, P < 0·01). Furthermore, IVIG treatment drastically ameliorated the dermal thickening effects of BLM-injected mice (241·1 ± 10·5 µm, BLM vs. IVIG, P < 0·01).
Fig. 1.
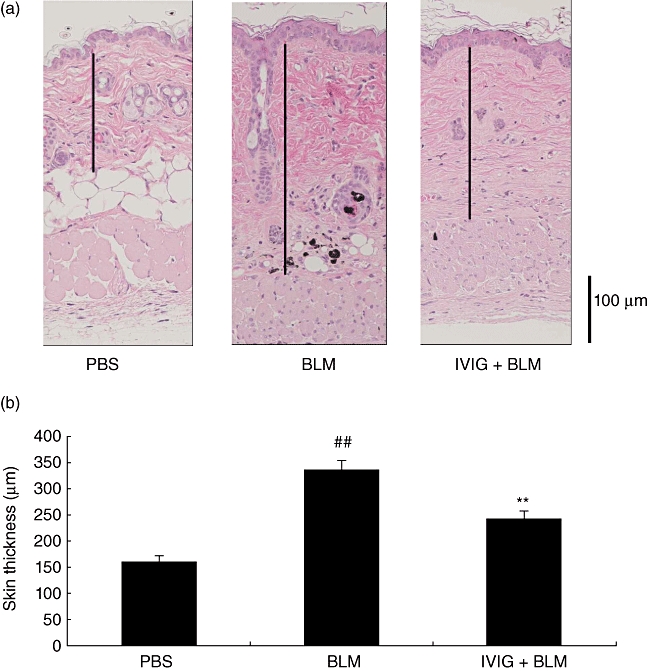
Effect of IVIG on BLM-induced fibrosis. The murine fibrosis model was induced by subcutaneously injecting mice with BLM for 35 days. Some mice were injected with PBS on the same schedule as negative control. IVIG (400 mg/kg/day) was administered intravenously for 5 consecutive days, and skin samples were taken 35 days after initiating BLM for pathological study. (a) Representative images of each group are shown, with bars and arrows representing the lengths of the dermis and dermal layer, respectively. (b) Thickness of the dermal layer was calculated using an image processor. Data are expressed as the mean ± SEM (n = 9–10). ##P < 0·01 (vs. PBS-treated mice, Student's-test), **P < 0·01 (vs. BLM-treated mice, Student's t-test).
Thickening in the dermal layer was corresponded to accumulation of collagen by Masson' s trichrome stain and immunohistochemistry of type I collagen(Fig. 2a). We determined the collagen content in skin samples from mice treated with BLM for 35 days (Fig. 2b), and found that they had significantly higher collagen content (6·30 ± 0·11 mg/g tissue) than the PBS group (5·80 ± 0·10 mg/g tissue, PBS vs. BLM, P < 0·01). Supplementary we confirmed that the similar results were obtained when the extent of fibrosis was determined with the contents of hydroxyproline, another index of fibrosis, in a different set of experiment. The content of the amino acid was significantly suppressed in the IVIG group (715 ± 11·8 µg/g tissue, n = 10) in the comparison with BLM group (775 ± 15·6 µg/g tissue, n = 10). These results show that collagen production was induced by BLM. In another group of mice, IVIG was given intravenously immediately after initiating BLM treatment. Their results revealed dermal collagen content significantly less than IVIG treatment (5·61 ± 0·09 mg/g tissue; BLM vs. IVIG, P < 0·01). In another post-onset experiment, IVIG was given intravenously 28 days after initiating BLM treatment. In addition, IVIG treatment suppressed collagen content in the dermis to a similar extent as the previous experiment (Fig. 2c). We discuss the effect of earlier treatment of IVIG later in this study.
Fig. 2.
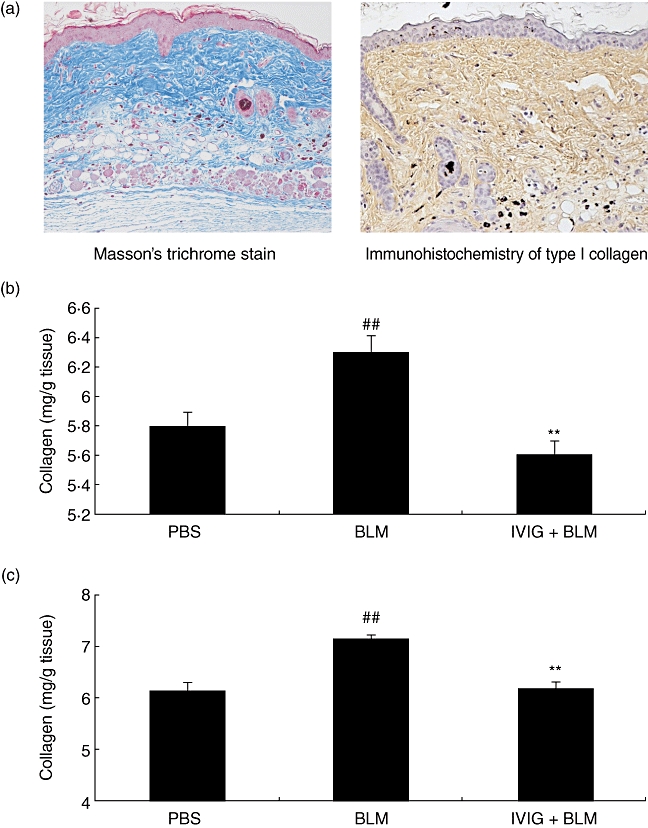
Collagen content and the effect of IVIG treatment at different time. (a)Collagen in the dermis was observed Masson's trichrome stains and immunohistochemistry of type I collagen in the BLM-treated mice. (b, c) IVIG was administered to mice intravenously (400 mg/kg/day for 5 consecutive days) either immediately (b) or 28 days (c) after initiating BLM treatment. In both experiments, skin samples were obtained 35 days after initiating BLM treatment to determine collagen content (Sircol™ Soluble Collagen Assay kit). Data are expressed as the mean ± SEM (n = 12). ##P < 0·01 (vs. PBS-treated mice, Student's t-test), **P < 0·01 (vs. BLM-treated mice, Student's t-test).
IVIG suppressed fibrogenic cytokine and chemokine in the skin
To examine the influence of TGF-β1 on IVIG mechanisms in early SSc, we obtained a skin sample 7 days after initiating the experiment. We subsequently determined mRNA expression levels. We found that TGF-β1 mRNA levels were upregulated in the BLM group compared to the PBS group, and were subsequently suppressed in the IVIG group (BLM vs. IVIG, P < 0·05) (Fig. 3).
Fig. 3.
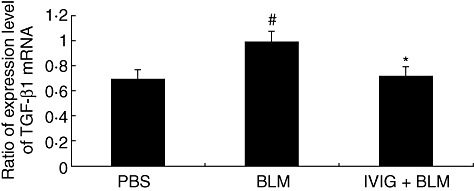
Expression of TGF-β1 mRNA in lesional skin of mice. Mice were locally treated with either PBS or BLM for 5 days and total mRNA was isolated from skin samples. IVIG was administered intravenously (400 mg/kg/day for 5 consecutive days) immediately after initiating BLM treatment. Skin samples were obtained after 7 days to determine TGF-β1 mRNA expression. Representative data are shown from 3 independent experiments. Relative amounts are expressed as arbitrary units after TGF-β1 m RNA normalized with GAPDH. Data are expressed as means ± SEM (n = 8). #P < 0·05 (vs. PBS-treated mice, Student's t-test), *P < 0·05 (vs. BLM-treated mice, Student's t-test).
We examined MCP-1 expression in this experimental in vivo model, and found that MCP-1 levels were significantly increased in the BLM group (2·49 ± 0·26 pg/mg tissue) compared to the PBS group (0·46 ± 0·04 pg/mg tissue) as seen for TGF-β1 mRNA expression. IVIG treatment lowered these levels (1·52 ± 0·19 pg/mg tissue; BLM vs. IVIG, P < 0·01) (Fig. 4).
Fig. 4.
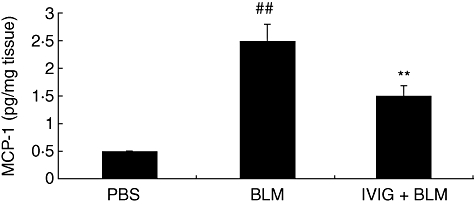
Immunoreactive MCP-1 levels determined by ELISA. Mice were locally treated with either PBS or BLM for 5 days, and IVIG was administered intravenously (400 mg/kg/day for 5 consecutive days) immediately after initiating BLM treatment. Skin samples were homogenized, centrifuged, and supernatants measured by ELISA. Data are expressed as means ± SEM (n = 12). ##P < 0·01 (vs. PBS-treated, Student's t-test), **P < 0·01 (vs. BLM-treated mice, Student's t-test).
In a preliminary study, we also measured concentrations of several inflammatory cytokines and chemokines (IL-2, IL-6, KC, MIP-1α, RANTES, GM-CSF, and TNFα) with BD FACSarray Bioanalyzer System and mRNA expression levels of IL-6 and IL-13, all of which might be involved in the IVIG mechanism in this model. Although some of these levels were increased in this model, IVIG treatment did not induce statistically significant changes (data not shown).
Immunohistochemical observation of macrophages and MCP-1
Immunohistochemistry was used to observe cellular infiltration in the dermal layer during early stages of BLM-induction. Staining with Iba-1 (anti-mouse macrophage antibody) revealed that most infiltrated cells were macrophages. Representative images of Iba-1 positive cells are shown in Fig. 5a and c. We also calculated the area of Iba-1 positive cells for each group, which revealed that IVIG treatment suppressed infiltration of macrophages (significantly increased by BLM treatment) in the dermis (Fig. 6). Furthermore, MCP-1 was mainly found in BLM-treated skin samples (Fig. 5b and d). These results suggest this chemokine may be expressed mainly in the infiltrated macrophages because most MCP-1-positive areas colocalized with Iba-1 staining. However, some MCP-1 staining was also shown in fibroblasts. MCP-1-positive area was less for the IVIG group than the BLM group (Fig. 5d).
Fig. 5.
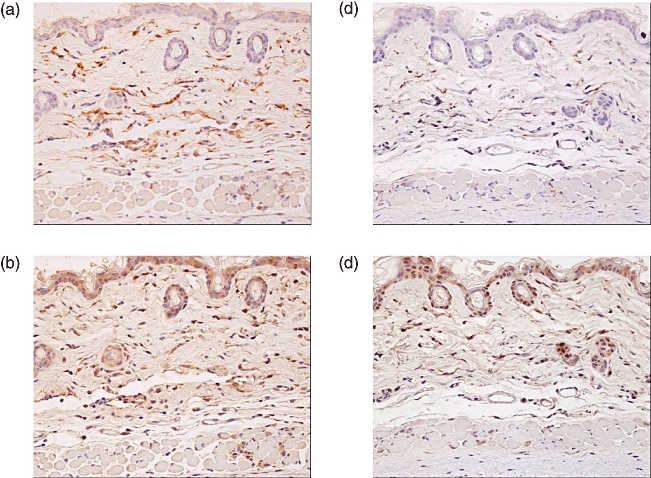
Immunohistochemical localization of MCP-1. Seven days after the first BLM treatment, immunohistochemistry was performed to identify cells that were positive for Iba-1 (a, c) and MCP-1 (b, d). IVIG was administered intravenously (400 mg/kg for 5 consecutive days) immediately after initiating BLM treatment (c, d). Positive staining for macrophages and MCP-1 were primarily found in BLM-treated skin.
Fig. 6.
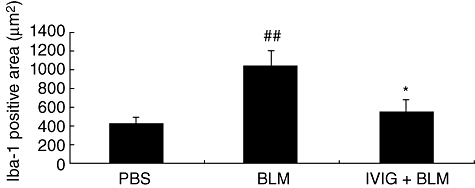
Immunohistochemical localization of macrophages. Seven days after initiating BLM treatment, immunoreactive cells for Iba-1 were correlated to mainly infiltrating macrophage-like cells in BLM-treated skin. IVIG was administered intravenously (400 mg/kg/day for 5 consecutive days) immediately after initiating BLM treatment. Data are expressed as means ± SEM (n = 12). ##P < 0·01 (vs. PBS-treated, Student's t-test), *P < 0·05 (vs. BLM-treated mice, Student's t-test).
Discussion
In this study, we used BLM-induced murine fibrotic models to investigate the efficacy and mechanisms of IVIG in treating SSc. In our first experiments, we observed dermal thickening and excess collagen production with infiltration of macrophages in the lesional skin of BLM-treated mice. We also examined how IVIG would affect the lesional skin if administered on the first day of BLM treatment. IVIG was found to almost completely suppress the increase in dermal thickness, as well as collagen content induced by BLM treatment. The concentration of IVIG and the time schedule used in this study was implemented to match typical clinical regimens. We then examined the effect of IVIG on various cytokines and chemokines, which may induce the first step toward fibrosis. Increase in expression of fibrosis-related cytokines, chemokines, and enzymes prior to the increase of collagens and other extracellular matrix proteins in the lesional area of dermis has been investigated [13]. Here we focused on TGF-β1 and MCP-1 because both are crucial factors in the induction of fibrosis. Their increased levels were drastically ameliorated by IVIG. We also examined other cytokines, such as IL-2, IL-6, KC, MIP-1α, RANTES, GM-CSF, and TNFα, but they were largely unaffected (data not shown). In addition, we successfully investigated MCP-1 suppression by IVIG using ELISA and immunohistochemical examination for the first time.
TGF-β is believed to influence the development of tissue fibrosis in SSc patients by directly inducing collagen formation from fibroblasts in the dermal layer [14]. We previously reported that local BLM injections induced collagen production and cellular infiltrates the skin, lung inflammation and fibrosis, and increased production of TGF-β1 in vivo[11]. TGF-β has been localized to the sites of mononuclear cell infiltration and fibroblast activation by immunostaining [11]. It is well known that activation of TGF-β cascade is regulated not only by the expression of the molecule but also molecular change from the latent molecule to the active one. Whether this kind of activation is occurred or not in this model and IVIG inhibition is observed at this point is interesting and should be elucidated.
The role of chemokines in BLM-induced scleroderma has been highlighted in literature [15,16]. MCP-1 is a multifunctional inflammatory chemokine belonging to the C-C chemokine superfamily. Upregulation of collagen expression is preceded by monocyte infiltration and increase in TGF-β mRNA expression. In addition, MCP-1 might influence modulation of extracellular matrix deposition by stimulating interstitial collagenase production in human fibroblasts [17]. Furthermore, fibrosis is not caused by BLM in MCP-1-deficient mice [18],. Other studies have shown that this chemokine is also important in other scleroderma models. MCP-1 is upregulated in growth factor-injected mice undergoing simultaneous treatment with bFGF and CTGF increased skin fibrosis [19]. By contrast, these treatments in MCP-1-deficient mice decreased collagen content in the skin suggesting the influence of MCP-1 in recruiting inflammatory cells [19]. These results suggest the MCP-1 may be a key determinant in the development of skin fibrosis induced by BLM [18]. Administration of an anti-MCP-1 neutralizing antibody reduced dermal sclerosis and decreased the skin collagen content usually seen in the pathogenesis of BLM-induced scleroderma. MCP-1 might contribute to the induction of dermal sclerosis through the infiltration of macrophages [16] or through the direct activation of the fibroblasts. This critical role played by the chemokine in SSc pathogenesis has also been demonstrated.
In this study, we mainly focused on effects of IVIG treatment on early fibrotic changes. However, we also showed that the drug significantly reduced the dermal thickness seen in BLM-treated mice, even when administered at the later stage of the experiment (Fig. 2b). This indicates that other mechanisms may exist for the action of IVIG in this study, with continued involvement of cytokines and chemokines resulting in scleroderma through an experiment. However, this point should be further elucidated in future studies. In various experimental models of diseases in mice and rats, pharmacological properties of IVIG have been reported. Indeed, immunological responsibility between human IgG and Fc receptor on the murine cells might be limited in this model. On the other hand, IVIG is considered to contain much more types of antibodies derived from several thousands of donors than that from mice bred in specific pathogen free conditions.
In conclusion, IVIG may act by inhibiting the recruitment of macrophages to the sites associated with fibrotic skin disorders. Furthermore, IVIG is thought to downregulate MCP-1 and TGF-β production by macrophages and monocytes, which is likely to activate fibroblasts, ultimately resulting in excessive accumulation of collagen in skin. This indicates that IVIG may become an important therapy for treating SSc patients.
Disclosure
Nothing to disclose.
References
- 1.Brandt D, Gershwin ME. Common variable immune deficiency and autoimmunity. Autoimmun Rev. 2006;5:465–70. doi: 10.1016/j.autrev.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 2.Ihn H, Mimura Y, Yazawa N, et al. High-dose intravenous immunoglobulin infusion as treatment for diffuse scleroderma. Br J Dermatol. 2007;156:1058–60. doi: 10.1111/j.1365-2133.2007.07777.x. [DOI] [PubMed] [Google Scholar]
- 3.Levy Y, Amital H, Langevitz P, et al. Intravenous immunoglobulin modulates cutaneous involvement and reduces skin fibrosis in systemic sclerosis: an open-label study. Arthritis Rheum. 2004;50:1005–7. doi: 10.1002/art.20195. [DOI] [PubMed] [Google Scholar]
- 4.Wada J, Shintani N, Kikutani K, Nakae T, Yamauchi T, Takechi K. Intravenous immunoglobulin prevents experimental autoimmune myositis in SJL mice by reducing anti-myosin antibody and by blocking complement deposition. Clin Exp Immunol. 2001;124:282–9. doi: 10.1046/j.1365-2249.2001.01499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sugihara T, Sekine C, Nakae T, et al. A new murine model to define the critical pathologic and therapeutic mediators of polymyositis. Arthritis Rheum. 2007;56:1304–14. doi: 10.1002/art.22521. [DOI] [PubMed] [Google Scholar]
- 6.Shintani N, Nakajima T, Okamoto T, Kondo T, Nakamura N, Mayumi T. Involvement of CD4+ T cells in the development of dextran sulfate sodium-induced experimental colitis and suppressive effect of IgG on their action. Gen Pharmacol. 1998;31:477–81. doi: 10.1016/s0306-3623(98)00004-4. [DOI] [PubMed] [Google Scholar]
- 7.Vani J, Elluru S, Negi V-S, et al. Role of natural antibodies in immune homeostasis: IVIG perspective. Autoimmun Rev. 2008;7:440–4. doi: 10.1016/j.autrev.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 8.Yamamoto T, Takagawa S, Katayama I, Nishioka K. Anti-sclerotic effect of transforming growth factor-β antibody in a mouse model of bleomycin-induced scleroderma. Clin Immunol. 1999;92:6–13. doi: 10.1006/clim.1999.4720. [DOI] [PubMed] [Google Scholar]
- 9.Matsushita M, Yamamoto T, Nishioka K. Plasminogen activator inhibitor-1 is elevated, but not essential, in the development of bleomycin-induced murine scleroderma. Clin Exp Immunol. 2005;139:429–38. doi: 10.1111/j.1365-2249.2005.02718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lakos G, Takagawa S, Varga J. Animal models of scleroderma. Method Mol Med. 2004;102:377–93. doi: 10.1385/1-59259-805-6:377. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto T, Kuroda M, Nishioka K. Animal model of sclerotic skin. III: histopathological comparison of bleomycin-induced scleroderma in various mice strains. Arch Dermatol Res. 2000;292:535–41. doi: 10.1007/s004030000183. [DOI] [PubMed] [Google Scholar]
- 12.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto T. The bleomycin-induced scleroderma model: what have we learned for scleroderma pathogenesis? Arch Dermatol Res. 2006;297:333–44. doi: 10.1007/s00403-005-0635-z. [DOI] [PubMed] [Google Scholar]
- 14.Charles C, Clements P, Furst DE. Systemic sclerosis: hypothesis-driven treatment strategies. The Lancet. 2006;367:1683–91. doi: 10.1016/S0140-6736(06)68737-0. [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto T. Chemokines and chemokine receptors in scleroderma. Int Arch Allergy Immunol. 2006;140:345–56. doi: 10.1159/000094242. [DOI] [PubMed] [Google Scholar]
- 16.Yamamoto T, Nishioka K. Role of monocyte chemoattractant protein-1 and its receptor, CCR-2, in the pathogenesis of bleomycin-induced scleroderma. Invest Dermatol. 2003;121:510–6. doi: 10.1046/j.1523-1747.2003.12408.x. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto T, Eckes B, Mauch C, Hartmann K, Krieg T. Monocyte chemoattractant protein-1 enhances gene expression and synthesis of matrix metalloproteinase-1 in human fibroblasts by an autocrine IL-1 alpha loop. J Immunol. 2000;164:6174–9. doi: 10.4049/jimmunol.164.12.6174. 15. [DOI] [PubMed] [Google Scholar]
- 18.Ferreira AM, Takagawa S, Fresco R, Zhu X, Varga J, Dipietro LA. Diminished induction of skin fibrosis in mice with MCP-1 deficiency. J Invest Dermatol. 2006;126:1900–8. doi: 10.1038/sj.jid.5700302. [DOI] [PubMed] [Google Scholar]
- 19.Chujo S, Shirasaki F, Kondo-Miyazaki M, Ikawa Y, Takehara K. Role of Connective tissue growth factor and its interaction with basic fibroblast growth factor and macrophage chemoattractant protein-1 in skin fibrosis. J Cell Physiol. 2009;220:189–95. doi: 10.1002/jcp.21750. [DOI] [PubMed] [Google Scholar]