

Abstract
BACKGROUND
The clinical success of the nucleoside analogs 5-aza-cytidine (5-azaC) and 5-aza-2′deoxycytidine (5-aza-dC) as DNA methyltransferase (DNMT) inhibitors has spurred interest in the development of non-nucleoside inhibitors with improved pharmacologic and safety profiles. Because DNMT catalysis features attack of cytosine bases by an enzyme thiol group, we tested whether disulfiram (DSF), a thiol-reactive compound with known clinical safety, demonstrated DNMT inhibitory activity.
METHODS
Inhibition of DNMT1 activity by DSF was assessed using methyltransferase activity assays with recombinant DNMT1. Next, prostate cancer cell lines were exposed to DSF and assessed for: i) reduction of global 5-methyl cytosine (5meC) content using liquid chromatography/tandem mass spectrometry (LC-MS/MS); ii) gene-specific promoter demethylation by methylation-specific PCR (MSP); and iii) gene-reactivation by real-time RT-PCR. DSF was also tested for growth inhibition using prostate cancer cell lines propagated in vitro in cell culture and in vivo as xenografts in nude mice.
RESULTS
Disulfiram showed a dose-dependent inhibition of DNMT1 activity on a hemimethylated DNA substrate. In prostate cancer cells in culture, DSF exposure led to reduction of global genomic 5meC content, increase in unmethylated APC and RARB gene promoters, and associated re-expression of these genes, but did not significantly alter prostate-specific antigen (PSA) expression. DSF significantly inhibited growth and clonogenic survival of prostate cancer cell lines in culture and showed a trend for reduced growth of prostate cancer xenografts.
CONCLUSIONS
Disulfiram is a non-nucleoside DNMT1 inhibitor that can reduce global 5meC content, reactivate epigenetically silenced genes, and significantly inhibit growth in prostate cancer cell lines.
Keywords: DNA methyltransferase inhibitor, DNA methylation, Prostate cancer, Disulfiram
INTRODUCTION
Alterations in DNA methylation, a key epigenetic process affecting chromatin structure and function without altering the underlying DNA base pairing, occur early in human prostate cancer and other cancers and can be conserved during cancer progression [1–6]. These DNA methylation changes are reversible, making them an interesting target for the treatment and prevention of prostate cancer [4]. Methylation of CpG dinucleotides in gene promoter regions can result in silencing of gene expression [7–9]. These CpG methylation marks are established and maintained by a group of DNA methyltransferases (DNMTs), which catalyze the transfer of a methyl group from the donor molecule S-adenosylmethionine (SAM) to a cytosine in the DNA. Inhibition of DNMT function can potentially reverse some of the cancer-associated methylation marks [10], and lead to reprogramming of the epigenetic make up of cancer cells and therefore represents an attractive therapeutic avenue [4,11].
In recent years several inhibitors of DNMTs have been developed and evaluated in pre-clinical models and in clinical trials [9,12–14]. Among these, 5-azacytidine (5-azaC) and 5-aza-deoxycytidine (5-aza-dC) have won Food and Drug Administration (FDA) approval for treatment of myelodysplastic syndromes (MDS) [15,16], and these agents and others are being tried alone and in combination with other drugs as cancer therapeutic agents. One major disadvantage of 5-azaC and 5-aza-dC is that they are nucleoside analogs, whose mechanism of action involves incorporation of the aza-modified base into DNA during DNA synthesis with subsequent covalent trapping of the DNMT [17,18]. As with other nucleoside analogs, these drugs can have significant cytotoxicity and can lead to major adverse effects, including myelosuppression, when administered to patients. The development of safe and efficacious non-nucleoside inhibitors of DNMTs has been of great interest because such agents might overcome the limitations of nucleoside analogs and allow prolonged inhibition of DNMTs without accompanying safety concerns.
Since the catalytic mechanism of DNMTs involves the covalent attack at the C6 position of cytosine by the thiol group of the catalytic cysteine on the DNMT enzyme [19–21], we hypothesized that known thiol-reactive compounds could be candidate DNMT non-nucleoside inhibitors. Disulfiram (DSF) is a drug that contains strong thiol-reactive functional groups and is known to attack the thiol group of the reactive cysteine in the active site of the aldehyde dehydrogenase enzyme [22]. We therefore hypothesized that DSF may have activity as a DNMT inhibitor. DSF has a long history of clinical use for the treatment of alcohol abuse [23,24]. Additionally, a recent screen of >3,000 clinical compounds in the Johns Hopkins Drug Library revealed that DSF can very potently inhibit prostate cancer cell growth at nanomolar concentrations (J.O. Liu, J.S. Shim, S. Yegnasubramanian, W.G. Nelson, unpublished data), a finding recently confirmed in an independent report [25]. The past 60 years of clinical use and research on DSF have provided valuable information about the safety, toxicity, and pharmacological properties of DSF [24,26]. DSF shows mild side-effects and is overall well-tolerated, making it an attractive candidate for “repurposing” for novel indications.
Here, we demonstrate that DSF inhibits DNMT1 activity, resulting in decreased genomic 5-methyl cytosine (5meC) content in cell lines. We further show that DSF treatment results in de-methylation of genes hypermethylated in prostate cancer with subsequent re-expression of these genes, suggesting that DSF can act as an epigenetic drug by inhibiting DNMT1. We also show that DSF inhibits prostate cancer cell line growth in vitro at nanomolar concentrations and shows a trend for xenograft growth inhibition in vivo.
MATERIALS AND METHODS
Cell Lines and Cell Culture
Human prostate cancer cell lines PC3, DU-145 cells were obtained from the American Type Culture Collection. CWR22Rv1, C4-2B cells were kindly provided by Dr. John T. Isaacs (Johns Hopkins University, Baltimore, MD). Normal prostate epithelial cells (PrEC) were purchased from Lonza (Basel, Switzerland) and were cultured in PrEGM medium (Lonza). All cell lines were maintained in a humidified incubator at 5% CO2 and 37°C. DSF (tetraethylthiuram disulfide) and dimethyl sulfoxide (DMSO) were purchased from Sigma (St. Louis, MO). DSF was dissolved in DMSO at stock concentration of 5 mM and stored at −20°C. The final concentrations of DMSO in the culture medium were below 0.1%.
MTT and Colony Formation Assay
Prostate cancer cells were seeded in 96-well plates at a density of 4,000 cells per well for DU145 and PC3 and 6,000 cells per well for CWR22R and C4-2B. Cell viability was quantified using the colorimetric 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazoliumbromide (MTT) assay. Cell viability IC50 values were calculated using CalcuSyn software (Biosoft, Cambridge, UK). For colony formation assays, cells were seeded in 6 cm dishes at a density of 300 cells per dish. Forty-eight hours later, cells were checked under the microscopy to confirm attachment and fresh medium containing 100 nM DSF was then added. After 12 and 14 days, colonies were fixed, stained with methylene blue, and counted.
DNA Methyltransferase Assay In Vitro
The in vitro DNMT assay was performed as described previously [13]. Briefly, recombinant His6-DNMT1 was incubated with hemimethylated oligonucleotides as substrates together with S-adenosyl-L-[methyl-3H]methionine (3H-AdoMet; Amersham Biosciences/GE, Piscataway, NJ) in the presence or absence of DSF. After incubation at 37°C, reactions were stopped by adding unlabeled S-adenosyl-L-methionine (Sigma) and samples were bound to a SAM2® 96 Biotin Capture plate (Promega, Madison, WI). The capture plate was washed five times with PBS +2 M NaCl and two times with dH2O to remove His6-DNMT and unreacted 3H-AdoMet. Tritium incorporation was quantified using the TopCount NXT liquid scintillation counter (PerkinElmer, Waltham, MA).
Quantification of Genomic 5-Methyl-2′-Deoxycytidine (5meC) Content
Quantification of genomic 5-methyl-2′-deoxycytidine (5meC) content in genomic and control DNAs was determined by an HPLC/Tandem Mass Spectrometry (LC/MS/MS) procedure as described in Ref. [3]. The degree of DNA methylation was expressed as the ratio of 5meC to total deoxycytidine (5meC/(5meC + 2dC)).
Bisulfite Modification and Real-Time Methylation-Specific PCR (MSP)
Cellular DNA was isolated by phenol–chloroform extraction. Sample DNA was subjected to sodium-bisulfite modification using EZ DNA methylation-Gold kit (Zymo research, Orange, CA). The bisulfite converted DNA was amplified by quantitative real-time PCR using iQ™ SYBR Green Supermix (Bio-Rad, Hercules, CA). Primer sequences are listed in Table I. All PCR reactions were carried out on an iCycler real-time thermal cycler (Bio-Rad) using the following temperature cycling conditions: 95°C for 5 min followed by 40 cycles of 95°C for 15 sec, 55–60°C (detailed in Table I) for 30 sec, and 72°C for 30 sec. The amplification products were confirmed by electrophoresis on a 2% agarose gel and visualized with staining by ethidium bromide.
TABLE I.
Primers Used in This Study
| Genes | Forward primer | Reverse primer | Annealing temperature (C) | |
|---|---|---|---|---|
| APC | U- | GAGGGTATATTTTTGAGGGGTATG | AATAAAAAACACCCTAATCCACA | 58 |
| M- | TTATATGTCGGTTACGTGCGTTTATAT | GAACCAAAACGCTCCCCAT | 58 | |
| RT- | GAGACAGAATGGAGGTGCTGC | GTAAGATGATTGGAATTATCTTCT | 55 | |
| RARB | U- | TTGGGATGTTGAGAATGTGAGTGATTT | CTTACTCAACCAATCCAACCAAA-ACAA | 60 |
| M- | TGTCGAGAACGCGAGCGATTC | CGACCAATCCAACCGAAACGA | 60 | |
| RT- | GACTGTATGGATGTTCTGTCA | ATTTGTCCTGGCAGACGAAGCA | 55 | |
| TBP | RT- | CACGAACCACGGCACTGATT | TTTTCTTGCTGCCAGTCTGGA | 55 |
U, unmethylated; M, methylated; RT, RT-PCR.
RNA Isolation, Reverse Transcription, and Quantitative Real-Time PCR
Total RNA was isolated by Trizol (Invitrogen, Carlsbad, CA) extraction. Reverse transcription was performed using the SuperScript III reverse transcriptase kit (Invitrogen) according the manufacturer’s instructions. To control for variations in RNA quality and quantity, expression of the gene of interest was normalized to the expression of TATA box binding protein (TBP). Relative mRNA expression levels were calculated and expressed as 2−ΔΔCT, where CT represents threshold cycle number. ΔCTsample was defined as (CTgene of interest − CTTBP) and ΔΔCT as (ΔCTsample − ΔCTnormalization sample).
In Vivo Growth Inhibition of C4-2B Xenograft by DSF
Xenografts of C4-2B cells were generated in athymic male mice with a median weight of 30 g. All animal experiments were carried out with approval of the Johns Hopkins Animal Care and Use Committee. One million C4-2B cells were resuspended in matrigel and inoculated subcutaneously into the hind flank of each animal through 25G5/8 needles. In each treatment arm, eight mice were used. Treatment was started about 2 weeks after implantation, at which point tumors were palpable. Mice were injected intraperitoneally (i.p.) with 10, 20, or 40 mg/kg DSF or solvent control every other day. Tumor size was measured weekly and tumor volume was calculated according to the formula 0.5326 × L × W × H [27]. The weights of all mice were measured weekly. Mice were sacrificed after 3 weeks of treatment by CO2 overdose.
Western Blot Analysis of DNMT1 Protein Expression
Prostate cancer cell lines PC3, DU145, CWR22Rv1, and C4-2B and normal PrEC were lysed as described previously [28]. Lysates were separated by SDS–PAGE and transferred to PVDF membranes. Membranes were blocked for 1 hr in Odyssey blocking buffer (LI-COR; Biosciences, Lincoln, NB). Primary antibodies anti-DNMT1 (HPA002694; Sigma) and anti-Actin (A5441; Sigma) were applied over night at 4° in Odyssey blocking buffer at 1:1,000 and 1:5,000 dilution, respectively. Membranes were then washed and incubated with anti-rabbit IR 800 (LI-COR) and anti-mouse IR 680 (LI-COR) secondary antibodies at 1:5,000 dilution. Immunoreactive bands were detected using a LICOR Odyssey Infrared Imager (LI-COR). Quantifications were performed using Odyssey Application Software Version 2.1 provided by LI-COR.
Prostate-Specific Antigen (PSA) Quantification
PSA producing C4-2B cells were seeded in 12-well plates at 105/well for 24 hr and then exposed to fresh medium containing DSF at different concentrations. The conditioned media as well as the total cell lysates were collected for PSA quantification after 48 hr. PSA was measured using Human PSA ELISA kit (Anogen, Ontario, Canada) according to the manufacturer’s instructions. For C4-2B PSA quantification when cells were exposed to DSF for 2 weeks, cells were seeded (105 cells/well) in 6 cm dish and treated with 0.5 μM DSF with change of growth medium every 2 days. The conditioned media were collected for PSA quantification and cell numbers were counted after trypsinization. Mice serum samples from xenograft mice were analyzed for PSA in the routine clinical chemistry laboratory at Johns Hopkins Hospital.
Statistics
The results of all in vitro and in vivo assays were expressed as mean ±SD. Student’s two-sided t-test was used to compare the values of the test and control samples. P <0.05 was considered statistically significant.
RESULTS
DSF Inhibits DNMT1 Catalytical Activity In Vitro and Results in Reduction of Global 5meC Contentin Prostate Cancer Cells
Previous reports have demonstrated that DSF can inhibit enzyme activity by reacting with thiol groups in the catalytically active site of the protein. Since the catalytical unit of DNMT1 uses a thiol group we hypothesized that DSF could also interfere with the catalytical activity of DNMT1. To investigate this, we tested the ability of DNMT1 to methylate a hemi-methylated DNA oligonucleotide substrate in an in vitro assay as described previously [13]. Recombinant DNMT1 was incubated with hemimethylated oligos, tritium labeled SAM, and increasing concentrations of DSF. DSF decreased the level of incorporated SAM in a dose-dependent manner showing a 95% reduction of activity at a concentration of 200 μM (Fig. 1A), indicating that DSF indeed inhibits DNMT1 catalytic activity.
Fig. 1.

Disulfiram inhibits DNMT1 in vitro and results in reduction of 5meC content in prostate cancer cells. A: DNMT1enzyme activity assays were performed by incubating recombinant His6-DNMT1 with hemimethylated oligonucleotide substrates and S-adenosyl-L-[methyl-3H] methionine as a methyl group donor in the presence of solvent control alone or DSF at increasing concentrations. Relative enzymatic activity is expressed as percentages of solvent control. B: DNMT1is expressed at high levels in prostate cancer cell lines but not in normal PrECs. Normal PrEC and prostate cancer cell lines (CWR22Rv1, PC3, C4-2B, DU145) were lysed and lysates were subjected to SDS^PAGE separation. Proteins were blotted onto PVDF membranes and were incubated with anti-DNMT1 and anti-Actin-specific antibodies. Desitometric analyses show DNMT1 band intensities normalized to Actin. C: LC-MS/MS analysis was used to determine 5meC as a fraction of total cytosine content in CWR22Rv1 and PC3 cells treated with DSF for indicated time points. Normal white blood cell (WBC) genomic DNA in which all CpG dinucleotides were methylated to completion with the M.SssI methyltransferase in vitro (WBC-SssI) was used as positive control. *P <0.05.
To assess DNMT1 expression levels in normal human PrEC and prostate cancer cell lines (CWR22Rv1, PC3, C4-2B, DU145) we performed Western blot analysis (Fig. 1B). Whereas PrEC cells showed very low DNMT1 expression, all prostate cancer cell lines expressed high levels of DNMT1. Since inhibition of DNMT1 could result in decreased maintenance methylation and therefore gradual loss of DNA methylation marks, we tested the effect of DSF treatment on the global 5meC content in androgen sensitive (CWR22Rv1) and androgen insensitive (PC3) prostate cancer cell lines. CWR22Rv1 and PC3 cells were treated with DSF or DMSO control for 3 and 10 days and 4, 8, and 21 days, respectively. DNA was extracted and global methylation status (5meC content) was determined as described previously [3]. Both cell lines showed a statistically significant reduction in 5meC content after 10 or 21 days of DSF exposure suggesting that DSF could also inhibit DNMT function in vivo (Fig. 1C).
DSF Restores Expression of Hypermethylated Genes in Prostate Cancer Cells
Hypermethylation of promoter regions can result in epigenetic silencing of genes [9]. Since DSF treatment affected maintenance methylation in prostate cancer cells, we asked whether DSF treatment could also reverse promoter CpG island methylation of genes known to be methylated in prostate cancer [2,29]. Conversion of DNA using sodium bisulfite results in a change of sequence composition dependent on the methylation status [30]. PCR amplification reactions using primers specific to either the methylated or unmethylated locus allow a qualitative assessment of the methylation status. We identified genes that were previously described to be methlyated in prostate cancers and assessed the methylation status using methylation-specific PCR (MSP) to monitor changes in promoter methylation upon DSF treatment [31]. Mock-treated cells showed presence of methylated RARβ promoter allele in C4-2B and APC promoter allele in CWR22Rv1 cells and absence of unmethylated alleles. DSF treatment resulted in an amplification product with unmethylated-specific primers, suggesting that DSF-induced de-methylation of APC and RARβ gene promoters in CWR22RV1 or C4-2B cells (Fig. 2A). To test whether this change in promoter methylation resulted in the re-expression of epigenetically silenced genes, we determined the mRNA expression levels of APC and RARβ in cells exposed to DSF by quantitative real-time PCR. DSF treatment for 2 or 4 weeks resulted in a strong increase in APC and RARβ transcript levels indicating that DSF treatment can revert epigenetic marks resulting in the re-expression of silenced genes (Fig. 2B).
Fig. 2.
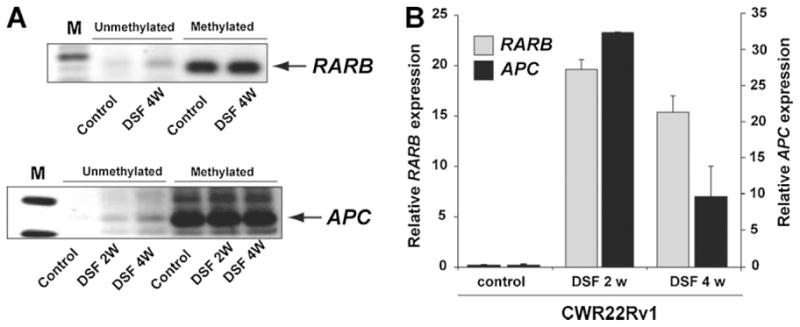
DSF treatment leads to de-methylation of methylated promoter regions in prostate cancer cells and results in re-expression of RARB and APC. A: C4-2B and CWR22Rv1 cells were cultured for 2 or 4 weeks in the presence of 0.5 μMDSF (C4-2B for RARB and CWR22Rv1 cells for APC). Total DNA was extracted and promoter methylation status was evaluated with methylation-specific PCR (MSP) using primers specific to the methylated (M) and unmethylated (U) sequence after bisulfite conversion. An increase in the signal with unmethylated-specific primers indicated de-methylation of the locus. B: RNA was extracted from CWR22Rv1 cells treated with 0.5 μMDSF or solvent control for 2 or 4 weeks. APC and RARB transcript levels were determined by quantitative real-time PCR and are shown as relative expression levels normalized to TBP expression.
DSF Inhibits PCa Cell Growth In Vitro and In Vivo
We further studied and confirmed the anticancer activity of DSF in prostate cancer cell lines by a colorimetric MTT assay. We exposed PC3, DU145, C4-2B, and CWR22Rv1 cells to various concentrations of DSF and calculated cell viability IC50 values (Fig. 3A,B). Cell viability IC50 values ranged from 26 nM in DU145 cells to 185 nM in C4-2B cells, indicating that DSF exerts cytotoxic effects at nano-molar concentrations in prostate cancer cell lines, a finding consistent with previous results [25]. We further investigated the ability of DSF to inhibit clonogenic survival. DU145, CWR22v1, and C4-2B were split to clonogenic density and treated with 100 nM DSF or solvent control. After 2 weeks, the control group showed significant colony formation, whereas colony formation was abolished in the presence of DSF (Fig. 3C). To further confirm the anti-neoplastic effects of DSF in vivo, we established xenografts of C4-2B cells in athymic nude mice. Mice in all DSF treatment groups showed a ~40% reduction in mean tumor volume as compared to control mice (comparing with control group, P-values were between 0.11 and 0.13; Fig. 3D). There was no difference between the high 40 mg/kg and low 10 mg/kg treatment groups. No major systemic toxicities were seen and mouse weights remained constant even in the 40 mg/kg DSF treatment group.
Fig. 3.

Disulfiram inhibits PCa cell growth in vitro and in vivo. A, B: Cells were exposed to DSF at indicated concentrations for 48 hr and cell viability was assessed by MTT and IC50 for DU145, CWR22R1, PC3, and C4-2B were calculated. C: DSF impairs clonogenic survival in prostate cancer cells. CWR22Rv1, DU145, and C4-2B cells were split to clonogenic density and cells were exposed to 100 nMDSF or solvent control. After 14 days, colony formation was assessed. *No colony formation was observed in any of the cell lines subjected to 100 nMDSF. D: Xenograft tumors of C4-2B cells were established in nude mice. When tumors became easily palpable (~0.1mm3) mice were injected i.p. with solvent control or indicated doses of DSF. Changes in tumor volume over time ±SD are shown for each treatment group (n = 8).
DSF Does Not Alter the Per-Cell Secretion of PSA
A critical aspect for any drug entering clinical development for the treatment of prostate cancer is its potential effect on the expression and secretion of the tumor marker PSA [32]. To test the effect of DSF on PSA expression and secretion, C4-2B cells were exposed for 48 hr to clinically relevant doses of DSF (dose range from 62.5 nM to 1 μM). DSF did not alter PSA expression in this cell line until at the concentration of 5 μM (Supplementary Fig. 1). PSA protein levels were analyzed in cell lysates and supernatants using a PSA-specific ELISA (Fig. 4A). Furthermore, C4-2B cells were exposed to 0.5 μM DSF and cell number and PSA concentration in the supernatant were determined after 2 weeks of exposure. Even though DSF greatly reduced the number of surviving cells, the per-cell secretion of PSA remained constant (Fig. 4B). This finding was further corroborated in a C4-2B cell animal xenograft model, which showed no change of tumor-weight-normalized PSA serum concentration upon DSF treatment (Fig. 4C).
Fig. 4.
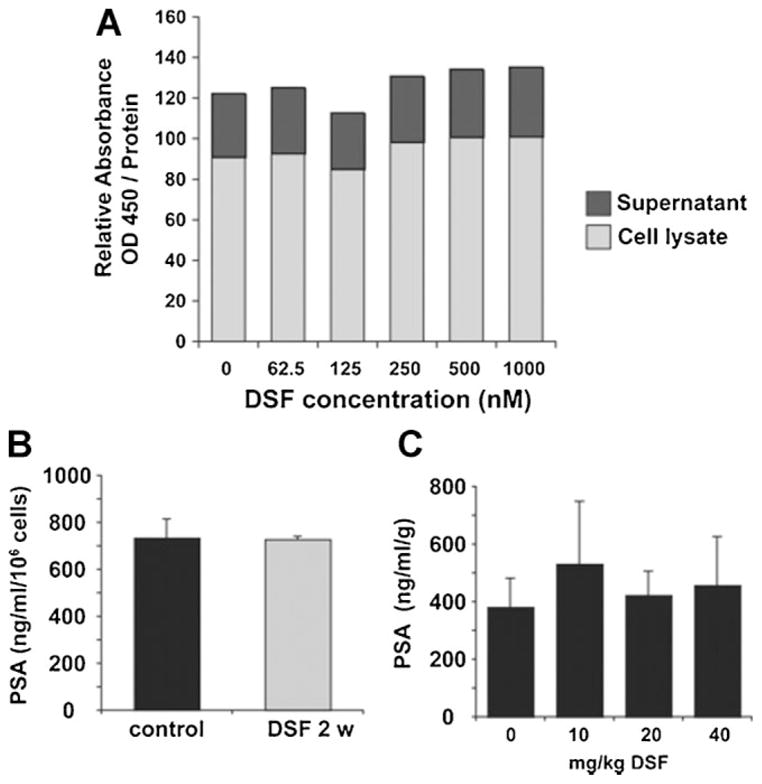
DSF does not affect per-cell PSA secretion. A: PSA producing C4-2B cells were exposed to indicated concentrations of DSF. The conditioned media as well as the total cell lysates were collected after 48 hr and PSA concentrations were determined by ELISA. B: C4-2B cells were exposed to 0.5 μM disulfiram. After 2 weeks PSA concentrations in conditioned medium as well as the cell number of surviving cells were determined. Graph shows PSA levels normalized to cell number in solvent control and DSF-treated cells. C: Serum samples from C4-2B xenograft mice (see Fig. 3D) were collected and PSA concentration was determined by ELISA. Graph shows relative PSA levels normalized to tumor weights.
DISCUSSION
Reverting epigenetic changes that are associated with tumor progression has been considered a valuable therapeutic approach [4,9,11]. The bulk of DNA methylation in dividing cells is maintained by DNMT1 [33,34]. DNMT1 is transcribed mostly during the S-phase of the cell cycle where it maintains methylation marks after replication [35]. In normal epithelial cells, DNMT1 levels are tightly controlled on the protein level and with lowest levels present in G1-phase. In cancer cells, however, this tight regulation of DNMT1 protein levels appears to be corrupted and cancer cell lines show increased stability of DNMT1 throughout the cell cycle [36]. Prostate cancer cell lines (Fig. 1B) as well as prostate cancer tissues show increased expression of DNMT1 compared to normal prostatic epithelial cells [37]. This is further supported by observations from an autochthonous murine model of prostate cancer (TRAMP), which show elevated expression and activity levels of DNMT1 during the entire cascade of prostate cancer progression [38,39]. Two nucleoside analogs 5-azaC and 5-aza-dC have been characterized as DNMT inhibitors. These nucleoside analogs are incorporated in the DNA during S-phase and inhibit DNMT by irreversibly trapping the enzyme to DNA [17,40]. 5-azaC and 5-aza-dC have been approved by the FDA for treatment of MDS and are investigated in further clinical trials for their efficacy in prostate cancer and other solid malignancies [41]. However, these drugs carry considerable concerns regarding toxicity and carcinogenesis for long-term use, since they are incorporated in the DNA which could result in DNA strand breaks and mutations [17], ultimately leading to severe side-effects such as myelosuppression. Therefore, non-nucleoside inhibitors of DNMTs may yield safer alternatives for prolonged epigenetic therapy.
In this manuscript, we show that DSF is a non-nucleoside DNMT inhibitor capable of reducing global genomic 5meC content, of demethylating gene promoters known to be hypermethylated in cancer cells, and of reactivating the expression of these epigenetically silenced genes. Although we do not yet know the precise mechanisms by which DSF may inhibit DNMT1, the mechanism of action of the DNMT enzymes and that of DSF in inhibiting aldehyde dehydrogenase can provide some insights. The mechanism of DNMT1 methyltransferase activity has been extensively studied and involves a nucleophilic attack at C6 of the cytosine ring via the thiol group of the catalytic cysteine of DNMT1 to form a covalently bonded intermediate between the enzyme and the base [17,40,42]. DSF is known to inhibit aldehyde dehyrogenase, the known target when DSF is used for treatment of alcohol abuse, by reacting with a catalytic cysteine thiol group leading to the formation of mixed disulfide or covalent adducts [22]. It is therefore possible that DSF could also interfere with the catalytic activity of DNMT1 via a similar mode of attack at the catalytic cysteine. Furthermore, since the active site of DNMT1 and DNMT3a/3b show high homology [43] and the enzymatic mechanism appears to be conserved between the different C5-cytosine methyltransferases [20], we would hypothesize that DSF also inhibits DNMT3a and DNMT3b.
DSF inhibited prostate cancer cell growth with IC50s ranging from 26 to 185 nM, almost completely abolished clonogenic survival in all cell lines tested, and inhibited tumor growth in xenograft models. These findings are highly concordant with a previous report [25]. The anti-neoplastic effect of DSF does not seem to be restricted to prostate cancer. Recent reports have shown in vitro and in vivo tumor cell killing effects of DSF in various cancer cell line models including melanoma, leukemia, small lung cell cancer, osteosacroma, cervical adenocarcinoma, and colorectal adenocarcinoma cell lines [44–48] and an anecdotal report from a patient with metastatic ocular melanoma described sustained regression of liver metastasis under DSF therapy [49].
It is possible that its epigenetic effects in inhibiting DNMT1 and causing re-expression of epigenetically silenced genes, as demonstrated in this report, are in part responsible for its activity as a potent inhibitor of cancer cell growth. Like other known non-nucleoside DNMT inhibitors, DSF has a significantly lower potency than 5-aza-C or 5-aza-dC for DNMT inhibition and DNA demethylation [50,51]. However, its favorable toxicity/safety profile could allow prolonged treatment with DSF as a demethylating agent to enhance the ultimate efficacy of this drug to levels comparable or even exceeding the efficacy of nucleoside analog drugs, which can only be given for short durations to avoid major toxicity [50,52]. Since DSF is an oral agent that is safe to administer daily, it may be more suitable than the existing nucleoside analog DNMT inhibitors for development of “epigenetic” treatment strategies in solid tumors. Clinical trials of single agent 5-azaC in late stage solid tumor malignancies have been disappointing with little clinical activity [53,54]. A phase II study of 5-azaC to restore responsiveness of prostate cancer to hormonal therapy is still ongoing [55] and evidence of the synergistic effects of DNMT1 inhibitors with cytotoxic agents are emerging [56,57]. Our study showed that prostate cancer growth inhibition by DSF occurred within 48 hr but that maximal DNA demethylation occurred in the remaining cells in 1–2 weeks. It might be possible to exploit this feature to kill the remaining cells via a combination approach in which DSF is used to re-express epigenetically silenced genes combined with agents targeting these genes [4]. Along these lines, in this study, DSF treatment resulted in the re-expression of RARB. Interestingly, retinoids, the ligands of RAR, have been shown to synergize with other epigenetic drugs in inhibiting prostate cancer growth [58].
Furthermore, several other non-epigenetic mechanisms have been proposed for the anti-neoplastic activity of DSF. It has been shown to inhibit NF-κB activation [48] and topoisomerase I and topoisomerase II catalytic activity at micromolar concentrations [59]. DSF can inhibit invasion and angiogenesis and a direct inhibitory effect of DSF on matrix metalloproteinases (MMP-2, MMP-9) was suggested [60]. DSF treatment leads to changes in the intracellular superoxide levels and decreases mitochondrial membrane polarization, suggesting that at least under certain circumstances DSF-induced apoptosis could be redox-dependent. Finally, DSF has been shown to inhibit proteasome activity in a variety of cancer cell line models [61,62]. In this context, it is also worth mentioning that DSF inhibits P-glycoprotein pumps involved in chemo-resistance through drug extrusion [63]. These pleiotropic mechanisms of action provide a potential for clinical activity via mechanisms in addition to those of DNMT inhibition.
It is interesting to note that the anti-proliferative effects of DSF were selective for prostate cancer cells, since normal PrECs showed only limited toxicity when exposed to concentrations of DSF that induced growth inhibition in cancer cells [25]. Given the pleiotropic effects of DSF on several key cellular pathways, one might wonder why DSF exerts such a tumor-specific toxicity, while normal cells appear to be unaffected. In this regard, it is worth noting that the presence of divalent cations appear to have an effect on the toxicity of DSF since Zn2+ supplementation can increase the antitumor activity of DSF [49,61]. Furthermore, DSF can be quickly converted into a great variety of metabolites, which can induce different modifications in target molecules [26]. For instance, inhibition of alcohol dehydrogenase by DSF in vitro involves oxidation of the active site, whereas the DSF metabolite Me-DTC diethylthiomethylcarbamate forms a covalent adduct with the protein resulting in cessation of enzyme activity [22], indicating the complexity of different mechanisms by which DSF and its metabolites can inhibit target enzyme function. Cell type specific accumulation of these different metabolites might further explain the increased sensitivity of cancer cells to DSF as well as differences in terms of DNMT1 inhibition seen between in vitro and in vivo experiments (Figs. 1A,B, 2).
PSA is commonly used as a biomarker for determining treatment response in the prostate cancer clinical trial setting [32]. However, using PSA to evaluate treatment response of experimental treatments that lead to increased per-cell PSA expression via signaling mechanisms would be confounded, and thus, it is important to know whether a given experimental therapeutic agent is capable of increasing per-cell PSA secretion. For instance, in the development of histone deacetylase (HDAC) inhibitors for the treatment of prostate cancer, vorinostat was found to be a potent growth inhibitor to several prostate cancer cell lines in vitro and in vivo but PSA mRNA expression in CWR22 xenograft was elevated, resulting in higher levels of serum PSA than predicted from tumor volume alone [64]. In a phase I study, another HDAC inhibitor sodium phenyl butyrate caused PSA elevations suggestive of the differentiating properties of phenyl butyrate or tumor progression [65]. To avoid these issues during clinical development of DSF for prostate cancer therapy, we tested the effect of DSF on PSA secretion in in vitro and in in vivo xenograft models (Fig. 4). We observed that DSF treatment did not change per-cell PSA secretion suggesting that PSA could potentially be used as a biomarker to monitor disease progression in patients treated with DSF in clinical trials.
CONCLUSIONS
Our results demonstrate that the anticancer activity of DSF could be mediated, at least in part, by its activity to inhibit DNMT activity and reactivate epigenetically silenced genes. The in vitro and in vivo growth inhibition effects are consistent with the literature. It is worth noting that DSF is a drug in clinical use for decades with mild or moderate side-effects even if used long term [24]. Based on these characteristics, DSF is well poised for “repurposing” as a demethylating and/or cancer therapeutic agent. That the pharmacokinetic and safety profiles are well-known and highly amenable to clinical development make this a very attractive approach, especially given that the clinical development of novel drugs is typically very time-consuming and costly. Clinical trials to test if DSF will give clinical benefits for prostate and other cancer patients are warranted.
Supplementary Material
Acknowledgments
Grant sponsor: NCI; Grant numbers: P50-CA58236, 5T32CA009071-27, R01CA070196, 5P30CA006973; Grant sponsor: Prostate Cancer Foundation; Grant sponsor: David Koch Foundation; Grant sponsor: Flight Attendant Medical Research Institute.
This study was supported by the NCI (P50-CA58236) to M.A.C., S.Y., and W.G.N., the Prostate Cancer Foundation to M.A.C., S.Y., and W.G.N., the NCI (5T32CA009071-27) to J.L., the David Koch Foundation to J.L., the NCI (R01CA070196) to W.G.N. and S.Y., NCI (5P30CA006973) to M.A.C., S.Y., and W.G.N., and the Flight Attendant Medical Research Institute (S.K.). The authors wish to thank Dr. James Herman for helpful discussion and providing the primers for RARB MSP and RT-PCR.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 31(1):27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yegnasubramanian S, Kowalski J, Gonzalgo ML, Zahurak M, Piantadosi S, Walsh PC, Bova GS, De Marzo AM, Isaacs WB, Nelson WG. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res. 2004;64(6):1975–1986. doi: 10.1158/0008-5472.can-03-3972. [DOI] [PubMed] [Google Scholar]
- 3.Yegnasubramanian S, Haffner MC, Zhang Y, Gurel B, Cornish TC, Wu Z, Irizarry RA, Morgan J, Hicks J, DeWeese TL, Isaacs WB, Bova GS, De Marzo AM, Nelson WG. DNA hypomethylation arises later in prostate cancer progression than CpG island hypermethylation and contributes to metastatic tumor heterogeneity. Cancer Res. 2008;68(21):8954–8967. doi: 10.1158/0008-5472.CAN-07-6088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelson WG, De Marzo AM, Yegnasubramanian S. Epigenetic alterations in human prostate cancers. Endocrinology. 2009;150(9):3991–4002. doi: 10.1210/en.2009-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson WG, Yegnasubramanian S, Agoston AT, Bastian PJ, Lee BH, Nakayama M, De Marzo AM. Abnormal DNA methylation, epigenetics, and prostate cancer. Front Biosci. 2007;12:4254–4266. doi: 10.2741/2385. [DOI] [PubMed] [Google Scholar]
- 6.Li LC, Carroll PR, Dahiya R. Epigenetic changes in prostate cancer: Implication for diagnosis and treatment. J Natl Cancer Inst. 2005;97(2):103–115. doi: 10.1093/jnci/dji010. [DOI] [PubMed] [Google Scholar]
- 7.Baylin SB, Esteller M, Rountree MR, Bachman KE, Schuebel K, Herman JG. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet. 2001;10(7):687–692. doi: 10.1093/hmg/10.7.687. [DOI] [PubMed] [Google Scholar]
- 8.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 9.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 10.Jung Y, Park J, Kim TY, Park JH, Jong HS, Im SA, Robertson KD, Bang YJ. Potential advantages of DNA methyltransferase 1 (DNMT1)-targeted inhibition for cancer therapy. J Mol Med. 2007;85(10):1137–1148. doi: 10.1007/s00109-007-0216-z. [DOI] [PubMed] [Google Scholar]
- 11.Mack GS. Epigenetic cancer therapy makes headway. J Natl Cancer Inst. 2006;98(20):1443–1444. doi: 10.1093/jnci/djj447. [DOI] [PubMed] [Google Scholar]
- 12.Brueckner B, Lyko F. DNA methyltransferase inhibitors: Old and new drugs for an epigenetic cancer therapy. Trends Pharmacol Sci. 2004;25(11):551–554. doi: 10.1016/j.tips.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 13.Lee BH, Yegnasubramanian S, Lin X, Nelson WG. Procainamide is a specific inhibitor of DNA methyltransferase 1. J Biol Chem. 2005;280(49):40749–40756. doi: 10.1074/jbc.M505593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brueckner B, Kuck D, Lyko F. DNA methyltransferase inhibitors for cancer therapy. Cancer J. 2007;13(1):17–22. doi: 10.1097/PPO.0b013e31803c7245. [DOI] [PubMed] [Google Scholar]
- 15.Kaminskas E, Farrell A, Abraham S, Baird A, Hsieh LS, Lee SL, Leighton JK, Patel H, Rahman A, Sridhara R, Wang YC, Pazdur R. Approval summary: Azacitidine for treatment of myelodysplastic syndrome subtypes. Clin Cancer Res. 2005;11(10):3604–3608. doi: 10.1158/1078-0432.CCR-04-2135. [DOI] [PubMed] [Google Scholar]
- 16.Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, Klimek V, Slack J, de Castro C, Ravandi F, Helmer R, III, Shen L, Nimer SD, Leavitt R, Raza A, Saba H. Decitabine improves patient outcomes in myelodysplastic syndromes: Results of a phase III randomized study. Cancer. 2006;106(8):1794–1803. doi: 10.1002/cncr.21792. [DOI] [PubMed] [Google Scholar]
- 17.Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21(35):5483–5495. doi: 10.1038/sj.onc.1205699. [DOI] [PubMed] [Google Scholar]
- 18.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 19.Chen L, MacMillan AM, Chang W, Ezaz-Nikpay K, Lane WS, Verdine GL. Direct identification of the active-site nucleophile in a DNA (cytosine-5)-methyltransferase. Biochemistry. 1991;30(46):11018–11025. doi: 10.1021/bi00110a002. [DOI] [PubMed] [Google Scholar]
- 20.O’Gara M, Klimasauskas S, Roberts RJ, Cheng X. Enzymatic C5-cytosine methylation of DNA: Mechanistic implications of new crystal structures for HhaL methyltransferase-DNA-AdoHcy complexes. J Mol Biol. 1996;261(5):634–645. doi: 10.1006/jmbi.1996.0489. [DOI] [PubMed] [Google Scholar]
- 21.Jeltsch A. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembio-chemistry. 2002;3(4):274–293. doi: 10.1002/1439-7633(20020402)3:4<274::AID-CBIC274>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 22.Veverka KA, Johnson KL, Mays DC, Lipsky JJ, Naylor S. Inhibition of aldehyde dehydrogenase by disulfiram and its metabolite methyl diethylthiocarbamoyl-sulfoxide. Biochem Pharmacol. 1997;53(4):511–518. doi: 10.1016/s0006-2952(96)00767-8. [DOI] [PubMed] [Google Scholar]
- 23.Eneanya DI, Bianchine JR, Duran DO, Andresen BD. The actions of metabolic fate of disulfiram. Annu Rev Pharmacol Toxicol. 1981;21:575–596. doi: 10.1146/annurev.pa.21.040181.003043. [DOI] [PubMed] [Google Scholar]
- 24.Chick J. Safety issues concerning the use of disulfiram in treating alcohol dependence. Drug Saf. 1999;20(5):427–435. doi: 10.2165/00002018-199920050-00003. [DOI] [PubMed] [Google Scholar]
- 25.Iljin K, Ketola K, Vainio P, Halonen P, Kohonen P, Fey V, Grafstrom RC, Perala M, Kallioniemi O. High-throughput cell-based screening of 4910 known drugs and drug-like small molecules identifies disulfiram as an inhibitor of prostate cancer cell growth. Clin Cancer Res. 2009;15(19):6070–6078. doi: 10.1158/1078-0432.CCR-09-1035. [DOI] [PubMed] [Google Scholar]
- 26.Johansson B. A review of the pharmacokinetics and pharmaco-dynamics of disulfiram and its metabolites. Acta Psychiatr Scand Suppl. 1992;369:15–26. doi: 10.1111/j.1600-0447.1992.tb03310.x. [DOI] [PubMed] [Google Scholar]
- 27.Shabbeer S, Kortenhorst MS, Kachhap S, Galloway N, Rodriguez R, Carducci MA. Multiple molecular pathways explain the anti-proliferative effect of valproic acid on prostate cancer cells in vitro and in vivo. Prostate. 2007;67(10):1099–1110. doi: 10.1002/pros.20587. [DOI] [PubMed] [Google Scholar]
- 28.Haffner MC, Jurgeit A, Berlato C, Geley S, Parajuli N, Yoshimura A, Doppler W. Interaction and functional interference of glucocorticoid receptor and SOCS1. J Biol Chem. 2008;283(32):22089–22096. doi: 10.1074/jbc.M801041200. [DOI] [PubMed] [Google Scholar]
- 29.Esteller M, Fraga MF, Paz MF, Campo E, Colomer D, Novo FJ, Calasanz MJ, Galm O, Guo M, Benitez J, Herman JG. Cancer epigenetics and methylation. Science. 2002;297(5588):1807–1808. doi: 10.1126/science.297.5588.1807d. discussion 1807–1808. [DOI] [PubMed] [Google Scholar]
- 30.Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89(5):1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93(18):9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scher HI, Halabi S, Tannock I, Morris M, Sternberg CN, Carducci MA, Eisenberger MA, Higano C, Bubley GJ, Dreicer R, Petrylak D, Kantoff P, Basch E, Kelly WK, Figg WD, Small EJ, Beer TM, Wilding G, Martin A, Hussain M. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: Recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26(7):1148–1159. doi: 10.1200/JCO.2007.12.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10(11):805–811. doi: 10.1038/nrg2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walsh CP, Bestor TH. Cytosine methylation and mammalian development. Genes Dev. 1999;13(1):26–34. doi: 10.1101/gad.13.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robertson KD, Keyomarsi K, Gonzales FA, Velicescu M, Jones PA. Differential mRNA expression of the human DNA methyl-transferases (DNMTs) 1, 3a and 3b during the G(0)/G(1) to S phase transition in normal and tumor cells. Nucleic Acids Res. 2000;28(10):2108–2113. doi: 10.1093/nar/28.10.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agoston AT, Argani P, Yegnasubramanian S, De Marzo AM, Ansari-Lari MA, Hicks JL, Davidson NE, Nelson WG. Increased protein stability causes DNA methyltransferase 1 dysregulation in breast cancer. J Biol Chem. 2005;280(18):18302–18310. doi: 10.1074/jbc.M501675200. [DOI] [PubMed] [Google Scholar]
- 37.Patra SK, Patra A, Zhao H, Dahiya R. DNA methyltransferase and demethylase in human prostate cancer. Mol Carcinog. 2002;33(3):163–171. doi: 10.1002/mc.10033. [DOI] [PubMed] [Google Scholar]
- 38.Morey SR, Smiraglia DJ, James SR, Yu J, Moser MT, Foster BA, Karpf AR. DNA methylation pathway alterations in an autochthonous murine model of prostate cancer. Cancer Res. 2006;66(24):11659–11667. doi: 10.1158/0008-5472.CAN-06-1937. [DOI] [PubMed] [Google Scholar]
- 39.McCabe MT, Low JA, Daignault S, Imperiale MJ, Wojno KJ, Day ML. Inhibition of DNA methyltransferase activity prevents tumorigenesis in a mouse model of prostate cancer. Cancer Res. 2006;66(1):385–392. doi: 10.1158/0008-5472.CAN-05-2020. [DOI] [PubMed] [Google Scholar]
- 40.Santi DV, Norment A, Garrett CE. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc Natl Acad Sci USA. 1984;81(22):6993–6997. doi: 10.1073/pnas.81.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muller CI, Ruter B, Koeffler HP, Lubbert M. DNA hyper-methylation of myeloid cells, a novel therapeutic target in MDS and AML. Curr Pharm Biotechnol. 2006;7(5):315–321. doi: 10.2174/138920106778521523. [DOI] [PubMed] [Google Scholar]
- 42.Santi DV, Garrett CE, Barr PJ. On the mechanism of inhibition of DNA-cytosine methyltransferases by cytosine analogs. Cell. 1983;33(1):9–10. doi: 10.1016/0092-8674(83)90327-6. [DOI] [PubMed] [Google Scholar]
- 43.Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9(16):2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 44.Wickstrom M, Danielsson K, Rickardson L, Gullbo J, Nygren P, Isaksson A, Larsson R, Lovborg H. Pharmacological profiling of disulfiram using human tumor cell lines and human tumor cells from patients. Biochem Pharmacol. 2007;73(1):25–33. doi: 10.1016/j.bcp.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 45.Chen D, Cui QC, Yang H, Dou QP. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006;66(21):10425–10433. doi: 10.1158/0008-5472.CAN-06-2126. [DOI] [PubMed] [Google Scholar]
- 46.Lovborg H, Oberg F, Rickardson L, Gullbo J, Nygren P, Larsson R. Inhibition of proteasome activity, nuclear factor-KappaB translocation and cell survival by the antialcoholism drug disulfiram. Int J Cancer. 2006;118(6):1577–1580. doi: 10.1002/ijc.21534. [DOI] [PubMed] [Google Scholar]
- 47.Cho HJ, Lee TS, Park JB, Park KK, Choe JY, Sin DI, Park YY, Moon YS, Lee KG, Yeo JH, Han SM, Cho YS, Choi MR, Park NG, Lee YS, Chang YC. Disulfiram suppresses invasive ability of osteosarcoma cells via the inhibition of MMP-2 and MMP-9 expression. J Biochem Mol Biol. 2007;40(6):1069–1076. doi: 10.5483/bmbrep.2007.40.6.1069. [DOI] [PubMed] [Google Scholar]
- 48.Wang W, McLeod HL, Cassidy J. Disulfiram-mediated inhibition of NF-kappaB activity enhances cytotoxicity of 5-fluorour-acil in human colorectal cancer cell lines. Int J Cancer. 2003;104(4):504–511. doi: 10.1002/ijc.10972. [DOI] [PubMed] [Google Scholar]
- 49.Brar SS, Grigg C, Wilson KS, Holder WD, Jr, Dreau D, Austin C, Foster M, Ghio AJ, Whorton AR, Stowell GW, Whittall LB, Whittle RR, White DP, Kennedy TP. Disulfiram inhibits activating transcription factor/cyclic AMP-responsive element binding protein and human melanoma growth in a metal-dependent manner in vitro, in mice and in a patient with metastatic disease. Mol Cancer Ther. 2004;3(9):1049–1060. [PubMed] [Google Scholar]
- 50.Lyko F, Brown R. DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J Natl Cancer Inst. 2005;97(20):1498–1506. doi: 10.1093/jnci/dji311. [DOI] [PubMed] [Google Scholar]
- 51.Lin X, Asgari K, Putzi MJ, Gage WR, Yu X, Cornblatt BS, Kumar A, Piantadosi S, DeWeese TL, De Marzo AM, Nelson WG. Reversal of GSTP1 CpG island hypermethylation and reactivation of pi-class glutathione S-transferase (GSTP1) expression in human prostate cancer cells by treatment with procainamide. Cancer Res. 2001;61(24):8611–8616. [PubMed] [Google Scholar]
- 52.Saiki JH, Bodey GP, Hewlett JS, Amare M, Morrison FS, Wilson HE, Linman JW. Effect of schedule on activity and toxicity of 5-azacytidine in acute leukemia: A Southwest Oncology Group Study. Cancer. 1981;47(7):1739–1742. doi: 10.1002/1097-0142(19810401)47:7<1739::aid-cncr2820470702>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 53.Momparler RL, Bouffard DY, Momparler LF, Dionne J, Belanger K, Ayoub J. Pilot phase I-II study on 5-aza-2′-deoxycytidine (Decitabine) in patients with metastatic lung cancer. Anticancer Drugs. 1997;8(4):358–368. doi: 10.1097/00001813-199704000-00008. [DOI] [PubMed] [Google Scholar]
- 54.Quagliana JM, O’Bryan RM, Baker L, Gottlieb J, Morrison FS, Eyre HJ, Tucker WG, Costanzi J. Phase II study of 5-azacytidine in solid tumors. Cancer Treat Rep. 1977;61(1):51–54. [PubMed] [Google Scholar]
- 55.Sonpavde G, Aparicio A, Guttierez I, Boehm KA, Hutson TE, Berry WR, Asmar L, von Hoff DD. Phase II study of azacitidine to restore responsiveness of prostate cancer to hormonal therapy. Clin Genitourin Cancer. 2007;5(7):457–459. doi: 10.3816/CGC.2007.n.036. [DOI] [PubMed] [Google Scholar]
- 56.Festuccia C, Gravina GL, D’Alessandro AM, Muzi P, Millimaggi D, Dolo V, Ricevuto E, Vicentini C, Bologna M. Azacitidine improves antitumor effects of docetaxel and cisplatin in aggressive prostate cancer models. Endocr Relat Cancer. 2009;16(2):401–413. doi: 10.1677/ERC-08-0130. [DOI] [PubMed] [Google Scholar]
- 57.Shang D, Liu Y, Liu Q, Zhang F, Feng L, Lv W, Tian Y. Synergy of 5-aza-2′-deoxycytidine (DAC) and paclitaxel in both androgen-dependent and -independent prostate cancer cell lines. Cancer Lett. 2009;278(1):82–87. doi: 10.1016/j.canlet.2008.12.034. [DOI] [PubMed] [Google Scholar]
- 58.Pili R, Kruszewski MP, Hager BW, Lantz J, Carducci MA. Combination of phenylbutyrate and 13-cis retinoic acid inhibits prostate tumor growth and angiogenesis. Cancer Res. 2001;61(4):1477–1485. [PubMed] [Google Scholar]
- 59.Yakisich JS, Siden A, Eneroth P, Cruz M. Disulfiram is a potent in vitro inhibitor of DNA topoisomerases. Biochem Biophys Res Commun. 2001;289(2):586–590. doi: 10.1006/bbrc.2001.6027. [DOI] [PubMed] [Google Scholar]
- 60.Shian SG, Kao YR, Wu FY, Wu CW. Inhibition of invasion and angiogenesis by zinc-chelating agent disulfiram. Mol Pharmacol. 2003;64(5):1076–1084. doi: 10.1124/mol.64.5.1076. [DOI] [PubMed] [Google Scholar]
- 61.Sauna ZE, Shukla S, Ambudkar SV. Disulfiram, an old drug with new potential therapeutic uses for human cancers and fungal infections. Mol Biosyst. 2005;1(2):127–134. doi: 10.1039/b504392a. [DOI] [PubMed] [Google Scholar]
- 62.Chen D, Peng F, Cui QC, Daniel KG, Orlu S, Liu J, Dou QP. Inhibition of prostate cancer cellular proteasome activity by a pyrrolidine dithiocarbamate-copper complex is associated with suppression of proliferation and induction of apoptosis. Front Biosci. 2005;10:2932–2939. doi: 10.2741/1749. [DOI] [PubMed] [Google Scholar]
- 63.Loo TW, Clarke DM. Blockage of drug resistance in vitro by disulfiram, a drug used to treat alcoholism. J Natl Cancer Inst. 2000;92(11):898–902. doi: 10.1093/jnci/92.11.898. [DOI] [PubMed] [Google Scholar]
- 64.Butler LM, Agus DB, Scher HI, Higgins B, Rose A, Cordon-Cardo C, Thaler HT, Rifkind RA, Marks PA, Richon VM. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 2000;60(18):5165–5170. [PubMed] [Google Scholar]
- 65.Carducci MA, Gilbert J, Bowling MK, Noe D, Eisenberger MA, Sinibaldi V, Zabelina Y, Chen TL, Grochow LB, Donehower RC. A Phase I clinical and pharmacological evaluation of sodium phenylbutyrate on an 120-h infusion schedule. Clin Cancer Res. 2001;7(10):3047–3055. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.