

Abstract
There has been growing evidence for extensive diversity of alternative splicing in human populations. Genetic variants within the 5′ splice site can cause splicing differences among human individuals and constitute an important class of human disease mutations. In this study, we explored whether natural variations of splicing could reveal important signals of 5′ splice site recognition. In seven lymphoblastoid cell lines of Asian, European and African ancestry, we identified 1174 single nucleotide polymorphisms (SNPs) within the consensus 5′ splice site. We selected 129 SNPs predicted to significantly alter the splice site activity, and quantitatively examined their splicing impact in the seven individuals. Surprisingly, outside of the essential GT dinucleotide position, only ∼14% of the tested SNPs altered splicing. Bioinformatic and minigene analyses identified signals that could modify the impact of 5′ splice site polymorphisms, most notably a strong 3′ splice site and the presence of intronic motifs downstream of the 5′ splice site. Strikingly, we found that the poly-G run, a known intronic splicing enhancer, was the most significantly enriched motif downstream of exons unaffected by 5′ splice site SNPs. In TRIM62, the upstream 3′ splice site and downstream intronic poly-G runs functioned redundantly to protect an exon from its 5′ splice site polymorphism. Collectively, our study reveals widespread context-dependent robustness to 5′ splice site polymorphisms in human transcriptomes. Consequently, certain exons are more susceptible to 5′ splice site mutations. Additionally, our work demonstrates that genetic diversity of alternative splicing can provide significant insights into the splicing code of mammalian cells.
INTRODUCTION
Alternative splicing is a prevalent mechanism of post-transcriptional gene regulation in multicellular eukaryotes. It allows a single gene to increase its functional and regulatory diversity, through the synthesis of multiple mRNA isoforms encoding structurally and functionally distinct protein products (1). High-throughput RNA sequencing reveals that over 90% of multi-exon genes in mammalian genomes undergo alternative splicing (2,3). The strikingly high frequency of alternative splicing underscores its contribution to the organismal complexity of higher eukaryotes.
The fidelity of splicing is tightly regulated by interactions between cis elements in exons and flanking introns and trans splicing regulators that recognize these elements (4,5). Disruption of normal splicing regulation, even a shift in the ratio of mRNA isoforms of the same gene sometimes can have major functional consequences and cause human diseases (6–8). The most conserved features of exon recognition are splice site signals known as the 5′ splice site (donor site) and the 3′ splice site (acceptor site). The splice sites define the boundaries between exons and introns, at which the spliceosome must assemble. Importantly, the recognition of the 5′ splice sites (i.e. the donor sites) represents the first and a critical step of spliceosome assembly (9). The vast majority (>99%) of 5′ splice sites in eukaryotic genomes are characterized by a highly conserved ‘GT’ dinucleotide in the intronic region immediately adjacent to the exon–intron boundaries (10–12). There are several additional conserved but degenerate nucleotide positions in the exonic and intronic regions surrounding the GT dinucleotide, which are part of the consensus 5′ splice site signal (12,13). Numerous disease-causing mutations within the consensus 5′ splice site disrupt splicing, leading to defective mRNA and protein products (14–16). However, there are also a large number of polymorphisms in the 5′ splice site with no effect on splicing (16). Given the prevalence of aberrant alternative splicing in human diseases, it is critical to obtain an improved understanding of the signals that determine the splicing impact of 5′ splice site mutations. Such knowledge could aid in the identification of pathogenic mutations among neutral variants in large-scale medical sequencing projects.
In recent years, there has been growing evidence for widespread natural variations of alternative splicing in humans (17–24). Single nucleotide polymorphisms (SNPs) are the major contributor of splicing variations in human populations (25). For example, an intronic SNP (rs3812718) in SCN1A, which encodes a neuronal sodium-channel alpha subunit, modulates the alternative splicing of its exon 5 and affects the dose-response to antiepileptic drugs (26). Another example is the low-density lipoproteins receptor (LDLR), in which a SNP (rs688) promotes skipping of its exon 12 in the liver of women (27). This exon skipping form is predicted to produce a truncated protein product lacking the transmembrane segment. Importantly, this SNP is strongly associated with an increased level of total and LDL-cholesterol in females especially in pre-menopausal women (27). Using high-density exon arrays or high-throughput RNA sequencing, several groups have performed genome-scale surveys of splicing differences among human individuals (17–19,21–23). For example, using the Affymetrix exon 1.0 array, Kwan et al. (18) examined alternative splicing patterns in lymphoblastoid cell lines (LCLs) of 57 unrelated HapMap CEU individuals. They identified 177 genes whose transcript isoform compositions (owing to alternative splicing, alternative promoter usage and alternative polyadenylation) correlated strongly with surrounding SNPs. Using a similar approach, Heinzen et al. (21) identified 80 high-confidence associations between SNP and alternative splicing in cortical brain samples and peripheral blood mononuclear cell samples.
In this study, we explored whether natural variations of alternative splicing among human individuals could reveal important signals of 5′ splice site recognition. In a panel of seven LCLs of Asian, European and African ancestry, for which extensive genotyping data were collected by the International HapMap project (28) and a recent genome-wide exome sequencing study (29), we identified 1174 SNPs within the consensus 5′ splice site (three exonic nucleotides and six intronic nucleotides surrounding the exon–intron boundary) (13). We selected 129 SNPs predicted to significantly alter the 5′ splice site activity according to the consensus splice site model in MAXENT (13), and examined their impacts on exon splicing using a fluorescently labeled RT–PCR assay. SNPs that disrupted the GT dinucleotide immediately downstream of the exon always altered splicing, consistent with the essential role of the GT dinucleotide in 5′ splice site recognition. Surprisingly, outside of the almost invariable GT dinucleotide, only ∼14% of tested SNPs affected splicing, while the vast majority (∼86%) of tested exons were unaffected by the 5′ splice site SNPs. Bioinformatic analysis identified signals that could modify the splicing impact of 5′ splice site polymorphisms, most notably a strong 3′ splice site upstream of the exon and the presence of particular intronic sequence motifs downstream of the 5′ splice site. The activity of these predicted sequence features was experimentally confirmed by minigene splicing reporter experiments. In an exon of TRIM62, the upstream 3′ splice site and poly-G runs in the downstream intron functioned redundantly to protect an exon from its 5′ splice site polymorphism. Collectively, our study provides genomic and experimental evidence for widespread context-dependent robustness to 5′ splice site polymorphisms in human transcriptomes.
RESULTS
Quantitative splicing analysis in seven human transcriptomes reveals widespread robustness to 5′ splice site polymorphisms
To identify 5′ splice site polymorphisms that may cause splicing differences among human individuals, we analyzed seven HapMap LCLs of Asian, European and African ancestry (Table S1). For these seven cell lines, extensive genotyping data were already collected by the International HapMap project (28) as well as a recent exome sequencing study using the targeted capture technology (29). In total, we identified 1174 SNPs (single base nucleotide substitutions only) within the nine nucleotides of the 5′ splice site among these seven individuals. Of these 1174 SNPs, 631 (53.7%) were supported by the HapMap data alone (Phase II + III), 293 (25.0%) were supported by the exome sequencing data alone and 250 (21.3%) were supported by both data sets. Ninety-five SNPs were located within the highly conserved GT dinucleotide. For the other 1079 SNPs outside of the GT dinucleotide position, we scored the 5′ splice sites of the major and minor alleles using the splice site model in MAXENT (13). MAXENT is a widely used computational tool for splice site analysis, which considers the dependencies among adjacent and nonadjacent nucleotide positions to evaluate the strength of 5′ splice sites (13). A higher MAXENT score indicates a stronger 5′ splice site. Among the 5′ splice site SNPs that kept the GT dinucleotide intact, 334 (30.9%) SNPs resulted in a difference of the MAXENT score of at least 2 (Fig. S1), representing a 4-fold reduction in the likelihood odds ratio of matching to the MAXENT 5′ splice site model. From these 334 SNPs, we removed exons in genes lowly expressed in LCLs (according to Affymetrix exon 1.0 array data, see Materials and Methods) (30,31). To facilitate RT–PCR primer design and analysis, we restricted our study to internal spliced exons no longer than 250 bp and flanked by constitutive exons. One hundred and fifteen exons remained after these selection steps. Additionally, we selected 14 exons whose GT dinucleotide was disrupted by SNPs for RT–PCR analysis. The entire procedure of our SNP analysis and exon selection is outlined in Figure 1.
Figure 1.

Overview of 5′ splice site (5′ ss) SNP analysis and RT–PCR assay of seven HapMap LCLs.
In order to determine which candidate 5′ splice site SNPs affected splicing, we used a fluorescently labeled RT–PCR assay to measure the exon inclusion levels of all 129 candidate exons in the seven individuals. Of the 129 tested exons, 40 were in genes without any detected band, indicating that the genes were lowly expressed or not expressed in LCLs. Another five tested exons were completely skipped in all seven individuals. We focused on the remaining 84 exons which were spliced into transcripts in at least one of the seven individuals (see Supplementary Material, Table S2 for RT–PCR gel pictures and Table S3 for primers). We identified 18 exons with a strong association between the genotypes of the 5′ splice site and the splicing patterns in seven individuals (Table 1). The predominant effect of SNPs on splicing in these 18 exons was exon skipping. For example, in exon 2 of HMSD (NM_001123366.1), we identified a G-to-A SNP (rs9945924) in +5 intronic position of the 5′ splice site (Table 1), which reduced the splice site score from 10.65 to 7.79. This exon was 100% included in the individual homozygous for the G allele. In contrast, its exon inclusion level was only 2% in the three individuals homozygous for the A allele. In the three individuals heterozygous for the G/A alleles, the exon had an intermediate inclusion level (33–38%) (Fig. 2A). Similarly, in exon 19 of WDR67 (NM_145647.3), a G-to-T SNP (rs10101626) disrupted the GT dinucleotide. While the three individuals homozygous for the G allele had a high exon inclusion level of 52–78%, the individual homozygous for the T allele showed complete skipping of the exon. The individuals heterozygous for the G/T alleles had intermediate exon inclusion levels (27–28%) (Fig. 2B). We also found cases where the SNPs resulted in activation of adjacent cryptic 5′ splice sites. For example, in exon 23 of PLD2 (NM_002663.3), an exonic A-to-G SNP (rs3764897) decreased the splice site score from 7.10 to 2.04. In the individual homozygous for the A allele, 100% of the transcripts utilized the canonical 5′ splice site. In contrast, in individuals homozygous for the G allele, only 73–80% of the transcripts utilized the canonical splice site. The remaining transcripts (20–27%) utilized an alternative cryptic 5′ splice site within the exon. In the heterozygous individuals, the canonical splice site was used at a frequency of 89–93% (Fig. 2C). A similar pattern was found for exon 5 of RCC2 (NM_001136204.1), in which a T-to-G SNP disrupted the GT dinucleotide and caused the activation of an intronic cryptic 5′ splice site (Fig. 2D). This SNP was a novel SNP identified by exome sequencing (29).
Table 1.
5′ Splice site SNPs affecting splicing patterns in seven human individuals
| Gene | Exon coordinate (hg18) | SNP ID | SNP source | 5′ Splice sitec | MaxEntScan scored | DAF of |
Fst | RS | ||
|---|---|---|---|---|---|---|---|---|---|---|
| YRI | CEU | ASN | ||||||||
| C3orf31 | −chr3:11861365–11861570 | rs392621 | HapMapa | gc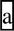 GTtagt GTtagt |
A: 0.14; G: 5.46 | 0.203 | 0.483 | 0.242 | 0.096 | NA |
| BC086863 | −chr1:217389904–217389996 | rs12079503 | HapMap | aagGTtgg |
A: 9.26; G: 5.28 | 0.178 | 0.117 | 0.350 | 0.076 | NA |
| CAST | +chr5:96102205–96102243 | rs7724759 | HapMap, exome sequencingb | tc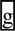 GTgagt GTgagt |
G: 11.11; A: 7.68 | 0.068 | 0.383 | 0.125 | 0.164 | 2.296 |
| DHRS1 | −chr14:23830602–23830671 | rs10134537 | HapMap, exome sequencing |
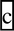 agGTgaag agGTgaag |
C: 6.66; T: 3.29 | 0.195 | 0.058 | 0.058 | 0.058 | 5.622 |
| TDG | +chr12:102890868–102890941 | rs17035056 | HapMap | gtgGTagt |
G: 7.23; A: 10.36 | 0.000 | 0.000 | 0.317 | 0.311 | NA |
| HMSD | +chr18:59771568–59771741 | rs9945924 | HapMap | cagGTact |
G: 10.65; A: 7.79 | 0.483 | 0.250 | 0.217 | 0.086 | NA |
| BC039374 | +chr2:97684049–97684199 | rs11894651 | HapMap | atcGTggt |
A: 8.88; C: 6.05 | 0.280 | 0.350 | 0.667 | 0.157 | 3.771 |
| DHRSX | −chrX:2336786–2336854 | rs5939175 | HapMap | aagGTacg |
C: 9.67; G: 6.91 | NA | NA | NA | NA | NA |
| EVC | +chr4:5797832–5797969 | rs2286343 | HapMap | aatGTggt |
C: 5.02; T: 2.42 | 0.288 | 0.325 | 0.708 | 0.198 | NA |
| AFTPH | +chr2:64650261–64650317 | rs2287531 | HapMap | ttgGTagt |
A: 10.47; C: 8.11 | 0.068 | 0.000 | 0.133 | 0.061 | 5.083 |
| PLD2 | +chr17:4669690–4669843 | rs3764897 | HapMap | cgGTagag |
A: 7.10; G: 2.04 | 0.678 | 0.825 | 0.925 | 0.089 | 3.490 |
| WDR67 | +chr8:124223683–124223877 | rs10101626 | HapMap, exome sequencing | aag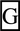 Taaaa Taaaa |
0.229 | 0.167 | 0.217 | 0.002 | 5.013 | |
| RCC2 | −chr1:17621286–17621374 | rs60580590 | Exome sequencing | cagG gacc gacc |
0.000 | 0.000 | 0.000 | 0.000 | 5.271 | |
| FGGY | +chr1:59695221–59695336 | Novel | Exome sequencing | tagTaaaa |
0.000 | 0.008 | 0.000 | 0.000 | 4.802 | |
| CRYZ | −chr1:74957481–74957644 | rs57504503 | Exome sequencing | cagTaata |
0.000 | 0.000 | 0.000 | 0.000 | 5.060 | |
| CASC5 | +chr15:38685883–38685942 | Novel | Exome sequencing | cagGaagt |
0.000 | 0.000 | 0.000 | 0.000 | 3.748 | |
| IFIH1 | −chr2:162844752–162844868 | rs35337543 | Exome sequencing | gaaTatgg |
0.000 | 0.008 | 0.000 | 0.000 | 5.458 | |
| ACAD11 | −chr3:133820168–133820306 | rs41272317 | Exome sequencing | cagTttact |
0.000 | 0.000 | 0.067 | 0.059 | 5.481 | |
DAF, derived allele frequency; YRI, African; CEU, European; ASN, Asian; RS, rejected substitution score calculated by the Genomic Evolutionary Rate Profiling (GERP) algorithm; NA, not available.
aSNPs from the HapMap project (Phase II + III).
bSNPs identified by targeted capture and exome sequencing.
cUpper-case bases mark the GT dinucleotide and boxes indicate the SNP position.
dMaximum entropy scores of the major and minor 5′ splice site alleles are calculated by MaxEntScan for SNPs outside of the GT dinucleotide position.
Figure 2.

Examples of 5′ splice site SNPs causing splicing differences among seven HapMap LCLs. The genotypes of seven individuals and the estimated exon inclusion levels are indicated below each gel picture. The exon inclusion level is calculated from the fluorescently labeled RT–PCR as the intensity of the exon inclusion band(s) over the total intensity of all exon inclusion and skipping bands. (A) HMSD, (B) WDR67, (C) PLD2, (D) RCC2.
For all 18 exons in which the SNPs caused splicing differences among individuals, the direction of change in the 5′ splice site score was always consistent with the shift in exon inclusion levels between different genotypes. To confirm that these SNPs were causal for the observed splicing differences, we randomly selected five exons from BC039374, TDG, CAST, DHRS1 and BC086863 for minigene experiments using the pI-11-H3 minigene reporter (32) (see Fig. 3A and Materials and Methods). In all five exons tested, the direction of splicing changes after introducing the SNP to the minigene construct was consistent with the endogenous exon splicing pattern observed in the LCLs of seven individuals (see Supplementary Material, Table S2 for endogenous splicing patterns and Fig. S2 for minigene splicing patterns).
Figure 3.

The effect of a given 5′ splice site SNP on splicing depends on its local sequence context. (A) Schematic diagram of the pI-11-H3 minigene splicing reporter and minigene analysis of the 5′ splice site polymorphism in CAST. (B) The maximum and minimum 5′ splice site scores of ‘splicing unaffected' exons and ‘splicing affected' exons. (C) Minigene analysis of TRIM62 and HMSD indicates that the identical set of 5′ splice site mutations has dramatically different impacts on splicing within two different exonic/intronic sequence contexts. In this experiment, we swapped in a series of 5′ splice site 9-mers (three exonic nucleotides and six intronic nucleotides) into the basic constructs of TRIM62 and HMSD, with a gradient of eight different splice site scores between 11.00 and 4.44. In all gel pictures, the number below each lane represents the percent exon inclusion level estimated by the fluorescently labeled RT–PCR. Asterisk denotes PCR products of unexpected sizes resulting from the usage of cryptic splice sites as confirmed by sequencing. 5′ ss, 5′ splice site.
In general, the 18 SNPs that affected splicing were located in evolutionarily constrained sites in the human genome and had low derived allele frequencies (DAFs). First, to assess the evolutionary conservation of these SNP sites, we obtained their rejected substitution (RS) scores as calculated by the Genomic Evolutionary Rate Profiling (GERP) algorithm on the UCSC 44-way genome alignments (33,34). An RS score of >2 is commonly used as the indication of evolutionary conservation (34). Of the 18 SNP sites, 12 had a sufficient number of aligned species for calculating the RS score. All 12 SNP sites had an RS score of >2 with an average score of 4.591, indicating that these SNPs were located at evolutionarily constrained sites. As expected, SNP sites within the GT dinucleotide had even higher RS scores (4.976 on average). Secondly, to estimate the DAF of these 18 SNP sites, we determined the ancestral status of each SNP based on the alignments of human, chimpanzee, orangutan and rhesus macaque genomes as in (35). We calculated the DAF of each SNP using the genotype data in the African (YRI), European (CEU) and Asian (ASN) populations from the pilot 1 study of the 1000 Genomes Project (35). As seen in Table 1, most SNPs had a DAF of <0.5 in all three populations. Three SNPs had a DAF of >0.5 in at least one population, including one SNP (rs3764897) in PLD2 with a DAF of >0.5 in all three populations. SNPs within the GT dinucleotide had particularly low DAFs. This result suggests that most of these 18 SNPs that affect splicing are either under strong purifying selection or evolutionarily too young to reach a high DAF in any population. Finally, we computed unbiased estimates of population differentiation statistic Fst averaged across the YRI, CEU and ASN populations (36,37). None of these 18 SNPs had a high Fst value (e.g. >0.5). The SNP rs17035056 in TDG appeared to be Asian-specific (see Table 1) and had the highest Fst (0.311) among the 18 SNPs. Additional tests for the reduction of SNP heterozygosity (38) or Fay and Wu's H statistic (39) also did not reveal convincing evidence of positive selection on these SNPs (data not shown). Nonetheless, despite the lack of detectable signatures of positive selection, some of these SNPs might have important functional impacts. A known example is the alternative splicing of HMSD resulting from the intronic SNP rs9945924 (Fig. 2A). The exon skipping isoform of HMSD produces a novel minor histocompatibility antigen, which has been proposed as a potential target for immunotherapy (40).
Among the 84 tested exons which were spliced in the seven LCLs, seven had SNPs disrupting the highly conserved GT dinucleotide. All seven SNPs affected exon splicing, consistent with the essential role of the GT dinucleotide in 5′ splice site recognition. In contrast, outside of the highly conserved GT dinucleotide, only 11 exons (out of 77 tested, 14.3%) showed splicing differences among individuals as caused by the SNP (Fig. 1 and Table 1). The remaining 66 exons (85.7%) did not show any difference in exon inclusion levels among the seven individuals. These results revealed widespread robustness of the splicing machinery to 5′ splice site polymorphisms.
Bioinformatic and experimental analyses indicate context-dependent robustness to 5′ splice site polymorphisms
We set out to investigate why certain exons were robust toward 5′ splice site polymorphisms. For the following analysis, we focused on the 77 exons whose SNPs kept the GT dinucleotide intact (Fig. 1). From the 11 exons whose splicing patterns were altered by SNPs, we removed a PLD2 exon in which the SNP caused activation of an adjacent cryptic 5′ splice site, and compiled a final group of 10 exons in which the SNP affected the inclusion/skipping of the entire exon. We referred to these exons as the ‘splicing affected' group. Similarly, from the 66 exons whose splicing patterns were not altered by SNPs, we removed 4 exons which were annotated as having alternative 5′ splice sites by the UCSC Genome database (41) and compiled a group of 62 ‘splicing unaffected' exons.
Our analysis of the ‘splicing affected' and ‘splicing unaffected' groups suggests that the 5′ splice site itself could not explain the observed robustness to 5′ splice site polymorphisms in many exons. As shown in Figure 3B, we plotted the maximum and minimum MAXENT 5′ splice site scores of each exon corresponding to its two alleles, but did not notice any difference in the magnitude of SNP-induced changes in 5′ splice site scores between these two groups of exons (Fig. 3B). Based on this result, we hypothesized that the sequence context in surrounding exonic and/or intronic regions could play a major role in determining the impact of 5′ splice site SNPs on splicing. To confirm the importance of the surrounding sequence context, we conducted a series of minigene experiments using exon 2 of TRIM62 (NM_018207.2) and exon 2 of HMSD as our test models. In the TRIM62 exon, a G-to-T SNP (rs2306257) reduced the 5′ splice site score from 9.60 to 5.61, but did not cause any change in either endogenous or minigene splicing patterns (see Supplementary Material, Table S2 and Fig. 3C). In the HMSD exon, the SNP reduced the splice site score from 10.65 to 7.79, which resulted in significant skipping of the exon (Fig. 2A). For these two exons, we cloned the entire exon and 500 bp from each side of flanking introns into the pI-11-H3 minigene reporter as our basic minigene constructs. We then swapped in a series of 5′ splice site 9-mers (three exonic nucleotides and six intronic nucleotides) into the basic constructs of TRIM62 and HMSD, with a gradient of eight different splice site scores between 11.00 and 4.44 (see Supplementary Material, Table S4 for splice site sequences). These minigene constructs allowed us to observe the impact of the identical set of 5′ splice site mutations within two different exonic/intronic sequence contexts (TRIM62, HMSD). The serial reduction in the 5′ splice site score had little impact on the splicing of the TRIM62 minigene (Fig. 3C). The exon was constitutively spliced with seven of the eight tested 5′ splice site 9-mers, which had a range of 5′ splice site scores between 11.00 and 5.61. Even with the weakest 5′ splice site tested (a score of 4.44), the inclusion level of the TRIM62 exon was still 98% with an additional minor transcript resulting from the usage of an adjacent cryptic 5′ splice site. In contrast, the reduction in the 5′ splice site score significantly affected the splicing of the HMSD minigene. While the minigene was constitutively spliced with a 5′ splice site score of 11.00 or 10.65, the exon became alternatively spliced with an inclusion level of 92% when the splice site score was reduced to 9.60. Its inclusion level dropped to 18% with a 5′ splice site score of 8.40, and to 2% with a 5′ splice site score of 5.61. With the weakest 5′ splice site (4.44), the exon was completely skipped (Fig. 3C). These data provide experimental evidence for context-dependent effects of 5′ splice site polymorphisms.
Strong upstream 3′ splice sites buffer 5′ splice site polymorphisms
To uncover the specific sequence context that contributed to the robustness to 5′ splice site polymorphisms, we compared the surrounding splicing signals between ‘splicing affected' and ‘splicing unaffected' exons. We did not observe any difference in the density of exonic splicing enhancers (ESEs) and silencers (ESSs) [collected by Burge and colleagues (42,43), see Materials and Methods] between these two groups of exons (Fig. 4A). However, we caution that due to our limited sample size (62 ‘splicing unaffected' exons versus 10 ‘splicing affected' exons), this result could not rule out the involvement of ESEs/ESSs. Also, our analysis considered all ESEs or all ESSs as a whole, while a single ESE or ESS may contribute to the robustness to 5′ splice site polymorphisms in individual exons.
Figure 4.

Strong upstream 3′ splice sites buffer 5′ splice site polymorphisms. (A) Box plots of splicing signals (ESE/ESS density, 3′ splice site score) of ‘splicing unaffected' exons and ‘splicing affected' exons. (B) Correlation between 3′ splice site score and 5′ splice site SNP density in 12 862 human constitutive exons. (C) The 3′ splice site strength of a CAST exon affects the impact of its 5′ splice site polymorphism on splicing. (D) The 3′ splice site strength of a TRIM62 exon affects the impact of its 5′ splice site polymorphism on splicing. In all gel pictures, the number below each lane represents the percent exon inclusion level estimated by the fluorescently labeled RT–PCR. Asterisk denotes PCR products of unexpected sizes resulting from the usage of cryptic splice sites as confirmed by sequencing. 5′ ss, 5′ splice site; 3′ ss, 3′ splice site.
Interestingly, we observed a trend for stronger 3′ splice sites upstream of ‘splicing unaffected' exons. For all exons in the ‘splicing affected' or the ‘splicing unaffected' groups, we scored the strength of their upstream 3′ splice sites using MAXENT. The median 3′ splice site score of ‘splicing unaffected' exons was 9.4, compared with 7.8 for ‘splicing affected' exons (Fig. 4A). Although the difference between these two groups was not statistically significant (P = 0.29, two-sided Wilcoxon rank sum test), possibly due to the relatively small sample size, the trend was consistent with previous studies suggesting coordinated evolution and compensatory effects between the 3′ and 5′ splice sites (44,45). To explore this observation further, we analyzed 12 862 high-confidence human constitutive exons which were 100% included in all human mRNA/EST sequences (see the criteria for selecting these constitutive exons in Materials and Methods). We hypothesized that if a stronger 3′ splice site could buffer the impact of 5′ splice site polymorphisms on exon splicing, constitutive exons with a stronger 3′ splice site would be more likely to tolerate SNPs in the 5′ splice site. This was indeed the case. We sorted all constitutive exons based on their 3′ splice site scores, and grouped them into 20 distinct bins. For each bin, we calculated the median 3′ splice site score and the average SNP density within the 5′ splice site, excluding SNPs that disrupted the GT dinucleotide (see Materials and Methods). Consistent with our hypothesis, we observed a positive correlation between the 3′ splice site strength and the density of 5′ splice site SNPs (Fig. 4B; Spearman correlation coefficient rho = 0.49, P = 0.029). On the other hand, we did not find any correlation between the 5′ splice site SNP density and the strength of the 3′ splice site of the downstream intron (data not shown). This is consistent with the prevalence of the ‘exon definition' model over the ‘intron definition' model in the splicing of mammalian exons (44).
To further confirm the role of the 3′ splice site in buffering 5′ splice site SNPs, we selected exon 13 of CAST (NM_001750.5) and exon 2 of TRIM62 (mentioned above) for minigene experiments. For each exon, we made two basic minigene constructs corresponding to the two 5′ splice site alleles. We then introduced a series of mutations to gradually increase (or decrease) the 3′ splice site score (see Supplementary Material, Table S4 for splice site sequences), and examined the impact on exon splicing of the two 5′ splice site alleles. In exon 13 of CAST, a G-to-A SNP (rs7724759) reduced the 5′ splice site score from 11.11 to 7.68, causing a 50–60% difference in the exon inclusion level between LCLs homozygous for the G allele and those heterozygous for the G/A alleles (Table S2). In the minigene experiment, the two basic minigene constructs corresponding to the G allele and the A allele had a difference in the exon inclusion level of 90% (Fig. 3A). The 3′ splice site score of this exon was 5.40. Reducing the score of the 3′ splice site to 3.39 or 1.59 resulted in complete exon skipping of both 5′ splice site alleles (Fig. 4C). On the other hand, when we strengthened the 3′ splice site, the inclusion levels of both 5′ splice site alleles increased, while the differences between the two alleles gradually decreased. For example, when we strengthened the 3′ splice site score to 8.84, the G allele (i.e. allele 1 in Fig. 4C) had an exon inclusion level of 100% while the A allele (i.e. allele 2 in Fig. 4C) had an exon inclusion level of 52%, a 48% difference. When the 3′ splice site score increased to 12.47, the differences in exon inclusion levels between the two 5′ splice site alleles decreased to 31% (Fig. 4C). We also tested exon 2 of TRIM62, a constitutive exon whose splicing pattern was not affected by a SNP (rs2306257) that reduced the 5′ splice site score from 9.60 to 5.61. When we reduced the score of the 3′ splice site to <5.93, the inclusion levels of both 5′ splice site alleles started to decrease, while there was a ≥20% difference in exon inclusion levels of the two alleles (Fig. 4D). Together, these experiments demonstrate that the strength of the upstream 3′ splice site could modify the influence of a given 5′ splice site polymorphism on exon splicing. A strong 3′ splice site could facilitate exon recognition, thus buffering the effect of a polymorphism weakening the 5′ splice site.
The upstream 3′ splice site and downstream intronic poly-G runs function redundantly to protect a TRIM62 exon from its 5′ splice site polymorphism
Motif enrichment analysis also identified putative intronic splicing elements that could contribute to the robustness to 5′ splice site polymorphisms. We sought to identify putative motifs that were significantly enriched within the 100 bp intronic region downstream of the ‘splicing unaffected' exons, using the ‘splicing affected' exons as the control. Considering the relatively small number of exons in these two groups, we focused our analysis on putative trinucleotide motifs (i.e. 3-mers). Of all 64 trinucleotides analyzed, four had a Bonferroni-corrected P-value of <0.05 (GGG, GAA, CCC, AGG; see Fig. 5A). We noted that all four motifs resembled the putative binding sites of splicing regulators [GGG by hnRNP F/H (HNRNPH1 and HNRNPHF); GAA by Tra2 (TRA2A and TRA2B); CCC by hnRNP K (HNRNPK); AGG by hnRNP A1/A2 (HNRNPA1 and HNRNPA2B1) (46–50)]. Some of these motifs were previously demonstrated to stimulate splicing when located in introns (50,51). Strikingly, the most significantly enriched motif was the GGG trinucleotide (P = 1.3e−5), an intronic splicing enhancer recognized by the hnRNP F/H family of splicing regulators (52–54). Previous studies demonstrated that the GGG motif (also referred to as the poly-G run) enhanced exon inclusion when located downstream of intermediate or weak 5′ splice sites, especially within the window of 11–70 bp downstream of the exon–intron boundary (50). Another enriched motif was the CCC trinucleotide (P = 7.0e−4). Several studies suggested a role of the CCC trinucleotide or C-rich sequences as intronic splicing enhancers downstream of the 5′ splice site (55–57).
To further assess the role of the enriched downstream intronic motifs (Fig. 5A), we selected exon 2 of TRIM62 as the model for our detailed minigene analysis. As mentioned above, a SNP (rs2306257) reduced the 5′ splice site score of this exon from 9.60 to 5.61, but had no impact on the level of exon inclusion (Fig. 3C and Supplementary Material, Table S2). The 100 bp intronic region downstream of the TRIM62 5′ splice site was remarkably G-rich, containing a total of 10 poly-G runs each with at least three consecutive G nucleotides (denoted as G1–G10, see Fig. 5B–C). We set out to test whether these poly-G runs were important for the observed robustness toward the 5′ splice site polymorphism. In the two basic minigene constructs corresponding to the two 5′ splice site alleles, we introduced a series of G-to-C substitutions to disrupt individual poly-G runs (Fig. 5B–C). Surprisingly, the disruption of poly-G runs in the minigene constructs had no impact on exon splicing. After all the poly-G runs within the 100 bp downstream intronic region were disrupted, the minigene constructs containing the two 5′ splice site alleles both remained constitutively spliced (Fig. 5D, left panel). Even when we deleted the entire downstream intron in TRIM62 and only kept the six intronic nucleotides within the 5′ splice site region for insertion into the minigene, the minigene construct containing the weaker 5′ splice site allele was still highly spliced with an estimated exon inclusion level of 98%.
Figure 5.

The upstream 3′ splice site and downstream intronic poly-G runs function redundantly to protect a TRIM62 exon from its 5′ splice site polymorphism. (A) Trinucleotide motifs enriched within the 100 bp intronic region downstream of the ‘splicing unaffected' exons as compared with the ‘splicing affected' exons. X-axis shows motif density in ‘splicing affected' exons. Y-axis shows motif density in ‘splicing unaffected' exons. The size of each dot is proportional to the negative log of the P-value for motif enrichment. The nucleotide sequences of motifs with Bonferroni-corrected P-values of <0.05 are shown. (B) The nucleotide sequence of the TRIM62 exon and its flanking intronic regions. Box frame indicates the exon. Ten poly-G runs in the 100 bp downstream intron are underlined and denoted as G1-G10. Filled diamonds represent the positions where single-nucleotide mutations are introduced to disrupt individual poly-G runs. (C) Schematic diagrams of mutant minigene constructs with serial disruptions of poly-G runs. (D) Results of TRIM62 minigene experiments by site-directed G-to-C disruptions of poly-G runs. Disruptions of poly-G runs in the minigene constructs containing the wild-type 3′ splice site have no impact on exon splicing (left panel). Disruptions of poly-G runs in the minigene constructs containing the mutant (weakened) 3′ splice site significantly reduce the exon inclusion level of the TRIM62 exon. In the ‘Intron Deletion' constructs, only the first 6 intronic nucleotides within the 5′ splice site of the TRIM62 exon are kept when inserted into the basic pI-11-H3 minigene vector. The vector sequence that now serves as the intronic sequence immediately downstream of the TRIM62 5′ splice site contains basic splicing signals, i.e. a strong branch point and the 3′ splice site for the downstream constitutive exon. In all gel pictures, the number below each lane represents the percent exon inclusion level estimated by the fluorescently labeled RT–PCR. Asterisk denotes PCR products of unexpected sizes resulting from the usage of cryptic splice sites as confirmed by sequencing. 5′ ss, 5′ splice site; 3′ ss, 3′ splice site.
We sought to elucidate why the site-directed disruption of poly-G runs was not sufficient to disrupt the robustness of this TRIM62 exon to its 5′ splice site polymorphism. We noticed that this exon carried a strong upstream 3′ splice site with a MAXENT score of 9.97. To assess the role of this strong 3′ splice site, we made a new set of minigene constructs, in which the wild-type 3′ splice site was replaced by a much weaker 3′ splice site with a score of 5.93. With this weakened 3′ splice site, the minigene construct containing the stronger 5′ splice site allele remained constitutively spliced, while the minigene construct containing the weaker 5′ splice site allele became alternatively spliced with an exon inclusion level of 97–98% (Figs 4D and 5D, right panel). In these minigene constructs, we again disrupted individual poly-G runs with G-to-C substitutions. In contrast to the results using the wild-type 3′ splice site, our minigene analysis using the mutant 3′ splice site revealed a strong impact of the downstream intronic poly-G runs on splicing of this TRIM62 exon. When the poly-G run closest to the 5′ splice site (i.e. G1, see Fig. 5B–C) was disrupted, the constructs containing the two 5′ splice site alleles had an exon inclusion level of 100 and 94% respectively, a 6% difference due to the 5′ splice site polymorphism (Fig. 5D, right panel). Disruptions of additional poly-G runs reduced the exon inclusion level of both minigene constructs, and increased the difference between these two 5′ splice site alleles (Fig. 5D, right panel). For example, when we disrupted all the poly-G runs within the 70 bp downstream intronic region (G1–G6), the constructs containing the two 5′ splice site alleles had an exon inclusion level of 96 and 51%, respectively, a 45% difference. When we disrupted all poly-G runs within the 100 bp downstream intronic region (G1–G10), the two 5′ splice site alleles had an exon inclusion level of 79 and 20% respectively, a 59% difference. Similarly, we observed a 56% difference in the exon inclusion levels of these two 5′ splice site alleles when we deleted the entire downstream intronic region of TRIM62 and only kept the six intronic nucleotides within the 5′ splice site for insertion into the minigene (73 versus 17%; Fig. 5D, right panel). To rule out the possibility that our minigene results were confounded by novel intronic splicing motifs created by the G-to-C substitutions in the downstream intronic region, we repeated all experiments by disrupting the poly-G runs with G-to-A substitutions, and obtained similar results (see Supplementary Material, Fig. S3).
To independently confirm the importance of the upstream 3′ splice site in controlling the dispensability of the poly-G runs, we used siRNA to knockdown the splicing factor hnRNP H (which recognizes intronic poly-G runs) (52–54) and examined the splicing patterns of minigene constructs containing the wild-type or the mutant 3′ splice site. The efficacy of the siRNA knockdown was confirmed by real-time PCR and western blot analysis of hnRNP H (see Supplementary Material, Fig. S4A and B). Consistent with the poly-G disruption experiments, the knockdown of hnRNP H did not alter the splicing of the minigene constructs containing the wild-type 3′ splice site (see Supplementary Material, Fig. S4C). However, in constructs containing the mutant 3′ splice site, siRNA knockdown of hnRNP H resulted in a 24% difference in the inclusion levels of the stronger and weaker 5′ splice site alleles (96 versus 72%), as compared with a 5% difference after treatment by a control siRNA (see Supplementary Material, Fig. S4C).
Together, these results indicate that the upstream 3′ splice site and downstream intronic poly-G runs provide redundant mechanisms to confer robustness of this TRIM62 exon to its 5′ splice site polymorphism. Although the large array of poly-G runs downstream of the TRIM62 exon indeed functioned as intronic splicing enhancers, their activities were masked by a strong 3′ splice site at the upstream intron–exon boundary. However, after the 3′ splice site was weakened, these poly-G runs became indispensable for protecting the exon from its 5′ splice site polymorphism (Fig. 5 and see Supplementary Material, Figs S3 and 4).
DISCUSSION
Genomic variations within cis splicing signals constitute a major source of alternative splicing events in higher eukaryotes (25). In this work, we systematically surveyed genetic polymorphisms affecting the 5′ splice sites of seven human individuals from three ancestral groups (Asian, African and European). Using statistical modeling of the 5′ splice site signal, we analyzed extensive genotype data generated by the HapMap project and exome sequencing to identify 5′ splice site SNPs that significantly altered the strength of the splice site. Our RT–PCR analysis of the LCLs revealed 18 exons whose splicing patterns were strongly associated with the 5′ splice site genotypes in these seven individuals. While in some cases the SNP caused a moderate shift in the exon inclusion level (e.g. PLD2, see Fig. 2C), we also found cases where the SNP resulted in an almost complete switch between exon inclusion and exon skipping (e.g. HMSD, see Fig. 2A). Together, these data indicate that genetic variation within the 5′ splice site is an important contributor to transcriptome differences within and between human populations.
Although 5′ splice site recognition during pre-mRNA splicing is known to be influenced by exonic and intronic signals (4), whether and how the sequence context surrounding the 5′ splice site shapes genetic diversity of alternative splicing has not been investigated before. Our study reveals surprisingly widespread robustness to 5′ splice site polymorphisms in human populations. Outside of the highly conserved GT dinucleotide, although all tested SNPs significantly reduced the 5′ splice site score, a very small fraction (∼14%) affected splicing. We demonstrated that the robustness to 5′ splice site polymorphisms was largely context dependent. For example, in our minigene analysis of exons in HMSD and TRIM62, we introduced the identical set of 5′ splice site mutations to the minigene constructs, and observed completely different consequences on the splicing pattern of the minigenes. Our detailed analysis of genomic sequences surrounding the 5′ splice sites reveals cis sequence signals that could buffer the reduction in the 5′ splice site strength, such as a strong 3′ splice site at the upstream intron–exon boundary, and intronic splicing enhancer motifs (particularly poly-G runs) downstream of the 5′ splice site. Consequently, exons lacking these signals are more susceptible to mutations within the 5′ splice site. Such knowledge should be valuable for prioritizing follow-up characterizations of 5′ splice site variants identified by targeted re-sequencing or whole-genome sequencing of patient samples.
Our study shows that natural variations of alternative splicing could reveal mechanisms of splicing regulation. Although the evolutionary conservation and genetic variation of genome sequences have been widely used to identify cis regulatory elements of gene expression and splicing (44,58–63), a unique feature of our approach is that we directly correlate transcriptome variations with genome variations among human individuals. Despite the relatively small number of exons in our study, by comparing exons unaffected by 5′ splice site polymorphisms to exons affected, we were able to discover important signals that could promote exon recognition and compensate for the reduction in 5′ splice site activity. In principle, our approach is analogous to conventional molecular studies of splicing regulation, in which the importance of a given regulatory motif is assessed by introducing specific mutations to the minigene constructs of selected exons (64,65). However, a major strength of our approach is that it utilizes the existing variations in the human population. This allows for an unbiased assessment of various signals associated with efficient exon splicing, which cannot be achieved by exon- or motif-specific studies. For example, a variety of intronic motifs was previously shown to promote 5′ splice site recognition (4,50,66,67). In our study, the poly-G run was recognized as the most significantly enriched intronic motif downstream of exons robust to 5′ splice site polymorphisms. These data suggest a widespread role of the poly-G run in promoting the recognition of weak 5′ splice sites. In one exon (TRIM62), we found that different types of splicing signals (i.e. 3′ splice site and downstream intronic poly-G runs) surrounding the 5′ splice site functioned redundantly to protect the exon from its 5′ splice site polymorphism. These data extend the current view of the 5′ splice site-dependent activity of downstream intronic poly-G runs (50), and suggest that the importance of these intronic motifs is also determined by the strength of the 3′ splice site across the exon.
Our approach could be applied to SNPs affecting other types of splicing signals, such as the 3′ splice site (i.e. the acceptor site). However, the consensus sequence of the 3′ splice site is much longer and more degenerate (13). As a result, a smaller percentage of SNPs significantly alters the strength of the 3′ splice site. In fact, in these seven individuals, we only found 145 SNPs that reduced the putative 3′ splice site score by at least 2, as compared with 334 SNPs in the 5′ splice site. Thus, a much larger panel of human individuals would be needed to obtain a sufficient number of exon–SNP pairs for studying context-dependent effects of 3′ splice site mutations.
It should be noted that the rapid advances in DNA/RNA sequencing technologies have greatly reduced the cost and improved the efficiency of genome-wide genotyping and transcriptome profiling (68). For example, RNA sequencing has emerged as a revolutionary technology for transcriptome analysis (69). Currently, RNA sequencing analysis of alternative splicing is still limited by false-negative and false-positive issues, with a strong bias toward the accurate discovery of splicing changes in highly expressed genes (69). Nevertheless, we anticipate that future improvements in the capacity and cost structure of these technologies will lead to a much more comprehensive catalog of splicing variations among human individuals for diverse tissues and cell types. This work demonstrates that globally correlating splicing variations with genomic variations in human populations provides a powerful tool for deciphering the splicing codes of mammalian cells.
MATERIALS AND METHODS
Seven HapMap LCLs and retrieval of their genotypes
Seven HapMap LCLs whose exon regions were re-sequenced by the targeted capture technique (29) were purchased from the Coriell Institute for Medical Research (Camden, NJ, USA). These seven LCLs are of European (CEU: GM12156 and GM12878), African (YRI: GM18517, GM19129 and GM19240) and Asian (CHB/JPT: GM18555 and GM18956) ancestry. The eighth LCL (GM18507) sequenced by Ng et al. (29) was not available from Coriell at the time of our study, thus was not included in this work. A complete list of the seven LCLs is provided in Supplementary Material, Table S1. All cell lines were cultured in Gibco RPMI 1640 containing 15% fetal bovine serum (FBS) (Invitrogen, Grand Island, NY, USA). Exponentially growing cells (viability was >85%) were harvested for RNA and DNA extraction.
The genotypes of the seven HapMap LCLs were obtained from the HapMap project (Phase II + III, February 2009, ftp://ftp.ncbi.nlm.nih.gov/hapmap/) as well as the targeted capture and exome sequencing data set generated by Ng et al. (29).
Collection of UCSC Known Genes exons and calculation of splice site scores
We collected 196 754 non-redundant human internal exons from the UCSC Known Genes database (70). For each exon, its 3′ and 5′ splice sites were scored using the maximum entropy splice site model in MAXENT (13). For 3′ splice sites, we analyzed 3 nucleotides in exons and 20 nucleotides in introns. For 5′ splice sites, we analyzed 3 nucleotides in exons and 6 nucleotides in introns.
Calculation of 5′ splice site SNP density
We collected 12 862 high-confidence constitutive in exons in the human genome from the Alternative Splicing Annotation Project 2 (ASAP2) database (71), using a series of stringent filtering criteria as described before (72). The SNP data were downloaded from the UCSC Genome Database (dbSNP 130, http://www.genome.ucsc.edu/) (41). Only single-nucleotide substitutions were kept for the SNP density analysis. SNPs within the highly conserved GT dinucleotide position were excluded from the calculation of 5′ splice site SNP density.
ESE and ESS analysis
The density of ESEs or ESSs was calculated as the number of nucleotides covered by ESEs or ESSs divided by the total length of the exons. Two hundred and thirty-eight ESEs were downloaded from the RESCUE-ESE database (http://genes.mit.edu/burgelab/rescue-ese/) (42) and 103 ESSs were downloaded from the FAS_ESS (FAS-hex3) database (http://genes.mit.edu/fas-ess/) (43).
Analysis of exon 1.0 array data
In our seven LCLs, five were previously profiled for gene expression by the Affymetrix exon 1.0 array (30,31). We used the exon array data to select genes expressed in LCLs for downstream splicing analysis. We downloaded the original CEL files of these five individuals (NCBI GEO: GSE7851) and calculated gene expression indexes using the background correction and iterative probe selection algorithms implemented in our GeneBASE program for exon 1.0 array analysis (73,74). We removed genes whose median gene expression indexes among five LCLs were <250 from consideration in the splicing analysis.
Identification of intronic trinucleotide motifs enriched downstream of exons unaffected by 5′ splice site polymorphisms
We enumerated all possible trinucleotide motifs (3-mers) to identify trinucleotide sequences enriched in intronic regions downstream of exons unaffected by 5′ splice site polymorphisms. Intronic nucleotides within the 5′ splice site region (i.e. the first six intronic nucleotides downstream of the exons) were excluded from the motif analysis. The enriched trinucleotide motifs were defined as motifs enriched within the 100 bp intronic region downstream of exons unaffected by 5′ splice site polymorphisms, as compared with the 100 bp intronic region downstream of exons affected by 5′ splice site polymorphisms. To rule out other confounding factors which may impact the 5′ splice site usage (e.g. a competing 5′ splice site in exons with alternative 5′ splice sites), we removed exons with alternative 5′ or 3′ splice sites (annotated by UCSC) from this analysis. For each trinucleotide motif analyzed, the P-value was calculated based on one-sided Fisher's exact test of the number of motif-containing sites versus non-motif sites in the two groups of exons.
Fluorescently labeled RT–PCR
Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA). The RNA samples were first treated by DNaseI (Fermentas, Hanover, MD, USA), then subjected to reverse transcription using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). For each tested exon, we designed a pair of regular forward and reverse PCR primers targeting flanking constitutive exons using PRIMER3 (75). A 22 nt universal tag sequence (5′-CGTCGCCGTCCAGCTCGACCAG-3′) was added to the 5′ end of the gene-specific forward primer during oligo synthesis. A fluorescently labeled universal primer (5′-FAM-CGTCGCCGTCCAGCTCGACCAG-3′) was used as the third primer in PCR (76). Then, regular PCR was carried out for 29–35 cycles (optimized for each exon). The reaction products were resolved on 5% urea (7.5 m)–TBE or regular 5% TBE–PAGE gels. The signal was captured by Typhoon 9200 (Molecular Dynamics, Sunnyvale, CA, USA) and quantified using the Quantity One 4.6.2 software (Bio-Rad, Hercules, CA, USA). For exons whose splicing was altered by 5′ splice site polymorphisms, we also used different passages of these seven cell lines and repeated the fluorescently labeled RT–PCR. Original gel pictures and RT–PCR primer sequences are shown in Supplementary Material, Tables S2 and S3. For RT–PCR products of unexpected sizes, we validated their identities by cloning (Invitrogen Zero Blunt TOPO PCR cloning kit) and sequencing (University of Iowa DNA facility).
Minigene construction and site-directed mutagenesis
We used the hybrid minigene construct pI-11-H3 (32) (kindly provided by Dr Russ P. Carstens, University of Pennsylvania, Philadelphia, PA, USA) for our minigene splicing assays. The In-Fusion™ Advantage PCR Cloning Kit was used to clone PCR products into vectors (Clontech, Mountain View, CA, USA). For the five exons from BC039374, TDG, CAST, DHRS1 and BC086863, we amplified the target exon and 150 bp from flanking introns. For the exons in TRIM62 and HMSD, we amplified the target exon and 500 bp from flanking introns for insertion into the minigene vectors. The primer sequences are listed in Supplementary Material, Table S3. The QuikChange method (Stratagene, Amsterdam, Netherlands) was used for site-directed mutagenesis following the manufacturer's instructions. The identities of all mutant constructs were confirmed by sequencing (University of Iowa DNA facility, Iowa City, IA, USA).
Minigene splicing assay
HEK293 cells were grown in Gibco DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen). Cells were transfected by Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. RNA extraction and reverse transcription were done ∼20 h after transfection. Fluorescently labeled RT–PCR was carried out for 25–27 cycles. The signal capture and quantification were carried out as described above. All transfections were repeated independently at least twice. The universal primer sequences targeting flanking exons on the minigene construct are shown in Supplementary Material, Table S3.
siRNA knockdown of hnRNP H (HNRNPH1)
We used double-stranded siRNA to knockdown hnRNP H (HNRNPH1). Synthetic duplex siRNA sequences for HNRNPH1 and negative control siRNA sequence were provided in (50). Cells were harvested 72 h after co-transfection of the minigene and 20 nm siRNA with Lipofectamine 2000 (Invitrogen). Quantitative real-time PCR of hnRNP H was carried out by Power SYBR green PCR Master Mix (Applied Biosystems). Relative amounts of mRNAs were measured by the comparative Ct 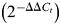 method (77). ACTB was used as the internal control. Primers are listed in Supplementary Material, Table S3. Total protein extract was analyzed by the NUGE® Bis–Tris Gel System (Invitrogen). HNRNPH1 (ab10374; Abcam, Cambridge, MA, USA) and ACTB (A5441; Sigma, St Louis, MO, USA) specific antibodies were used for western blot.
method (77). ACTB was used as the internal control. Primers are listed in Supplementary Material, Table S3. Total protein extract was analyzed by the NUGE® Bis–Tris Gel System (Invitrogen). HNRNPH1 (ab10374; Abcam, Cambridge, MA, USA) and ACTB (A5441; Sigma, St Louis, MO, USA) specific antibodies were used for western blot.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This work was supported by grants from the National Institutes of Health (R01HG004634, R01GM088342) and a junior faculty grant from the Edward Mallinckrodt Jr Foundation.
ACKNOWLEDGEMENTS
We thank Peter Stoilov for discussions on this work and Russ Carstens for providing the pI-11-H3 minigene construct. We thank David Eichmann, Ben Rogers and the University of Iowa Institute for Clinical and Translational Science (NIH grant UL1 RR024979) for computer support.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Nilsen T.W., Graveley B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., Burge C.B. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pan Q., Shai O., Lee L.J., Frey B.J., Blencowe B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 4.Wang Z., Burge C.B. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barash Y., Calarco J.A., Gao W., Pan Q., Wang X., Shai O., Blencowe B.J., Frey B.J. Deciphering the splicing code. Nature. 2010;465:53–59. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- 6.Cartegni L., Chew S.L., Krainer A.R. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat. Rev. Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 7.Wang G.S., Cooper T.A. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 8.Cooper T.A., Wan L., Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Black D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 10.Sheth N., Roca X., Hastings M.L., Roeder T., Krainer A.R., Sachidanandam R. Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Res. 2006;34:3955–3967. doi: 10.1093/nar/gkl556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burset M., Seledtsov I.A., Solovyev V.V. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000;28:4364–4375. doi: 10.1093/nar/28.21.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roca X., Olson A.J., Rao A.R., Enerly E., Kristensen V.N., Borresen-Dale A.L., Andresen B.S., Krainer A.R., Sachidanandam R. Features of 5′-splice-site efficiency derived from disease-causing mutations and comparative genomics. Genome Res. 2008;18:77–87. doi: 10.1101/gr.6859308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeo G., Burge C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004;11:377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 14.Krawczak M., Reiss J., Cooper D.N. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum. Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 15.Stenson P.D., Ball E.V., Mort M., Phillips A.D., Shiel J.A., Thomas N.S., Abeysinghe S., Krawczak M., Cooper D.N. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 16.Krawczak M., Thomas N.S., Hundrieser B., Mort M., Wittig M., Hampe J., Cooper D.N. Single base-pair substitutions in exon–intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum. Mutat. 2007;28:150–158. doi: 10.1002/humu.20400. [DOI] [PubMed] [Google Scholar]
- 17.Kwan T., Benovoy D., Dias C., Gurd S., Serre D., Zuzan H., Clark T.A., Schweitzer A., Staples M.K., Wang H., et al. Heritability of alternative splicing in the human genome. Genome Res. 2007;17:1210–1218. doi: 10.1101/gr.6281007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwan T., Benovoy D., Dias C., Gurd S., Provencher C., Beaulieu P., Hudson T.J., Sladek R., Majewski J. Genome-wide analysis of transcript isoform variation in humans. Nat. Genet. 2008;40:225–231. doi: 10.1038/ng.2007.57. [DOI] [PubMed] [Google Scholar]
- 19.Kwan T., Grundberg E., Koka V., Ge B., Lam K.C., Dias C., Kindmark A., Mallmin H., Ljunggren O., Rivadeneira F., et al. Tissue effect on genetic control of transcript isoform variation. PLoS Genet. 2009;5:e1000608. doi: 10.1371/journal.pgen.1000608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hull J., Campino S., Rowlands K., Chan M.S., Copley R.R., Taylor M.S., Rockett K., Elvidge G., Keating B., Knight J., et al. Identification of common genetic variation that modulates alternative splicing. PLoS Genet. 2007;3:e99. doi: 10.1371/journal.pgen.0030099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heinzen E.L., Ge D., Cronin K.D., Maia J.M., Shianna K.V., Gabriel W.N., Welsh-Bohmer K.A., Hulette C.M., Denny T.N., Goldstein D.B. Tissue-specific genetic control of splicing: implications for the study of complex traits. PLoS Biol. 2008;6:e1. doi: 10.1371/journal.pbio.1000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pickrell J.K., Marioni J.C., Pai A.A., Degner J.F., Engelhardt B.E., Nkadori E., Veyrieras J.B., Stephens M., Gilad Y., Pritchard J.K. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464:768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montgomery S.B., Sammeth M., Gutierrez-Arcelus M., Lach R.P., Ingle C., Nisbett J., Guigo R., Dermitzakis E.T. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coulombe-Huntington J., Lam K.C., Dias C., Majewski J. Fine-scale variation and genetic determinants of alternative splicing across individuals. PLoS Genet. 2009;5:e1000766. doi: 10.1371/journal.pgen.1000766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graveley B.R. The haplo-spliceo-transcriptome: common variations in alternative splicing in the human population. Trends Genet. 2008;24:5–7. doi: 10.1016/j.tig.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heinzen E.L., Yoon W., Tate S.K., Sen A., Wood N.W., Sisodiya S.M., Goldstein D.B. Nova2 interacts with a cis-acting polymorphism to influence the proportions of drug-responsive splice variants of SCN1A. Am. J. Hum. Genet. 2007;80:876–883. doi: 10.1086/516650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu H., Tucker H.M., Grear K.E., Simpson J.F., Manning A.K., Cupples L.A., Estus S. A common polymorphism decreases low-density lipoprotein receptor exon 12 splicing efficiency and associates with increased cholesterol. Hum. Mol. Genet. 2007;16:1765–1772. doi: 10.1093/hmg/ddm124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.International-HapMap-Consortium. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 29.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E., et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duan S., Huang R.S., Zhang W., Bleibel W.K., Roe C.A., Clark T.A., Chen T.X., Schweitzer A.C., Blume J.E., Cox N.J., et al. Genetic architecture of transcript-level variation in humans. Am. J. Hum. Genet. 2008;82:1101–1113. doi: 10.1016/j.ajhg.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W., Duan S., Bleibel W.K., Wisel S.A., Huang R.S., Wu X., He L., Clark T.A., Chen T.X., Schweitzer A.C., et al. Identification of common genetic variants that account for transcript isoform variation between human populations. Hum. Genet. 2009;125:81–93. doi: 10.1007/s00439-008-0601-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Warzecha C.C., Sato T.K., Nabet B., Hogenesch J.B., Carstens R.P. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol. Cell. 2009;33:591–601. doi: 10.1016/j.molcel.2009.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooper G.M., Stone E.A., Asimenos G., Green E.D., Batzoglou S., Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goode D.L., Cooper G.M., Schmutz J., Dickson M., Gonzales E., Tsai M., Karra K., Davydov E., Batzoglou S., Myers R.M., et al. Evolutionary constraint facilitates interpretation of genetic variation in resequenced human genomes. Genome Res. 2010;20:301–310. doi: 10.1101/gr.102210.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Durbin R.M., Abecasis G.R., Altshuler D.L., Auton A., Brooks L.D., Gibbs R.A., Hurles M.E., McVean G.A. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weir B.S., Hill W.G. Estimating F-statistics. Annu. Rev. Genet. 2002;36:721–750. doi: 10.1146/annurev.genet.36.050802.093940. [DOI] [PubMed] [Google Scholar]
- 37.Akey J.M., Zhang G., Zhang K., Jin L., Shriver M.D. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002;12:1805–1814. doi: 10.1101/gr.631202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith J.M., Haigh J. The hitch-hiking effect of a favourable gene. Genet. Res. 1974;23:23–35. [PubMed] [Google Scholar]
- 39.Fay J.C., Wu C.I. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kawase T., Akatsuka Y., Torikai H., Morishima S., Oka A., Tsujimura A., Miyazaki M., Tsujimura K., Miyamura K., Ogawa S., et al. Alternative splicing due to an intronic SNP in HMSD generates a novel minor histocompatibility antigen. Blood. 2007;110:1055–1063. doi: 10.1182/blood-2007-02-075911. [DOI] [PubMed] [Google Scholar]
- 41.Rhead B., Karolchik D., Kuhn R.M., Hinrichs A.S., Zweig A.S., Fujita P.A., Diekhans M., Smith K.E., Rosenbloom K.R., Raney B.J., et al. The UCSC Genome Browser database: update 2010. Nucleic Acids Res. 2010;38:D613–619. doi: 10.1093/nar/gkp939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fairbrother W.G., Yeh R.F., Sharp P.A., Burge C.B. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297:1007–1013. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- 43.Wang Z., Rolish M.E., Yeo G., Tung V., Mawson M., Burge C.B. Systematic identification and analysis of exonic splicing silencers. Cell. 2004;119:831–845. doi: 10.1016/j.cell.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 44.Xiao X., Wang Z., Jang M., Burge C.B. Coevolutionary networks of splicing cis-regulatory elements. Proc. Natl Acad. Sci. USA. 2007;104:18583–18588. doi: 10.1073/pnas.0707349104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carothers A.M., Urlaub G., Grunberger D., Chasin L.A. Splicing mutants and their second-site suppressors at the dihydrofolate reductase locus in Chinese hamster ovary cells. Mol. Cell. Biol. 1993;13:5085–5098. doi: 10.1128/mcb.13.8.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Venables J.P., Koh C.S., Froehlich U., Lapointe E., Couture S., Inkel L., Bramard A., Paquet E.R., Watier V., Durand M., et al. Multiple and specific mRNA processing targets for the major human hnRNP proteins. Mol. Cell. Biol. 2008;28:6033–6043. doi: 10.1128/MCB.00726-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Auweter S.D., Oberstrass F.C., Allain F.H. Sequence-specific binding of single-stranded RNA: is there a code for recognition? Nucleic Acids Res. 2006;34:4943–4959. doi: 10.1093/nar/gkl620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stoilov P., Daoud R., Nayler O., Stamm S. Human tra2-beta1 autoregulates its protein concentration by influencing alternative splicing of its pre-mRNA. Hum. Mol. Genet. 2004;13:509–524. doi: 10.1093/hmg/ddh051. [DOI] [PubMed] [Google Scholar]
- 49.Tacke R., Tohyama M., Ogawa S., Manley J.L. Human Tra2 proteins are sequence-specific activators of pre-mRNA splicing. Cell. 1998;93:139–148. doi: 10.1016/s0092-8674(00)81153-8. [DOI] [PubMed] [Google Scholar]
- 50.Xiao X., Wang Z., Jang M., Nutiu R., Wang E.T., Burge C.B. Splice site strength-dependent activity and genetic buffering by poly-G runs. Nat. Struct. Mol. Biol. 2009;16:1094–1100. doi: 10.1038/nsmb.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martinez-Contreras R., Fisette J.F., Nasim F.U., Madden R., Cordeau M., Chabot B. Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol. 2006;4:e21. doi: 10.1371/journal.pbio.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caputi M., Zahler A.M. Determination of the RNA binding specificity of the heterogeneous nuclear ribonucleoprotein (hnRNP) H/H'/F/2H9 family. J. Biol. Chem. 2001;276:43850–43859. doi: 10.1074/jbc.M102861200. [DOI] [PubMed] [Google Scholar]
- 53.McCullough A.J., Berget S.M. G triplets located throughout a class of small vertebrate introns enforce intron borders and regulate splice site selection. Mol. Cell. Biol. 1997;17:4562–4571. doi: 10.1128/mcb.17.8.4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marcucci R., Baralle F.E., Romano M. Complex splicing control of the human Thrombopoietin gene by intronic G runs. Nucleic Acids Res. 2007;35:132–142. doi: 10.1093/nar/gkl965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Majewski J., Ott J. Distribution and characterization of regulatory elements in the human genome. Genome Res. 2002;12:1827–1836. doi: 10.1101/gr.606402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yeo G., Hoon S., Venkatesh B., Burge C.B. Variation in sequence and organization of splicing regulatory elements in vertebrate genes. Proc. Natl Acad. Sci. USA. 2004;101:15700–15705. doi: 10.1073/pnas.0404901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang X.H., Heller K.A., Hefter I., Leslie C.S., Chasin L.A. Sequence information for the splicing of human pre-mRNA identified by support vector machine classification. Genome Res. 2003;13:2637–2650. doi: 10.1101/gr.1679003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Louie E., Ott J., Majewski J. Nucleotide frequency variation across human genes. Genome Res. 2003;13:2594–2601. doi: 10.1101/gr.1317703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang C., Zhang Z., Castle J., Sun S., Johnson J., Krainer A.R., Zhang M.Q. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev. 2008;22:2550–2563. doi: 10.1101/gad.1703108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yeo G.W., Van Nostrand E.L., Liang T.Y. Discovery and analysis of evolutionarily conserved intronic splicing regulatory elements. PLoS Genet. 2007;3:e85. doi: 10.1371/journal.pgen.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woolfe A., Mullikin J.C., Elnitski L. Genomic features defining exonic variants that modulate splicing. Genome Biol. 2010;11:R20. doi: 10.1186/gb-2010-11-2-r20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kheradpour P., Stark A., Roy S., Kellis M. Reliable prediction of regulator targets using 12 Drosophila genomes. Genome Res. 2007;17:1919–1931. doi: 10.1101/gr.7090407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lomelin D., Jorgenson E., Risch N. Human genetic variation recognizes functional elements in noncoding sequence. Genome Res. 2010;20:311–319. doi: 10.1101/gr.094151.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cooper T.A. Use of minigene systems to dissect alternative splicing elements. Methods. 2005;37:331–340. doi: 10.1016/j.ymeth.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 65.Singh G., Cooper T.A. Minigene reporter for identification and analysis of cis elements and trans factors affecting pre-mRNA splicing. Biotechniques. 2006;41:177–181. doi: 10.2144/000112208. [DOI] [PubMed] [Google Scholar]
- 66.Voelker R.B., Berglund J.A. A comprehensive computational characterization of conserved mammalian intronic sequences reveals conserved motifs associated with constitutive and alternative splicing. Genome Res. 2007;17:1023–1033. doi: 10.1101/gr.6017807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aznarez I., Barash Y., Shai O., He D., Zielenski J., Tsui L.C., Parkinson J., Frey B.J., Rommens J.M., Blencowe B.J. A systematic analysis of intronic sequences downstream of 5′ splice sites reveals a widespread role for U-rich motifs and TIA1/TIAL1 proteins in alternative splicing regulation. Genome Res. 2008;18:1247–1258. doi: 10.1101/gr.073155.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shendure J., Ji H. Next-generation DNA sequencing. Nat. Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 69.Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hsu F., Kent W.J., Clawson H., Kuhn R.M., Diekhans M., Haussler D. The UCSC Known Genes. Bioinformatics. 2006;22:1036–1046. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- 71.Kim N., Alekseyenko A.V., Roy M., Lee C. The ASAP II database: analysis and comparative genomics of alternative splicing in 15 animal species. Nucleic Acids Res. 2007;35:D93–98. doi: 10.1093/nar/gkl884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lin L., Shen S., Tye A., Cai J.J., Jiang P., Davidson B.L., Xing Y. Diverse splicing patterns of exonized Alu elements in human tissues. PLoS Genet. 2008;4:e1000225. doi: 10.1371/journal.pgen.1000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kapur K., Xing Y., Ouyang Z., Wong W.H. Exon arrays provide accurate assessments of gene expression. Genome Biol. 2007;8:R82. doi: 10.1186/gb-2007-8-5-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xing Y., Kapur K., Wong W.H. Probe selection and expression index computation of Affymetrix Exon Arrays. PLoS One. 2006;1:e88. doi: 10.1371/journal.pone.0000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rozen S., Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 76.Schuelke M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000;18:233–234. doi: 10.1038/72708. [DOI] [PubMed] [Google Scholar]
- 77.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.