

Abstract
A number of studies have shown that the polyol pathway, consisting of aldose reductase (AR) and sorbitol dehydrogenase (SDH), contributes to ischemia–reperfusion (I/R)-induced myocardial infarction due to depletion of ATP. In this report we show that the polyol pathway in I/R heart also contributes to the impairment of sacro/endoplasmic reticulum Ca2+-ATPase (SERCA) and ryanodine receptor (RyR), two key players in Ca2+ signaling that regulates cardiac contraction. Rat hearts were isolated and retrogradely perfused with either Krebs’ buffer containing 1 μM AR inhibitor, zopolrestat, or 200 nM SDH inhibitor, CP-170,711, and challenged by 30 min of regional ischemia and 45 min of reperfusion. We found that post-ischemic contractile function of the isolated perfused hearts was improved by pharmacological inhibition of the polyol pathway. I/R-induced contractile dysfunction is most likely due to impairment in Ca2+ signaling and the activities of SERCA and RyR. All these abnormalities were significantly ameliorated by treatment with ARI or SDI. We showed that the polyol pathway activities increase the level of peroxynitrite, which enhances the tyrosine nitration of SERCA and irreversibly modify it to form SERCAC674-SO3H. This leads to reduced level of S-glutathiolated SERCA, contributing to its inactivation. The polyol pathway activities also deplete the level of GSH, leading to decreased active RyR, the S-glutathiolated RyR. Thus, in I/R heart, inhibition of polyol pathway improved the function of SERCA and RyR by protecting them from irreversible oxidation.
INTRODUCTION
Contractile dysfunction often occurs after acute myocardial infarction, cardiac bypass surgery, heart transplantation, and coronary angioplasty (1). It has been shown that early reperfusion after coronary occlusion improves heart functions and reduces infarct size (2). However, reperfusion after a certain time period of ischemia may exacerbate cardiac contractile dysfunction, ultrastructural damage, and changes in myocardial metabolism (3).
During ischemia-reperfusion (I/R), cardiac contractile dysfunction is attributed to the impairment of calcium (Ca2+) handling activities of the cardiomyocyte. Under normal condition, Ca2+ homeostasis is exquisitely controlled by regulatory proteins in sarcolemmal and sarcoplasmic reticulum (SR) membranes. Ca2+ enters the cardiomyocyte via the L-type Ca2+ channels when the sarcolemmal membrane is depolarized. Entry of Ca2+ triggers further release of Ca2+ through the ryanodine receptor (RyR) of the SR, leading to a large increase in cytosolic Ca2+ concentration, known as the intracellular [Ca2+] transient ([Ca2+]i) (4). The elevated [Ca2+]i, which stimulates contraction of the myofilaments, is removed mainly to the SR by the Ca2+-ATPase (SERCA) and out of the cell by the Na+/Ca2+ exchanger (NCX) to initiate relaxation. These periodic changes in [Ca2+] between cytosol and SR control the cycles of excitation-contraction (EC) coupling and relaxation. Abnormalities in Ca2+ handling leading to cytosolic [Ca2+] overload, has been suggested to explain contractile dysfunction of the heart following I/R in the heart (3). However, the mechanism is not entirely clear.
Apart from the impairment in Ca2+ homeostasis, the increase in reactive oxygen species (ROS) within the first few minutes of reperfusion has been proposed to explain the I/R-induced contractile changes in the heart (5). In fact, exposure of the heart to different species of ROS has been shown to cause functional alterations (6) similar to that observed in the I/R heart. More importantly, these changes have been demonstrated to be attributed to abnormalities in Ca2+ handling by the SR (7) and sarcolemma (8). Therefore it is likely that, during I/R, release of ROS impaired the Ca2+ handling activities in the cardiomyocytes. In this report we demonstrated that polyol pathway contributes to the increased ROS during I/R leading to impairment of two key calcium handling proteins, SERCA and RyR, in the rat heart.
Polyol pathway has been implicated in the pathogenesis of various diabetic complications (9, 10). In this metabolic pathway, glucose is reduced to sorbitol by aldose reductase (AR; EC 1.1.1.21) with the oxidation of its co-factor NADPH to NADP, and sorbitol is then converted to fructose by sorbitol dehydrogenase (SDH: EC 1.1.1.14) with the concomitant reduction of NAD+ to NADH (11). Under hyperglycemia, increased flux of glucose through the polyol pathway leads to the depletion of NADPH and NAD+. Decrease in the level of NADPH is thought to lead to decreased level of reduced glutathione (GSH) because NADPH is also the co-factor for glutathione reductase (GR) that regenerates GSH from oxidized glutathione (GSSG) (12). Further, increased level of NADH, a substrate for NAD(P)H oxidase, would increase ROS. Thus, increased polyol pathway activity would decrease antioxidation defense and increase ROS, resulting in increased oxidative stress.
Importantly, it has been demonstrated that the polyol pathway is activated in I/R heart even in non-diabetic animals (13). It has been shown to play a key role in I/R induced injury of the heart (13–15) and brain (16). The protective effect of inhibition of AR or SDH against myocardial I/R injury is thought to be due to normalization of cytosolic NADH/NAD+ ratio, thereby preventing the depletion of ATP and redox imbalance. Thus, AR and SDH present novel targets for pharmacological protection against I/R-induced injuries of the heart.
A recent study in our laboratory demonstrated that in the I/R hearts of non-diabetic rats polyol pathway-mediated depletion of NAD+ leads to the induction of HIF-1α, which increases the expression of TfR and consequently, increases Tf-bound Fe uptake, contributing to increased Fe-catalyzed oxidative damage (17). Thus, together with depletion of GSH and increase in ROS, it is likely that activation of polyol pathway in I/R heart contributes to the impairment of SERCA and RyR, leading to cardiac dysfunction.
EXPERIMENTAL PROCEDURES
Animal model
Sprague-Dawley male rats weighing between 200g to 250g were supplied by the Laboratory Animal Unit, University of Hong Kong. The study protocol was approved by the Committee on the Use of Experimental Animals for Teaching and Research of The University of Hong Kong. Also, the investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Isolated perfused heart preparation
Hearts were isolated and perfused as described previously (18). Rats were anaesthetized with sodium pentobarbital (60 mg kg−1, i.p.) and given heparin (200 IU, i.v.) before decapitation. Hearts were excised immediately and, placed in ice-cold Krebs–Henseleit perfusion buffer before mounting on the Langendorff apparatus for perfusion. Isolated hearts were perfused retrogradely with Krebs–Henseleit buffer equilibrated with 95% O2+5% CO2 at a constant pressure of 80 cm H2O and a temperature of 37 °C. Hearts exhibiting arrhythmias during stabilization were discarded. A latex balloon was inserted into the left ventricle and the end-diastolic pressure (LVEDP) was adjusted to 4–8 mm Hg. The cardiac parameters, such as heart rate, left ventricular developed pressure (LVDP), and + dp/dtmax and − dp/dtmax representing, respectively, maximum derivatives of the ventricular pressure (± dp/dtmax) were monitored continuously by a PowerLab/4SD system (AD Instruments, Castle Hill, Australia).
Ischemia/reperfusion (I/R) protocol of isolated perfused rat hearts
Regional ischemia was induced by ligation of the left anterior descending artery in the left ventricles with a surgical suture for 30 min. Reperfusion was achieved by releasing the ligation, and the hearts were harvested after 45 min. After stabilization period of 30 min, hearts were perfused with modified Krebs–Henseleit buffer containing 5 μmol/L ARI (zopolrestat) or 1 μmol/L SDH inhibitor (CP-470,711) starting 10 min before ischemia and continued throughout reperfusion. Both zopolrestat and CP-470,711 were gifts from Pfizer Global Research and Development. The concentration of zoplrestat and CP-470,711 used in the present study was based on the previous findings that this dose was sufficient to protect heart from ischemic injury (13, 19). Dose response for these inhibitors was performed to determine the optimal dose (see the supplementary figure 5). Myocardial injury was assessed by lactate dehydrogenase (LDH) efflux. Effluent from isolated perfused rat heart was collected at 40 min after reperfusion and LDH was assayed spectrophotometrically by using a kit from Sigma-Aldrich (St. Louis, MO, USA). LDH activity measured was expressed as units per liter.
Isolation of rat ventricular myocytes and I/R of rat cardiomyocytes
Ventricular myocytes were isolated from the hearts by the collagenase method described previously (20). After isolation, they were allowed to stabilize for 30 min before experiments. The yield of myocytes was determined microscopically using a haemocytometer. Myocyte viability was assessed by Cell Titer Blue reagent (Promega, Madison, WI, USA). Preparations were considered satisfactory only if rod-shaped cells accounted for >80% of the counted cells at the beginning of each experiment. Myocytes were first subjected to 60-min pretreatment with an AR inhibitor (5 μmol/L zopolrestat) or SDH inhibitor (1 μmol/L CP-470,711) in normal Krebs’ solution. After incubation, myocytes were subjected to metabolic inhibition and anoxia with glucose-free Krebs’ solution for 10 min containing 10-glucose mmol/L 2-deoxy-D(2-DOG), an inhibitor of glycolysis, and 10 mmol/L sodium dithionite (Na2S2O4), an oxygen scavenger (21). Finally, the myocytes were transferred back to normal Krebs’ solution for 10-min reperfusion. LDH release was determined using a cytotoxicity detection kit (Roche, Indiana-polis, IN, USA) according to the manufacturer’s instructions.
Measurement of [Ca2+]i transients in the single cardiomyocyte
[Ca2+]i transients were measured using a spectrofluorometric method with fura-2 AM as the Ca2+ indicator as described previously (22). Ventricular myocytes were incubated with 5 μM fura-2 AM for 30 min. Fluorescent signals obtained at 340-nm and 380-nm excitation wavelengths were recorded and stored in computer for data processing and analysis. Cardiomyocytes were subjected to 0.2 Hz electrical stimulation with a 15-ms pulse at 60 V through two platinum wires in the bathing chamber. The amplitude of electrically induced [Ca2+]i transient (E[Ca2+]i) was determined as the difference between the resting and the peak [Ca2+]i levels; the time for 50% decay of the transient (t50) was used to quantitate the decay of transients.
Isolation of SR vesicles and measurement of 45Ca2+ uptake
SR vesicles were obtained by a method described previously with some modifications (23, 24), and the ATP-dependent transport of Ca2+ to SR was measured at room temperature (22 °C) with a method described previously (25). SR protein (50–100 μg) was added to 1 ml of a medium that contained 40 mmol/L imidazole-HCl (pH 7.0), 100 mmol/L KCl, 20 mmol/L NaCl, 5 mmol/L MgCl2, 4 mmol/L ATP-Na2, 1.3 μCi 45 CaCl2, 5 μmol/L Ru-360, an inhibitor of Ca2+ uptake in mitochondria (26), and 5 μmol/L calmidazolium, an inhibitor of sarcolemmal Ca2+-ATPase (27). The concentration of free Ca2+ in this solution (5 μmol/L) was determined by a Ca2+-EGTA buffer and calculated according to Fabiato and Fabiato (28). To measure the oxalate-supported Ca2+ uptake, 5 mmol/L K-oxalate was added to the aforementioned solution. After 2–20 min, aliquots of 0.9 ml were filtered through Millipore filters (0.45 μm; Bedford, MA). Filters were washed three times with 4 ml of cold (2–4 °C) solution containing 40 mmol/L imidazole-HCl (pH 7.0), 100 mmol/L KCl, and 0.1 mmol/L EGTA. After being washed, the Millipore filters were placed into vials containing 10 ml of scintillation cocktail (Universal LSC cocktail for aqueous samples, Sigma) and left for about 40 min. The radioactivity was then counted in scintillation counter (LS 6500, Beckman).
The 45Ca2+ uptake by SERCA was defined as the difference between the rate of 45Ca2+ uptake in the K-oxalate containing solution in the presence and absence of 10 μmol/L cyclopiazonic acid, a specific inhibitor of SERCA (29). The difference between uptake in the presence and absence of 50 μmol/L ryanodine, a specific blocker of RyR, was defined as the 45Ca2+ release via the RyR receptor. The concentration of ryanodine used in this study was based on a previous report (25).
Biochemical assay for lactate and pyruvate
To determine changes in the cytosolic redox state (i.e. NADH/NAD+), parallel experiments were performed with hearts freeze-clamped before ischemia; at the end of 30 min of ischemia; and after 45 min of reperfusion. Lactate and pyruvate were extracted from the freeze-clamped tissue using perchloric acid and measured using standard biochemical assays as we published previously (17).
Measurement of sorbitol and fructose content
High performance liquid chromatography (HPLC) was performed to determine the levels of sorbitol and fructose in rat hearts as previously described (17). Briefly, approximately 300 mg of frozen samples were homogenized in 1 ml of ice cold 5% trichloroacetic acid with 5.5 μl of 10 mmol/L xylitol added as an internal standard. Twenty five μl of sample was injected into HPLC MA-1 analytical column (Dionex) by AS-50 autosampler (Dionex), and the components in the sample were separated at a flow rate of 0.4 ml/min. The level of sorbitol and fructose content was determined by integrated amperometry, and the peaks were analyzed by Dionex Peaknet software. The amount of sorbitol and fructose was normalized to the wet weight of the tissue.
Determination of oxidative stress
The oxidative stress was determined by the method described previously (30). Briefly, about 100 mg of the heart tissue was minced and then incubated in 1 ml of Krebs-Ringer’s solution containing 0.5 μmol/L lucigenin at pH 7.4. The chemiluminescence elicited by oxidative stress in the presence of lucigenin was measured in a luminometer (Berthold Lumat LB9507). The level of oxidative stress was reported as RLU after background luminescence subtraction and normalized to milligram wet tissue weight. To validate that the chemiluminescence signals were derived from oxidative stress, the presence of 10 mM Tiron, a superoxide scavenger, was added as a control.
Determination of GSH level
Approximately 100 mg of the heart tissue was minced and then homogenized in 1 ml of 6 % perchloric acid. The homogenate was centrifuged at 13,000 rpm for 10 mins at 4 °C. The supernatant was collected and added to the solution containing 85.7 μl of 70 % perchloric acid, 14.3 μl of dddH2O and 107 μl of 5 mol/L K2CO3, and then centrifuged at 4,000 rpm for 5 minutes under 4 °C. After centrifugation, 100 μl of supernatant was added to 890 μl of Tris-EDTA (pH 8.1) and 10 μl of O-pathaldialdehyde and the reaction mixture was kept on ice for 15 min. After that, the absorbance of the mixture was measured with a fluorescent spectrometer (425 nm; excitation at 345 nm). The concentration of GSH in the tissue was calculated from the GSH standard curve.
Immunohistochemistry
Paraffin sections of the hearts were deparaffinized in xylene and rehydrated with a graded series of ethanol. After washing, sections were blocked with 0.3% hydrogen peroxide for 15 min. The sections were then blocked for 1 h with 1.5% normal goat serum (Vector Laboratories, Burlingame, CA). Sections were then incubated with rabbit anti-nitrotyrosine (NT) (1:200; Santa Cruz Biotechnology) overnight at 4°C. Immunoreactivity was detected with biotinated goat anti-rabbit secondary antibodies and the avidin-biotin-peroxidase complex (Vector Laboratories). Immunoreactive signal was developed using diaminobenzidine as a substrate (Zymed Laboratories, San Francisco, CA) for 2 min. Photomicrographs were taken with an Olympus IX71 microscope system. All histological and immunohistochemical samples were coded and examined and graded in a blinded fashion.
Immunohistochemical staining of ROS modification of SERCA was performed using antibodies against SERCAC674-SO3H (31). Briefly, nonspecific binding was blocked with 10% normal goat serum in phosphate-buffered saline (PBS, pH 7.4) for 30 min before incubation with individual primary antibodies. Anti-SERCA C674-SO3H antibody was used at 2 μg/mL. The secondary antibody, a biotinylated anti-rabbit IgG secondary antibody was used at 1:200. Vector Red alkaline phosphatase substrate (Vector) was used to visualize positive immunoreactivity.
Immunoprecipitation with anti-SERCA2 or anti-RyR antibody
The immunoprecipitation was performed as described (21). Briefly, one milligram of protein extract was diluted in 500 μl of lysis buffer. After preclearing with protein G agarose, the supernatant was mixed with 10 μl of anti-SERCA2 or anti-RyR, and then incubated at 4 °C for 4 hrs. Prewashed protein G (50μl) was added to the samples, and further incubated for 1 hr. For detecting the tyrosine nitration of SERCA, the immunocomplex was resuspended in Laemmli’s buffer containing mercaptoethanol. For detecting the S-glutathiolation of RyR and SERCA, the immunocomplex was resuspended in non-reducing loading buffer containing 5mM N-ethylmaleimide. The samples with the loading buffer were then boiled at 5 min at 90 °C. The samples were separated by SDS-PAGE and transferred electrophoretically. After blocking, the membrane was incubated overnight at 4 °C with rabbit anti-nitrotyrosine (1:200; Santa Cruz Biotechnology), goat anti-SERCA (1:400; Santa Cruz Biotechnology), mouse anti-glutathione (1:1000; Virogen) or mouse anti-RyR (1:3,330 dilution; Affinity BioReagent, Golden, CO) in blocking solution. The second antibody was either anti-goat or anti-mouse antibody conjugated to horseradish peroxidase for 1 h at room temperature, followed by detection using the chemiluminescence method.
Statistical Analysis
All data were expressed as means ± SE. One-way analysis of variance, followed by Newman-Keuls multiple comparison tests, was used to assess differences between the mean values within the same study. A difference of p<0.05 was considered significant.
RESULTS
Polyol pathway activities in pre-ischemic, ischemic and reperfused rat hearts
To assess the polyol pathway activities in the I/R rat hearts, the levels of sorbitol and fructose, the enzymatic products of AR and SDH, respectively, were determined in the pre-ischemic, ischemic and post-ischemic reperfused hearts (Fig. 1). There was no significant change in sorbitol and fructose level after 30 mins of ischemia. However, there was a significant increase in fructose level in the reperfused hearts, indicating increased polyol pathway activity during reperfusion. This was confirmed by large increase in sorbitol in the reperfused heart treated with SDH inhibitor that blocks the conversion of sorbitol to fructose. Treatment with AR or SDH inhibitor prevented the increase in fructose level in the reperfused hearts, indicating that the increased fructose was synthesized by the polyol pathway and that the AR and SDH inhibitors were effective in blocking the polyol pathway enzyme activities.
Figure 1.
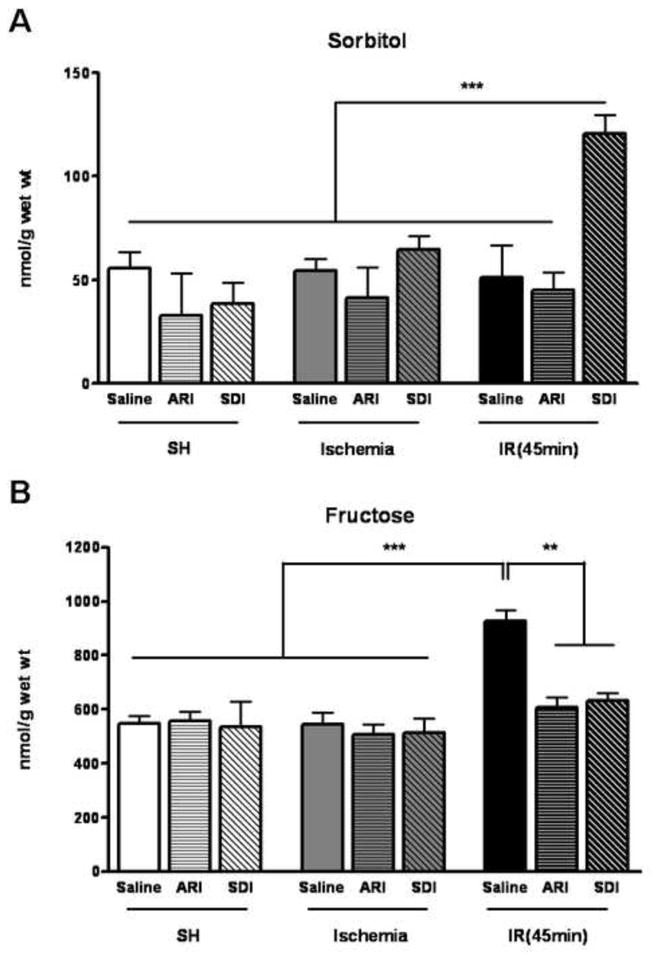
A. Changes in levels of sorbitol in the isolated hearts challenged with 30 minutes of ischemia (Ischemia) and followed by 45 minutes of reperfusion (IR). Within each group some were injected with saline (Saline), some treated with ARI (ARI) and some treated with SDI (SDI). Data are means ± SE (n=5). ***p<0.001 & **p<0.01, compared with SDH-inhibited IR group. B. Changes in levels of fructose the isolated hearts challenged with 30 minutes of ischemia (Ischemia) and followed by 45 minutes of reperfusion (IR). Within each group, some were injected with saline (saline), some treated with ARI (ARI) and some treated with SDI (SDI). Data are means ± SE (n=4). ***p<0.001 & **p<0.01, compared with control group after reperfusion.
Inhibition of the polyol pathway improved contractile dysfunction of the I/R hearts
The ischemic insult caused marked decrease in LVDP, LVSP and ±dP/dtmax, and reperfusion led to marked elevation in LVEDP. To determine the role of polyol pathway in the contractile function, we used two specific inhibitors, AR inhibitor (ARI), zopolrestat and SDH inhibitor (SDI), CP-470,711. These two inhibitors have been extensively studied in the previous reports (13, 17, 19, 32, 33), with no unintended effect on myocardial function. To further test the effect of these two inhibitors in our model, the pharmacological control was done, and no effect was found on myocardial function under normal condition (data not shown). During I/R, inhibition of AR with 5 μM zopolrestat significantly attenuated the reduction in LVDP, ±dP/dtmax and the elevation in LVEDP compared to the vehicle group. Similar effect was also observed in the hearts treated with 1 μM SDH inhibitor CP-470,711. Among all the groups, no difference was found in LVSP (Fig. 2), and the heart rate remained steady during I/R (see the supplementary figure 4). To determine the extent of myocardial injury during I/R and the effect of inhibition of AR and SDH, LDH release was measured at 45 min after reperfusion. As shown in Figure 2F, there was a significant increase in LDH release in the I/R group compared to SH group. Inhibition of AR or SDH slightly reduced the LDH release but the difference was not statistically significant. These results indicated that inhibition of polyol pathway improved the contractile function during I/R, but the I/R-induced myocardial injury could not be protected by AR and SDH inhibitors.
Figure 2.
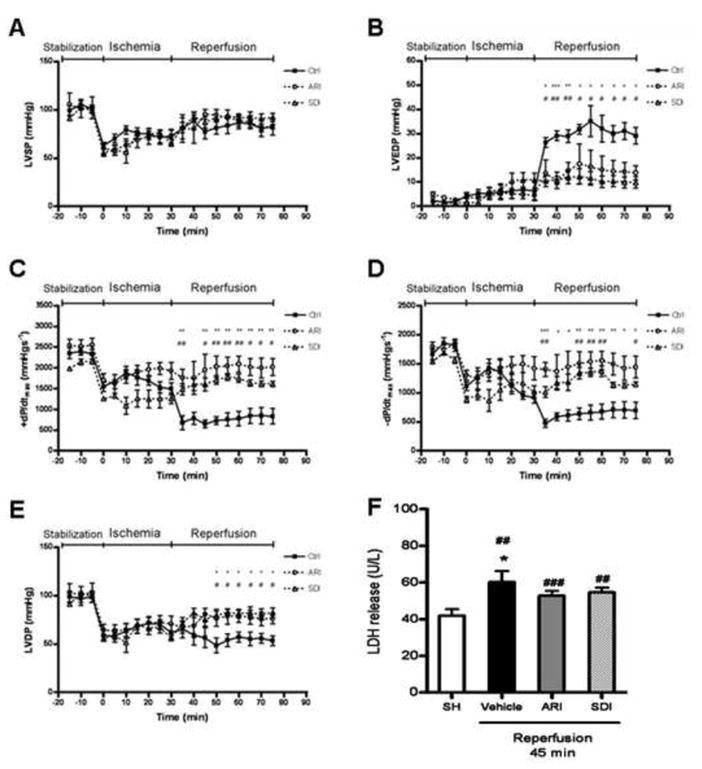
Effect of ischemic insults on (A) LVSP, (B) LVEDP, (C & D) ±dP/dtmax and (E) LVDP in control (vehicle group), ARI-treated and SDI-treated hearts from rats. All these parameters were obtained from isolated perfused hearts in preischemic conditions (stabilization), during ischemia and then during reperfusion. Data are means ± SE (n=5). ***p<0.001, **p<0.01 & *p<0.05, compared with corresponding ARI group; ###p<0.001, ##p<0.01 & #p<0.05, compared with corresponding SDI group. (F) LDH release from SH (sham-operated), vehicle, ARI-treated and SDI-treated hearts from rats during post-ischemic reperfusion (IR). Data are means ± SE (n=4). ###p<0.001, ##p<0.01, compared with SH group; *p<0.05, compared with vehicle group with I/R.
Inhibition of the polyol pathway attenuated the increase in cytosolic lactate/pyruvate ratio in I/R hearts
Previous findings indicate that the rise in cytosolic NADH/NAD+ ratio, as represented by increased cytosolic lactate/pyruvate (L/P) ratio, is an important indicator of AR-mediated ischemic injury (17, 34). As shown in Table 1, the cytosolic L/P ratio was increased significantly during I/R, and the absolute amount of lactate and pyruvate was shown in the supplementary figure 3. Similar to previous reports, inhibition of AR or SDH both attenuated the increase in cytosolic L/P ratio in the I/R heart in our experimental protocol.
Table 1.
Lactate/pyruvate ratios in control (vehicle group), AR-inhibited and SDH-inhibited rat hearts under various conditions. Lactate/pyruvate ratios were measured under baseline conditions, at the end of 30 min of ischemia, and after 45 min of reperfusion.
| Groups | Lactate/Pyruvate ratio | ||
|---|---|---|---|
| Baseline | Ischemia | Reperfusion | |
| Control (n=4) | 24.25 ± 3.50 | 424.54 ± 49.39 | 39.13 ± 6.34 |
| ARI (n=4) | 12.57 ± 0.94*** | 167.05 ± 13.23*** | 14.02 ± 0.80*** |
| SDI (n=4) | 12.82 ± 1.34*** | 174.79 ± 16.71*** | 16.93 ± 1.86*** |
Data are means ± SE (n=4).
p<0.001,
p<0.01 &
p<0.05, compared with control group.
Inhibition of the polyol pathway attenuated the reduction in GSH level and the increase in the oxidative stress in I/R hearts
I/R significantly reduced the level of GSH in the heart. Inhibition of AR attenuated the I/R-induced decrease in GSH level, and similar effect was found in the SDI-treated group. Further, oxidative stress was significantly elevated in I/R hearts, and such increase was attenuated by the inhibition of AR and SDH (Fig. 3).
Figure 3.
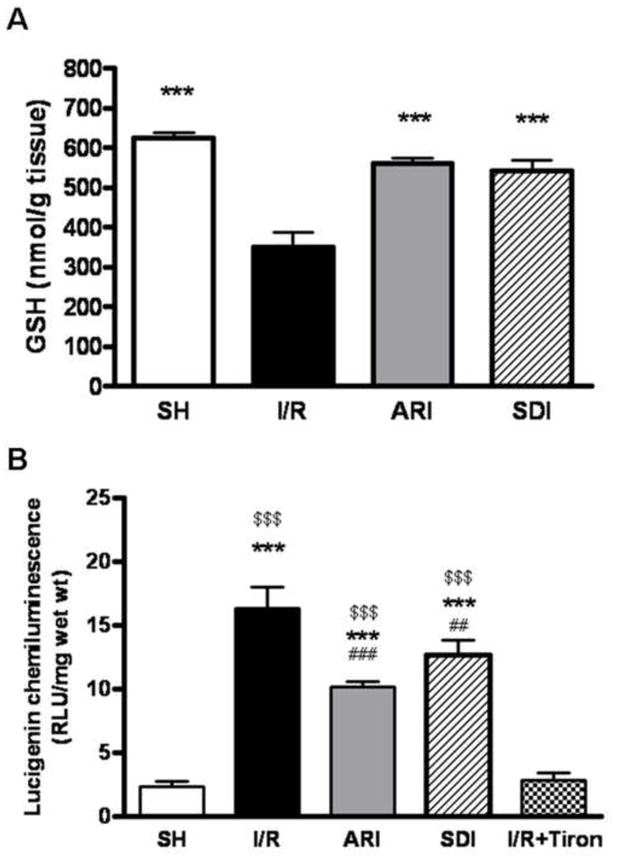
A. Effect of ischemic insults on GSH level in sham-operated (SH), ARI-treated and SDI-treated hearts isolated from rats. GSH level was measured in the heart homogenates after I/R. Data are means ± SE (n=8). **p<0.01 & *p<0.05, compared with I/R group. B. Effect of ischemic insults on superoxide level in SH, ARI-treated and SDI-treated hearts isolated from rats. Superoxide level was measured by lucigenin-elicited chemiluminescence after I/R. Data are means ± SE (n=6). ***p<0.001, **p<0.01 & *p<0.05, compared with normal group; $$$p<0.001, $$p<0.01 & $p<0.05, compared with I/R+Tiron group; ###p<0.001, ##p<0.01 & #p<0.05, compared with I/R group.
Inhibition of polyol pathway reduced the level of peroxynitrite in I/R hearts
Formation of nitrotyrosine (NT), reflecting the level of peroxynitrite, is an important oxidative stress marker. Immunohistochemical staining of the heart slices showed that NT level was much higher in the I/R heart than that in the control group, indicating that I/R led to large increases in peroxynitrite. Inhibition of AR or SDH effectively reduced the level of nitrotyrosine (Fig. 4), demonstrating that polyol pathway contributed to the increased level of peroxynitrite. This observation further confirmed that the polyol pathway is an important contributor to I/R-induced oxidative stress in the heart.
Figure 4.
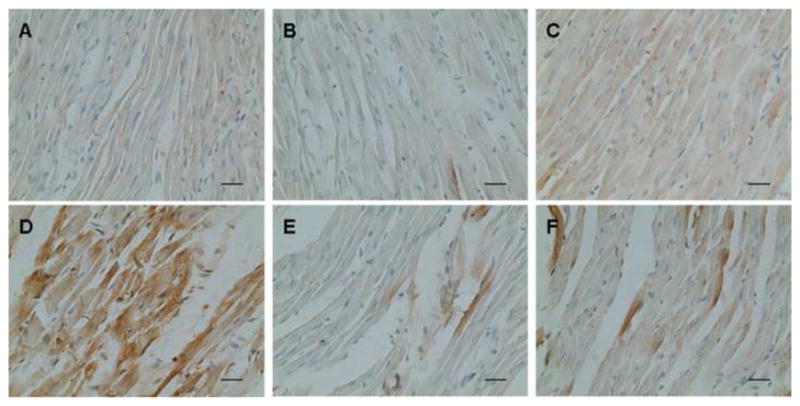
Effect of ischemic insult on nitrotyrosine level in left ventricles from normal, ARI-treated and SDI-treated rat hearts. Representative staining with sham operation was taken from (A) normal, (B) ARI-treated and (C) SDI-treated group at 40X magnification; Representative staining challenged by I/R was taken from (D) normal, (E) ARI-treated & (F) SDI-treated groups (n=6).
Inhibition of polyol pathway improved the impairment of Ca2+ homeostasis in cardiomyocytes induced by I/R
To further understand the role of polyol pathway in I/R-induced contractile dysfunction we investigated the effect of inhibition of AR and SDH activities on Ca2+ signaling of cardiomyocytes under simulated I/R conditions. No significant increase in LDH release was found in cardiomyocytes subjected to I/R, indicating no significant cell death in the short period of ischemia and reperfusion (see the supplementary figure 1). Contraction of the cells was induced by electrical pacer at 0.2 Hz. The representative tracings of electrically stimulated [Ca2+]i transients were shown in Figure 5E. The amplitude of the electrically-induced Ca2+ signal (E[Ca2+]i) that has been shown to be directly correlated with contraction (24), was greatly reduced after I/R. It was restored in the presence of AR or SDH inhibitors (Fig. 5A).
Figure 5.
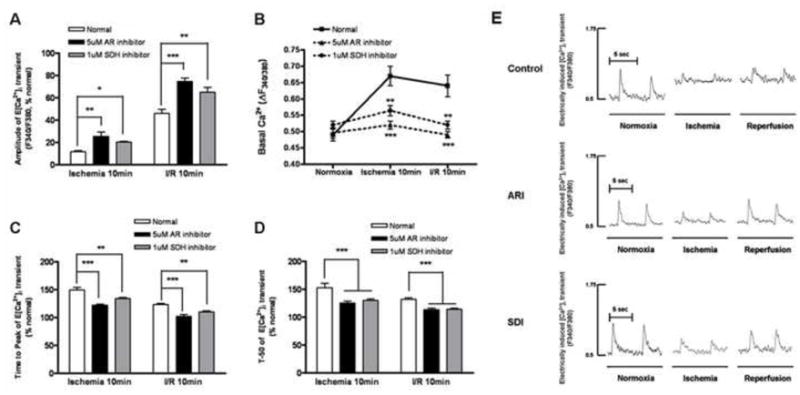
Changes in the (A) peak amplitude, (B) basal cytosolic Ca2+ level, (C) time-to-peak and (D) decay time of E[Ca2+]i transients of normal, ARI-treated and SDI-treated cardiomyocytes subjected to I/R. Data are means ± SE (n=6 cells from 5 rats in each group). ***p<0.001, **p<0.01 & *p<0.05, compared with corresponding normal group. (E) The representative tracings of electrically stimulated intracellular [Ca2+] transients of normal, ARI-treated & SDI-treated cardiomyocytes subjected to I/R.
The basal Ca2+ level, representing the diastolic cytosolic Ca2+ content, was increased from 0.5 AU to 0.675 AU after 10 min of ischemia, and to 0.65 AU after 10 min of reperfusion. Inhibition of AR maintains the basal Ca2+ level close to the pre-ischemic level after ischemia and reperfusion. For SDH-inhibited group, after 10-min ischemia, the basal Ca2+ level was slightly increased to 0.55 AU, and returned to normal level after 10-min reperfusion (Fig. 5B).
Time to peak of E[Ca2+]i (TP) represents the rate of Ca2+ release from the SR, mainly via RyR. After 10 min of ischemia, TP was increased by 150%, suggesting that ischemia reduced RyR activity. Inhibition of AR or SDH reduced TP to 120% and 130% respectively (Fig. 5C). In the control cells, 10 min of reperfusion reduce TP from 150% to 125% of cells before ischemia. In cells treated with AR or SDH inhibitors, TP was further reduced to 100% and 110%, respectively. These results indicated that blocking the polyol pathway protects the RyR against I/R-induced damages.
The decay of E[Ca2+]i is mainly determined by Ca2+ uptake via SERCA, which is responsible for the removal of approximately 90% of Ca2+ from the cytoplasm (35). We therefore measured the time to reduce 50% of the peak E[Ca2+]i (t50) as an indicator of SERCA activity. In the control cells after 10-min ischemia and 10-min reperfusion, t50 was increased to 150% and 130% respectively, of the pre-ischemic cells. Inhibition of AR reduced t50 of the ischemic and reperfused cells to 125% and 110% respectively, of the pre-ischemic cells. Inhibition of SDH also reduced t50 of the I/R cells to a similar extent (Fig. 5D).
Inhibition of polyol pathway restored the activities of SERCA and RyR during I/R
To better understand how polyol pathway contributes to I/R-induced impairment in Ca2+ homeostasis, we measured 45Ca2+ uptake in the isolated SR vesicles as described in the methods. We first determined the 45Ca2+ uptake via SERCA by SR. After 10 min of ischemia, Ca2+ uptake was decreased from 35 nmol mg−1 min−1 to 20 nmol mg−1 min−1, and it was not changed by 10 min reperfusion (Fig. 6B&D). Inhibition of AR restored Ca2+ uptake to 30 nmol mg−1 min−1 after ischemia as well as after reperfusion. Similarly, inhibition of SDH improved Ca2+ uptake to 30 nmol mg−1 min−1 after ischemia and 26 nmol mg−1 min−1 after reperfusion. However, the level of SERCA protein was not changed after 10-min ischemia and 10-min reperfusion. In addition, inhibition of AR or SDH did not alter its protein level (see the supplementary figure 2).
Figure 6.
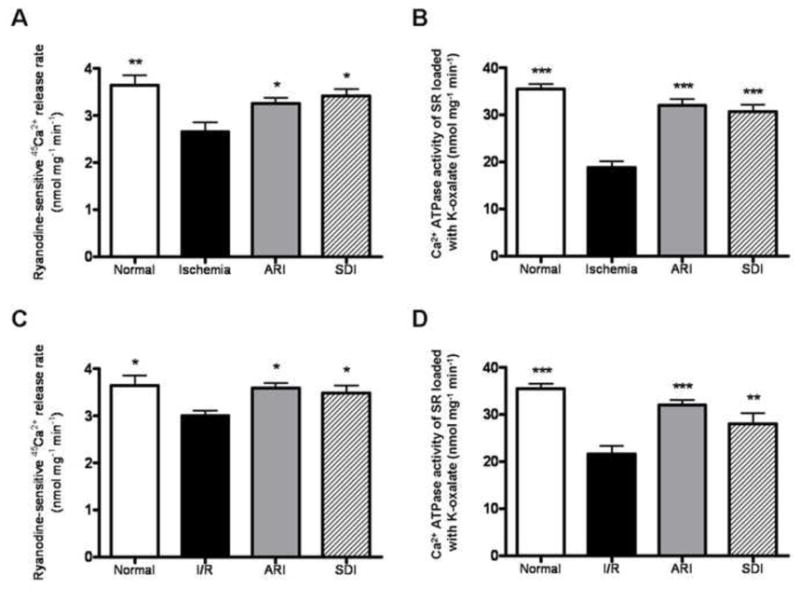
Activities of cardiac RyR and SERCA in the normal, ARI-treated and SDI-treated groups subjected to ischemia (A & B) and reperfusion (C & D). Data are means ± SE (n=5). ***p<0.001, **p<0.01 & *p<0.05, compared with corresponding I/R group.
Ryanodine-sensitive 45Ca2+ release rate, an indication of SR Ca2+ release through the RyR, was decreased from 3.5 nmol mg−1 min−1 to 2.5 nmol mg−1 min−1 after 10 min of ischemia; and decreased to 3 nmol mg−1 min−1 after 10 min reperfusion (Fig. 6A&C). Inhibition of AR or SDH restored the uptake to 3 nmol mg−1 min−1 after ischemia; and restored to 3.5 nmol mg−1 min−1 after reperfusion. Similar to SERCA protein the level of RyR protein was not altered by I/R, and also inhibition of AR or SDH also had no effect on its protein level (see the supplementary figure 2).
Inhibition of polyol pathway attenuated the tyrosine nitration of SERCA2 and maintained the S-glutathiolation of SERCA2 in I/R rat hearts
Previous studies have shown that SERCA2 is sensitive to oxidative stress and its activity is inhibited by both ONOO− and nitrotyrosine formation, and these multiple oxidations block the S-glutathiolation of SERCA (i.e. GSS-SERCA), which increases SERCA activity (21, 36–38). As stated above, we found that the activity of SERCA2 was impaired, and superoxide anion and ONOO− production increased in I/R hearts. To determine if SERCA2 in I/R hearts have increased nitrotyrosine, the protein was immunoprecipitated from heart extracts using anti-SERCA2 antibody and analyzed by western blot using anti-nitrotyrosine antibody (Fig. 7). The level of nitrotyrosine on SERCA2 was significantly increased in I/R hearts, and inhibition of either AR or SDH attenuated tyrosine nitration of SERCA2, suggesting that blockade of polyol pathway restored the activity of SERCA2 by attenuating the formation of nitrotyrosine.
Figure 7.
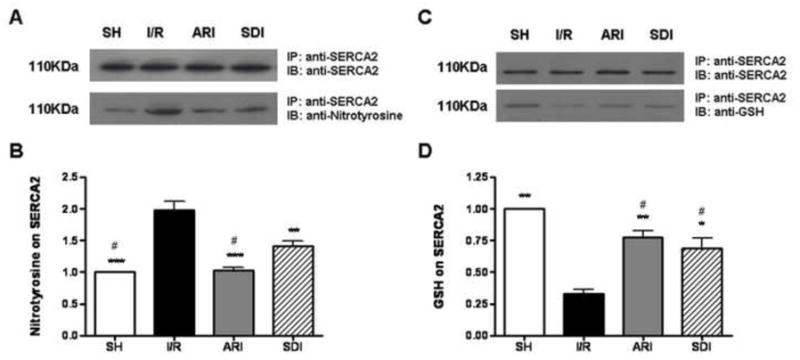
A. Effect of ischemic insult on the level of tyrosine nitration on SERCA2 in sham-operated (SH), ARI & SDI-treated hearts isolated from rats. For detection of nitrotyrosine in SERCA2 protein, (A, upper) an immunoprecipitate (IP) was obtained using anti-SERCA2 antibody and then immunoblotted with anti-SERCA2 antibody. (A, lower) An IP was obtained using anti-SERCA2 antibody and then immunoblotted with anti-nitrotyrosine antibody. B. Quantitative analysis of nitrotyrosine by scanning densitometry. Data are means ± SE (n=3). ***p <0.001 & **p<0.01, compared with I/R group; #p<0.01, compared with SDI group. C. Effect of ischemic insult on the level of glutathiolation on SERCA2 in sham-operated (SH), ARI & SDI-treated hearts isolated from rats. (C, lower) An IP was obtained using anti-SERCA2 antibody and then immunoblotted with anti-GSH antibody. D. Quantitative analysis of GSH by scanning densitometry. Data are means ± SE (n=3). **p<0.01 & *p<0.05, compared with I/R group; #p<0.01, compared with SH group.
Besides tyrosine nitration of SERCA, S-glutathiolation of SERCA also affects its activity. As shown in Figure 7 the amount of GSH on the immunoprecipitated SERCA was significantly decreased in I/R heart, indicating that the level of GSS-SERCA was lowered in I/R hearts. Inhibition of either AR or SDH restored the level of S-glutathiolation, suggesting that the blockade of polyol pathway restored the activity of SERCA by normalizing S-glutathiolation.
Inhibition of polyol pathway attenuated the I/R-induced oxidation of cystein-674 in SERCA
Previous studies demonstrated that high level of peroxynitrite irreversibly oxidized cysteine-674 of SERCA to the sulfonic acid form, contributing to its inactivation (31, 39). Therefore, antibodies specific for SERCAC674-SO3H, was used to examine the role of polyol pathway in the I/R inactivation of SERCA. Compared to control, the left ventricle from the I/R group showed large increase in the staining of SERCAC674-SO3H, and the staining was substantially reduced in tissues treated with ARI or SDI (Fig. 8), indicating that polyol pathway is a major contributor to I/R inactivation of SERCA.
Figure 8.
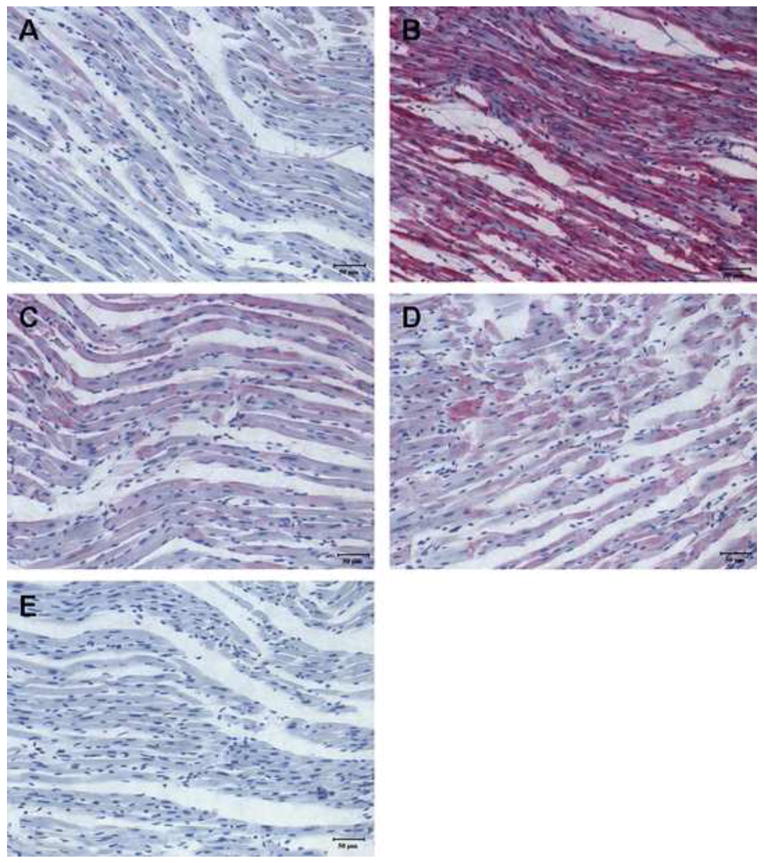
Immunohistochemical staining with anti-SERCA C674-SO3H of left ventricles from post-ischemic reperfused rat hearts. Representative staining was taken from (A) normal, (B) I/R, (C) I/R+ARI, (D) I/R+SDI group and (E) non-specific IgG control.
Inhibition of polyol pathway maintained the S-glutathiolation of RyR in I/R hearts
It has been reported that GSH acts as a cofactor for the stimulation of H2O2-mediated calcium release by RyR in cardiac myocytes (40). S-glutathionation of cysteine residues of RyR was found in normal hearts, and the level was increased under myocardial preconditioning (41). Therefore, we investigated if the polyol pathway influenced the level of S-glutathiolation of RyR in I/R hearts. To assess the effect of polyol pathway on the level of S-glutathiolation of RyR in I/R hearts, RyR was immunoprecipitated and analyzed by western blotting with anti-GSH antibody (Fig. 9). The level of RyR S-glutathiolation was significantly decreased in I/R hearts, and inhibition of either AR or SDH restored the level of S-glutathiolation, suggesting that the blockade of polyol pathway restored the activity of RyR by S-glutathiolation.
Figure 9.
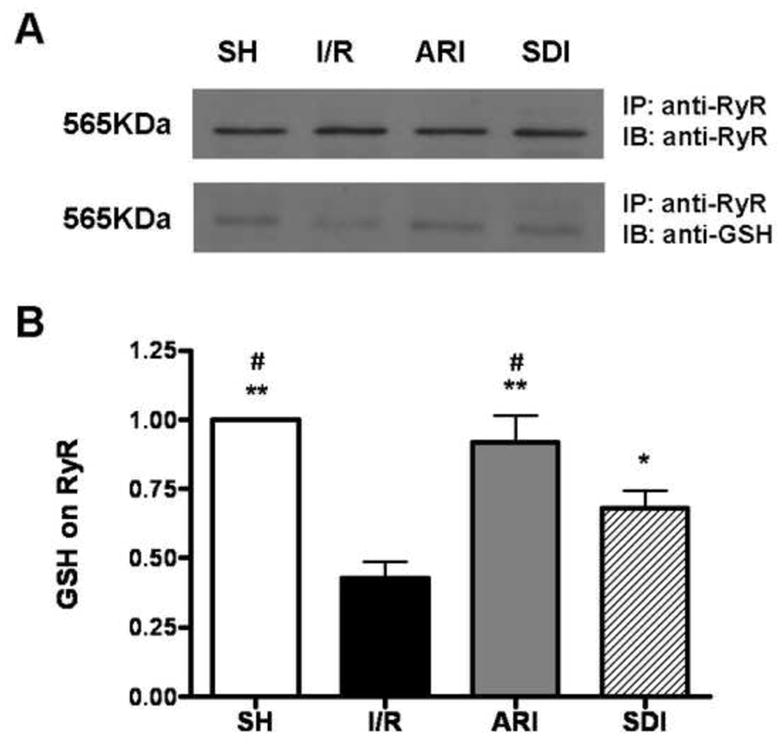
A. Effect of ischemic insult on the level of glutathiolation on RyR in sham-operated (SH), ARI & SDI-treated hearts isolated from rats. For detection of GSH in RyR protein, (A, upper) an immunoprecipitate (IP) was obtained using anti-RyR antibody and then immunoblotted with anti-RyR antibody. (A, lower) An IP was obtained using anti-RyR antibody and then immunoblotted with anti-GSH antibody. B. Quantitative analysis of GSH by scanning densitometry. Data are means ± SE (n=3). **p<0.01 & *p<0.05, compared with I/R group; #p<0.01, compared with SDI group.
DISCUSSION
The isolated rat hearts subjected to 30 min of ischemia followed by 45 min of reperfusion exhibited a significant impairment in their contractile function. This includes the decrease in LVDP, LVSP and ±dP/dtmax, and the elevation in LVEDP. These contractile abnormalities were significantly attenuated by the inhibition of AR or SDH, indicating that polyol pathway is an important contributor to I/R-induced abnormalities in cardiac contraction. To better understand the mechanism, intracellular Ca2+ transients in cardiomyocytes were analyzed. Rat cardiomyocytes subjected to simulated I/R showed significant alternations in Ca2+ signal profile. This includes increases in base to peak time and decay time of Ca2+ transient, accompanied by decrease in Ca2+ peak amplitude and increase in basal Ca2+ level. All these changes are most likely consequences of the alternations in the function of RyR and SERCA.
Indeed we found that during I/R, the RyR activity was significantly decreased, and this reduction was attenuated by inhibition of AR or SDH. Similarly SERCA activity was reduced during I/R, and inhibition of AR or SDH restored its activity. We showed that polyol pathway activity was increased during I/R, consistent with previous studies. During ischemia, no significant increases in sorbitol and fructose were found due to low level of glucose available. During post-ischemic reperfusion, increase in the availability of glucose for the polyol pathway led to a significant rise in sorbitol level was found in the SDH inhibited group.
The polyol pathway most likely impaired the RyR and SERCA activity during post-ischemic reperfusion by increasing oxidative stress. We showed that GSH level was reduced and superoxide level elevated in I/R hearts, and these changes were significantly attenuated by the administration of ARI and SDI. GSH is required for glutathione peroxidase to remove ROS. Thus low level of GSH would lead to increased level of ROS. Furthermore, one of the major sources of ROS in cardiomyocytes is superoxide produced by the membrane associated NADH oxidase, an enzyme controlled by cytosolic NADH/NAD+ ratio and oxygen pressure (30). During I/R, the activation of the polyol pathway increased the level of NADH as the consequence of oxidation of sorbitol into fructose by SDH. The increased NADH stimulates NADH oxidase, which is in close proximity to SR, to generate ROS (30, 42). Increased ROS inhibits SERCA by oxidizing the cysteine thiols, interfering with the ATP binding site, making it unable to hydrolyze the ATP (43). Recently, it has been demonstrated that, in the presence of GSH, NO-derived peroxynitrite reversibly modifies SERCA by S-glutathiolation into the activated form, GSS-SERCA (38). However, too much peroxynitrite irreversibly inactivates SERCA.
Multiple oxidations were previously found to be associated with a 50% decrease in SERCA activity in atherosclerotic rabbit aorta (21). In the I/R hearts, the increased level of peroxynitrite led to increased tyrosine nitration of SERCA, and accompanied by decreased GSS-SERCA level. We showed that inhibition of AR or SDH normalized the levels of nitrotyrosine in SERCA and GSS-SERCA. Moreover, inhibition of the polyol pathway also attenuated the I/R-induced increase in SERCAC674-SO3H, the irreversibly oxidized form of SERCA, which has been shown to contribute to the inactivation of SERCA (31, 39).
RyR activity also appeared to be reduced by I/R and ameliorated by inhibition of AR or SDH. Polyol pathway-mediated increased ROS could irreversibly inactivates RyR by distorting its protein structure, although initial stimulation of its activity by oxidative modification was also observed (44). It has been shown that the gating property of cardiac RyR is modulated by the cytosolic NADH/NAD+ ratio. NADH inhibits the current through RyR channels by a mechanism that required oxidation of NADH and reversed by NAD+ (45). It is well established that SDH activity increased cytosolic NADH/NAD+ ratio in diabetic tissues. Here we showed that NADH/NAD+ ratio, as reflected by L/P ratio, was also increased in I/R hearts, contributing to the impairment of RyR. It has been shown that S-glutathiolation of RyR causes a significant stimulation of channel activity in skeletal RyR (46, 47), and S-glutathiolation of cardiac RyR was observed under normal and myocardial preconditioning (41). We found that S-glutathiolation of the cardiac RyR2 was reduced during I/R, and inhibition of the polyol pathway attenuated the decrease in S-glutathiolation of cardiac RyR2. This provides another explanation for polyol pathway’s contribution to contractile dysfunction of I/R hearts. However, the decreased RyR activity we measured may also be due to decreased Ca2+ content in SR. This is because the measurement of RyR activity was based on the difference between SR Ca2+ uptake in the presence and absence of ryanodine, and the amount of Ca2+ release via RyR is dependent on the amount of Ca2+ load in SR. Therefore, the reduced Ca2+ load in SR due to depressed SERCA activity, may lead to the apparent reduction in RyR activity.
The present study demonstrated that the decreased LVEDP during reperfusion was caused by the impairment of cardiac SERCA and RyR due to the irreversible modification of SERCA and RyR by the polyol pathway-induced oxidative stress. It is possible that the alteration of calcium binding affinity and function of other contractile proteins may also contribute to the decreased LVEDP under reperfusion. Recently, it was found that the decreased of cardiac function after ischemia/reperfusion was contributed by the proteolytic degradation of cardiac troponin I, resulting in alterations in cross-bridge cycling (48).
Although inhibition of the polyol pathway has been shown to reduce infract size of the 2 hour I/R protocol, it is unlikely that protection of SERCA and RyR activity by AR or SDH inhibitors is a non-specific consequence of protection against I/R-induced cell damage in our protocol of 30 minutes ischemia followed by 45 minutes reperfusion, where previous studies (49) and unpublished studies of our own showed little infarct. Moreover, the protective effect of AR and SDH inhibitors were also demonstrated in the cardiac cell studies, where there were no significant increase in cell death or leakage of LDH after 10-minute ischemia followed by ischemia. This is consistent with the previous finding that a short period (up to 20 minutes) of simulated ischemic condition similar to that reported here, followed by reperfusion, causes no significant decrease in cell viability (50).
In conclusion we demonstrated that during I/R, the polyol pathway activities increase oxidative stress, leading to increased tyrosine nitration of SERCA, irreversible modification to form SERCAC674-SO3H, reduction of GSS-SERCA. Polyol pathway activities also decrease S-glutathiolation of RyR in I/R hearts. The increased oxidation of the two major Ca2+ handling proteins contributes to the impairment of contractile dysfunction. Thus inhibiting AR or SDH in the I/R heart not only would reduce infarct size as demonstrated earlier (15, 17), but would also improve post-ischemic contractile function.
Acknowledgments
We thank C.P. Mok and Dr S. Wu for their technical assistance. XT and XH were supported by NIH grant RO1 HL31607-25. We would like to thank Prof Richard A. Cohen for critical reading of the manuscript.
LIST OF ABBREVATIONS
- AR
aldose reductase
- ARI
aldose reductase inhibitor
- ATP
adenosine triphosphate
- E[Ca2+]i
electrically induced calcium transient
- GAPDH
glyceraldehydes-3-phosphate dehydrogenase
- GSH
glutathione
- HR
heart rate
- [Ca2+]i
intracellular calcium
- I/R
ischemia-reperfusion
- LDH
lactate dehydrogenase
- LVEDP
left ventricular end-diastolic pressure
- LVDP
left ventricular developed pressure
- LVSP
left ventricular systolic pressure
- NAD+
nicotinamide adenine dinucleotide
- NADH
reduced nicotinamide adenine dinucleotide
- NADP+
nicotinamide adenine dinucleotide phosphate
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- NCX
Na+/Ca2+ exchanger
- NT
nitrotyrosine
- RLU
relative light unit
- ROS
reactive oxygen species
- RyR
ryanodine receptor
- SERCA
sacro/endoplasmic reticulum Ca2+-ATPase
- SDH
sorbitol dehydrogenase
- SDI
sorbitol dehydrogenase inhibitor
- SH
sham-operated
- TP
time-to-peak of E[Ca2+]i
- T50
time to reduce 50% of E[Ca2+]i
References
- 1.Bolli R. Myocardial ‘stunning’ in man. Circulation. 1992 Dec;86(6):1671–91. doi: 10.1161/01.cir.86.6.1671. [DOI] [PubMed] [Google Scholar]
- 2.Kloner RA, Ellis SG, Lange R, Braunwald E. Studies of experimental coronary artery reperfusion. Effects on infarct size, myocardial function, biochemistry, ultrastructure and microvascular damage. Circulation. 1983 Aug;68(2 Pt 2):I8–15. [PubMed] [Google Scholar]
- 3.Dhalla NS, Panagia V, Singal PK, Makino N, Dixon IM, Eyolfson DA. Alterations in heart membrane calcium transport during the development of ischemia-reperfusion injury. J Mol Cell Cardiol. 1988 Mar;20( Suppl 2):3–13. doi: 10.1016/0022-2828(88)90327-6. [DOI] [PubMed] [Google Scholar]
- 4.Guatimosim S, Dilly K, Santana LF, Saleet Jafri M, Sobie EA, Lederer WJ. Local Ca(2+) signaling and EC coupling in heart: Ca(2+) sparks and the regulation of the [Ca(2+)](i) transient. J Mol Cell Cardiol. 2002 Aug;34(8):941–50. doi: 10.1006/jmcc.2002.2032. [DOI] [PubMed] [Google Scholar]
- 5.Zweier JL. Measurement of superoxide-derived free radicals in the reperfused heart. Evidence for a free radical mechanism of reperfusion injury. J Biol Chem. 1988 Jan 25;263(3):1353–7. [PubMed] [Google Scholar]
- 6.Blaustein AS, Schine L, Brooks WW, Fanburg BL, Bing OH. Influence of exogenously generated oxidant species on myocardial function. Am J Physiol. 1986 Apr;250(4 Pt 2):H595–9. doi: 10.1152/ajpheart.1986.250.4.H595. [DOI] [PubMed] [Google Scholar]
- 7.Rowe GT, Manson NH, Caplan M, Hess ML. Hydrogen peroxide and hydroxyl radical mediation of activated leukocyte depression of cardiac sarcoplasmic reticulum. Participation of the cyclooxygenase pathway. Circ Res. 1983 Nov;53(5):584–91. doi: 10.1161/01.res.53.5.584. [DOI] [PubMed] [Google Scholar]
- 8.Kaneko M, Beamish RE, Dhalla NS. Depression of heart sarcolemmal Ca2+-pump activity by oxygen free radicals. Am J Physiol. 1989 Feb;256(2 Pt 2):H368–74. doi: 10.1152/ajpheart.1989.256.2.H368. [DOI] [PubMed] [Google Scholar]
- 9.Chung SS, Chung SK. Aldose reductase in diabetic microvascular complications. Curr Drug Targets. 2005 Jun;6(4):475–86. doi: 10.2174/1389450054021891. [DOI] [PubMed] [Google Scholar]
- 10.Srivastava SK, Ramana KV, Bhatnagar A. Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocr Rev. 2005 May;26(3):380–92. doi: 10.1210/er.2004-0028. [DOI] [PubMed] [Google Scholar]
- 11.Chung SS, Chung SK. Genetic analysis of aldose reductase in diabetic complications. Curr Med Chem. 2003 Aug;10(15):1375–87. doi: 10.2174/0929867033457322. [DOI] [PubMed] [Google Scholar]
- 12.Lee AY, Chung SS. Contributions of polyol pathway to oxidative stress in diabetic cataract. Faseb J. 1999 Jan;13(1):23–30. doi: 10.1096/fasebj.13.1.23. [DOI] [PubMed] [Google Scholar]
- 13.Ramasamy R, Oates PJ, Schaefer S. Aldose reductase inhibition protects diabetic and nondiabetic rat hearts from ischemic injury. Diabetes. 1997 Feb;46(2):292–300. doi: 10.2337/diab.46.2.292. [DOI] [PubMed] [Google Scholar]
- 14.Ramasamy R, Trueblood N, Schaefer S. Metabolic effects of aldose reductase inhibition during low-flow ischemia and reperfusion. Am J Physiol. 1998 Jul;275(1 Pt 2):H195–203. doi: 10.1152/ajpheart.1998.275.1.H195. [DOI] [PubMed] [Google Scholar]
- 15.Tracey WR, Magee WP, Ellery CA, MacAndrew JT, Smith AH, Knight DR, et al. Aldose reductase inhibition alone or combined with an adenosine A(3) agonist reduces ischemic myocardial injury. Am J Physiol Heart Circ Physiol. 2000 Oct;279(4):H1447–52. doi: 10.1152/ajpheart.2000.279.4.H1447. [DOI] [PubMed] [Google Scholar]
- 16.Lo AC, Cheung AK, Hung VK, Yeung CM, He QY, Chiu JF, et al. Deletion of aldose reductase leads to protection against cerebral ischemic injury. J Cereb Blood Flow Metab. 2007 Aug;27(8):1496–509. doi: 10.1038/sj.jcbfm.9600452. [DOI] [PubMed] [Google Scholar]
- 17.Tang WH, Wu S, Wong TM, Chung SK, Chung SS. Polyol pathway mediates iron-induced oxidative injury in ischemic-reperfused rat heart. Free Radic Biol Med. 2008 May 15; doi: 10.1016/j.freeradbiomed.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Ford WR, Clanachan AS, Jugdutt BI. Opposite effects of angiotensin AT1 and AT2 receptor antagonists on recovery of mechanical function after ischemia-reperfusion in isolated working rat hearts. Circulation. 1996 Dec 15;94(12):3087–9. doi: 10.1161/01.cir.94.12.3087. [DOI] [PubMed] [Google Scholar]
- 19.Hwang YC, Bakr S, Ellery CA, Oates PJ, Ramasamy R. Sorbitol dehydrogenase: a novel target for adjunctive protection of ischemic myocardium. Faseb J. 2003 Dec;17(15):2331–3. doi: 10.1096/fj.03-0128fje. [DOI] [PubMed] [Google Scholar]
- 20.Wu S, Li HY, Wong TM. Cardioprotection of preconditioning by metabolic inhibition in the rat ventricular myocyte. Involvement of kappa-opioid receptor. Circ Res. 1999 Jun 25;84(12):1388–95. doi: 10.1161/01.res.84.12.1388. [DOI] [PubMed] [Google Scholar]
- 21.Adachi T, Matsui R, Xu S, Kirber M, Lazar HL, Sharov VS, et al. Antioxidant improves smooth muscle sarco/endoplasmic reticulum Ca(2+)-ATPase function and lowers tyrosine nitration in hypercholesterolemia and improves nitric oxide-induced relaxation. Circ Res. 2002 May 31;90(10):1114–21. doi: 10.1161/01.res.0000019757.57344.d5. [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Kam KW, Borchert GH, Kravtsov GM, Ballard HJ, Wong TM. Further study on the role of HSP70 on Ca2+ homeostasis in rat ventricular myocytes subjected to simulated ischemia. Am J Physiol Cell Physiol. 2006 Feb;290(2):C583–91. doi: 10.1152/ajpcell.00145.2005. [DOI] [PubMed] [Google Scholar]
- 23.Temsah RM, Netticadan T, Chapman D, Takeda S, Mochizuki S, Dhalla NS. Alterations in sarcoplasmic reticulum function and gene expression in ischemic-reperfused rat heart. Am J Physiol. 1999 Aug;277(2 Pt 2):H584–94. doi: 10.1152/ajpheart.1999.277.2.H584. [DOI] [PubMed] [Google Scholar]
- 24.Zucchi R, Ronca-Testoni S, Yu G, Galbani P, Ronca G, Mariani M. Effect of ischemia and reperfusion on cardiac ryanodine receptors--sarcoplasmic reticulum Ca2+ channels. Circ Res. 1994 Feb;74(2):271–80. doi: 10.1161/01.res.74.2.271. [DOI] [PubMed] [Google Scholar]
- 25.Kravtsov GM, Pokudin NI, Orlov SN. Ca2+-accumulating capacity of mitochondria, sarcolemma and sarcoplasmic reticulum of rat heart. Biokhimiia. 1979 Nov;44(11):2058–65. [PubMed] [Google Scholar]
- 26.Ying WL, Emerson J, Clarke MJ, Sanadi DR. Inhibition of mitochondrial calcium ion transport by an oxo-bridged dinuclear ruthenium ammine complex. Biochemistry. 1991 May 21;30(20):4949–52. doi: 10.1021/bi00234a016. [DOI] [PubMed] [Google Scholar]
- 27.Nakazawa K, Higo K, Abe K, Tanaka Y, Saito H, Matsuki N. Blockade by calmodulin inhibitors of Ca2+ channels in smooth muscle from rat vas deferens. Br J Pharmacol. 1993 May;109(1):137–41. doi: 10.1111/j.1476-5381.1993.tb13543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fabiato A, Fabiato F. Effects of magnesium on contractile activation of skinned cardiac cells. J Physiol. 1975 Aug;249(3):497–517. doi: 10.1113/jphysiol.1975.sp011027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaefer A, Magocsi M, Stocker U, Kosa F, Marquardt H. Early transient suppression of c-myb mRNA levels and induction of differentiation in Friend erythroleukemia cells by the [Ca2+]i-increasing agents cyclopiazonic acid and thapsigargin. J Biol Chem. 1994 Mar 25;269(12):8786–91. [PubMed] [Google Scholar]
- 30.Mohazzab HK, Kaminski PM, Wolin MS. Lactate and PO2 modulate superoxide anion production in bovine cardiac myocytes: potential role of NADH oxidase. Circulation. 1997 Jul 15;96(2):614–20. doi: 10.1161/01.cir.96.2.614. [DOI] [PubMed] [Google Scholar]
- 31.Ying J, Sharov V, Xu S, Jiang B, Gerrity R, Schoneich C, et al. Cysteine-674 oxidation and degradation of sarcoplasmic reticulum Ca(2+) ATPase in diabetic pig aorta. Free Radic Biol Med. 2008 Sep 15;45(6):756–62. doi: 10.1016/j.freeradbiomed.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwata K, Matsuno K, Nishinaka T, Persson C, Yabe-Nishimura C. Aldose reductase inhibitors improve myocardial reperfusion injury in mice by a dual mechanism. J Pharmacol Sci. 2006 Sep;102(1):37–46. doi: 10.1254/jphs.fp0060218. [DOI] [PubMed] [Google Scholar]
- 33.Li Q, Hwang YC, Ananthakrishnan R, Oates PJ, Guberski D, Ramasamy R. Polyol pathway and modulation of ischemia-reperfusion injury in Type 2 diabetic BBZ rat hearts. Cardiovasc Diabetol. 2008;7:33. doi: 10.1186/1475-2840-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hwang YC, Kaneko M, Bakr S, Liao H, Lu Y, Lewis ER, et al. Central role for aldose reductase pathway in myocardial ischemic injury. Faseb J. 2004 Aug;18(11):1192–9. doi: 10.1096/fj.03-1400com. [DOI] [PubMed] [Google Scholar]
- 35.Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res. 2000 Aug 18;87(4):275–81. doi: 10.1161/01.res.87.4.275. [DOI] [PubMed] [Google Scholar]
- 36.Viner RI, Ferrington DA, Williams TD, Bigelow DJ, Schoneich C. Protein modification during biological aging: selective tyrosine nitration of the SERCA2a isoform of the sarcoplasmic reticulum Ca2+-ATPase in skeletal muscle. Biochem J. 1999 Jun 15;340( Pt 3):657–69. [PMC free article] [PubMed] [Google Scholar]
- 37.Knyushko TV, Sharov VS, Williams TD, Schoneich C, Bigelow DJ. 3-Nitrotyrosine modification of SERCA2a in the aging heart: a distinct signature of the cellular redox environment. Biochemistry. 2005 Oct 4;44(39):13071–81. doi: 10.1021/bi051226n. [DOI] [PubMed] [Google Scholar]
- 38.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, et al. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004 Nov;10(11):1200–7. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 39.Tong X, Ying J, Pimentel DR, Trucillo M, Adachi T, Cohen RA. High glucose oxidizes SERCA cysteine-674 and prevents inhibition by nitric oxide of smooth muscle cell migration. J Mol Cell Cardiol. 2008 Feb;44(2):361–9. doi: 10.1016/j.yjmcc.2007.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suzuki YJ, Cleemann L, Abernethy DR, Morad M. Glutathione is a cofactor for H2O2-mediated stimulation of Ca2+-induced Ca2+ release in cardiac myocytes. Free Radic Biol Med. 1998 Jan 15;24(2):318–25. doi: 10.1016/s0891-5849(97)00227-x. [DOI] [PubMed] [Google Scholar]
- 41.Sanchez G, Pedrozo Z, Domenech RJ, Hidalgo C, Donoso P. Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle. J Mol Cell Cardiol. 2005 Dec;39(6):982–91. doi: 10.1016/j.yjmcc.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 42.Bassenge E, Sommer O, Schwemmer M, Bunger R. Antioxidant pyruvate inhibits cardiac formation of reactive oxygen species through changes in redox state. Am J Physiol Heart Circ Physiol. 2000 Nov;279(5):H2431–8. doi: 10.1152/ajpheart.2000.279.5.H2431. [DOI] [PubMed] [Google Scholar]
- 43.Xu KY, Zweier JL, Becker LC. Hydroxyl radical inhibits sarcoplasmic reticulum Ca(2+)-ATPase function by direct attack on the ATP binding site. Circ Res. 1997 Jan;80(1):76–81. doi: 10.1161/01.res.80.1.76. [DOI] [PubMed] [Google Scholar]
- 44.Holmberg SR, Cumming DV, Kusama Y, Hearse DJ, Poole-Wilson PA, Shattock MJ, et al. Reactive oxygen species modify the structure and function of the cardiac sarcoplasmic reticulum calcium-release channel. Cardioscience. 1991 Mar;2(1):19–25. [PubMed] [Google Scholar]
- 45.Zima AV, Copello JA, Blatter LA. Effects of cytosolic NADH/NAD(+) levels on sarcoplasmic reticulum Ca(2+) release in permeabilized rat ventricular myocytes. J Physiol. 2004 Mar 16;555(Pt 3):727–41. doi: 10.1113/jphysiol.2003.055848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aracena P, Sanchez G, Donoso P, Hamilton SL, Hidalgo C. S-glutathionylation decreases Mg2+ inhibition and S-nitrosylation enhances Ca2+ activation of RyR1 channels. J Biol Chem. 2003 Oct 31;278(44):42927–35. doi: 10.1074/jbc.M306969200. [DOI] [PubMed] [Google Scholar]
- 47.Aracena P, Tang W, Hamilton SL, Hidalgo C. Effects of S-glutathionylation and S-nitrosylation on calmodulin binding to triads and FKBP12 binding to type 1 calcium release channels. Antioxid Redox Signal. 2005 Jul–Aug;7(7–8):870–81. doi: 10.1089/ars.2005.7.870. [DOI] [PubMed] [Google Scholar]
- 48.Tachampa K, Kobayashi T, Wang H, Martin AF, Biesiadecki BJ, Solaro RJ, et al. Increased cross-bridge cycling kinetics after exchange of C-terminal truncated troponin I in skinned rat cardiac muscle. J Biol Chem. 2008 May 30;283(22):15114–21. doi: 10.1074/jbc.M801636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palmer BS, Hadziahmetovic M, Veci T, Angelos MG. Global ischemic duration and reperfusion function in the isolated perfused rat heart. Resuscitation. 2004 Jul;62(1):97–106. doi: 10.1016/j.resuscitation.2003.12.027. [DOI] [PubMed] [Google Scholar]
- 50.Saini HK, Dhalla NS. Defective calcium handling in cardiomyocytes isolated from hearts subjected to ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2005 May;288(5):H2260–70. doi: 10.1152/ajpheart.01153.2004. [DOI] [PubMed] [Google Scholar]