

Abstract
Transient receptor potential vanilloid 1 (TRPV1) is implicated in cisplatin ototoxicity. Activation of this channel by cisplatin increases reactive oxygen species generation, which contribute to loss of outer hair cells in the cochlea. Knockdown of TRPV1 by short interfering RNA protected against cisplatin ototoxicity. In this study, we examined the mechanism underlying TRPV1-mediated ototoxicity using cultured organ of Corti transformed cells (UB/OC-1) and rats. Trans-tympanic injections of capsaicin produced transient hearing loss within 24 h, which recovered by 72 h. In UB/OC-1 cells, capsaicin increased NOX3 NADPH oxidase activity and activation of signal transducer and activator of transcription 1 (STAT1). Intratympanic administration of capsaicin transiently increased STAT1 activity and expression of downstream proinflammatory molecules. Capsaicin produced a transient increase in CD14-positive inflammatory cells into the cochlea, which mimicked the temporal course of STAT1 activation but did not alter the expression of apoptotic genes or damage to outer hair cells. In addition, trans-tympanic administration of STAT1 short interfering RNA protected against capsaicin-induced hearing loss. These data suggest that activation of TRPV1 mediates temporary hearing loss by initiating an inflammatory process in the cochlea via activation of NOX3 and STAT1. Thus, these proteins represent reasonable targets for ameliorating hearing loss. Antioxid. Redox Signal. 14, 999–1010.
Introduction
Reactive oxygen species (ROS) play a prominent role in mediating drug- and noise-induced hearing loss. For example, ROS contribute to paraquat (3) and cisplatin-mediated (23) hearing loss as well as noise-induced hearing loss (14, 26, 46). The NOX3 isoform of NADPH oxidase contributes significantly to the generation of ROS by cisplatin (23) and possibly by noise (30). As such, antioxidant therapy has been shown to protect against both drug- (34) and noise-induced (25) hearing loss. In addition, selective knockdown of NOX3 in the cochlea by trans-tympanic delivery of short interfering (si)RNA protects against cisplatin ototoxicity (22). Activation of NOX3 has been shown to activate and induce transient receptor potential vanilloid 1 (TRPV1) channel in the cochlea. These channels, in turn, appear to regulate the activity and expression of NOX3, suggesting the existence of a positive feedback between these proteins (23).
ROS mediates ototoxicity through generation of inflammatory cytokines. For example, ROS promotes age-related hearing loss in CD/1 mice by increasing the production of tumor necrosis factor-α (TNF-α) (31). Increase in immune activation has been linked to hearing loss in some individuals (20), which could explain the clinical utility of steroids to restore cochlear function in some patients with acute hearing loss (13). The induction of inflammatory cytokines has been linked to cisplatin-induced ototoxicity (38). Downregulation of the inflammatory response by flunarizine through activation of NF-E2-related factor 2/heme oxygenase-1 protects against cisplatin ototoxicity (38). These studies suggest that pharmacological approaches that are geared to the suppression of inflammation should be useful in combating drug- and noise-induced hearing loss.
TRPV1 is a nonselective cationic channel normally present in sensory neurons that mediates thermal perception and inflammatory pain. The demonstration of this receptor in other tissues suggests additional functions of TRPV1 besides heat and pain perception. TRPV1 present on afferent nerve terminals and epithelial cells lining the bladder lumen regulate normal bladder function (4). TRPV1 expressed in the abdominal viscera regulates body temperature (39). An N-terminal splice variant of TRPV1 in the hypothalamus regulates the release of arginine-vasopressin (37). Although TRPV1 is expressed in the cochlea, its exact physiological role in this organ is unclear. We have recently implicated TRPV1 in cisplatin ototoxicity (23). Cisplatin activates and induces TRPV1 expression by increasing ROS generation through NOX3. Increases in TRPV1 activity and expression are believed to enhance Ca2+ influx in cochlear tissues expressing this ion channel, leading to cell death. As such, downregulation of this channel by siRNA protects against cisplatin ototoxicity.
TRPV1 has been linked to inflammation in various tissues (1, 6, 19). We hypothesized that activation of TRPV1 could mediate inflammation in the cochlea, and thereby contribute to hearing loss produced by cisplatin and noise. Our data demonstrate that ROS serve as important signaling molecules downstream of TRPV1, which promote inflammation and transient hearing loss by activating signal transducer and activator of transcription 1 (STAT1) in the rat cochlea. Moreover, knockdown of STAT1 by trans-tympanic delivery of siRNA abrogated the increases in inflammatory mediators and protected against capsaicin-induced transient hearing loss. These studies provide novel insights into the role of TRPV1 in mediating inflammation in the cochlea and hearing loss. They also provide evidence for the utility trans-tympanic delivery of siRNA in the treatment of ototoxicity.
Materials and Methods
Reagents
Capsaicin, capsazepine, diphenyleneiodonium, 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis (acetoxymethyl ester) (BAPTA-AM), and TRI reagent were purchased from Sigma-Aldrich; 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) dye was from EMD Biosciences. Various antibodies used were as follows: inducible nitric oxide synthase (iNOS), cyclooxygenase 2 (COX-2), TNF-α, CD14 (Santa Cruz Biotechnology), TRPV1 antibody (Neuromics), p-STAT1 and STAT1 (Cell Signaling Technology Inc.), goat anti-rabbit, donkey anti-goat and goat anti-mouse secondary antibodies (Santa Cruz Biotechnology), and fluorescent tagged (dylight 488 and TRITC) secondary antibodies (Jackson Immuno Laboratories).
Animal procedures
Male Wistar rats (250–300 g) were used for this study. Pretreatment auditory brainstem responses (ABRs) were performed immediately before trans-tympanic application of siRNA against STAT1 or NOX3, or a scrambled siRNA sequence that served as a control. Fifty microliters of capsaicin (0.1 μM) was administered trans-tympanically 48 h after siRNA administration. The tympanic membrane was checked for perforation, and it was observed that the puncture wound had healed completely by 72 h postcapsaicin (or day 5 post siRNA injection in no treatment group) in all the rats. There was no evidence of middle ear effusion or infection in these animals. Post-treatment ABRs were then performed 24 or 72 h after capsaicin administration. The cochleae were dissected and used for the extraction of total RNA, or perfused with 2.5% glutaraldehyde for morphological studies by scanning electron microscopy (SEM) or with 4% paraformaldehyde for immunohistochemical analyses. All animal procedures were approved by the SIU Laboratory Animal Care and Use Committee.
Trans-tympanic administration of siRNA
Rats were anesthetized with ketamine/xylazine mixture. With the help of a Zeiss operating microscope, 28G–30G needles, 1/2–5/8 of an inch in length were used to make a single puncture of the tympanic membrane in the antero-inferior region, taking care not to touch the malleus or the facial nerve, and 50 μl of solution was injected into the middle ear (siRNA was resuspended in 50 μl of sterile water to get the desired concentration). The rat was then left undisturbed for 15 min with the treated ear facing up. This procedure was then repeated in the other ear.
Evoked potentials
ABRs were measured before administration and 24 or 72 h after capsaicin administration, as described previously (41). Animals were tested with a stimulus intensity series that was initiated at 0 dB sound pressure level and reached a maximum at 90 dB sound pressure level. The stimulus intensity levels were increased in 10 dB increments, and the evoked ABR waveforms were observed on a video monitor. The auditory stimuli included tone bursts at 8, 16, and 32 kHz with a 10 ms plateau and a 1 ms rise/fall time presented at a rate of 5/s. Threshold was defined as the lowest intensity capable of evoking a reproducible, visually detectable response with two distinct waveforms and minimum amplitude of 0.5 μV.
Scanning electron microscopy
Immediately after completion of post-treatment ABRs, deeply sedated rats were euthanized, and their cochleae were harvested and processed as described previously (12). Sputter-coated cochleae were then viewed and photographed with a Hitachi S-500 scanning electron microscope (Hitachi Ltd.).
Hair cell count
Hair cell counts were performed using a modified version of the method described previously (17). Two representative areas of the basal turn and hook portion were photographed. In each area, outer hair cells (OHCs) were counted in an area that was 10 pillar cell heads in length. The results are presented as the percent hair cell damage per cochlear turn.
Processing of cochleae for immunohistochemistry
Cochleae were perfused with 4% paraformaldehyde, decalcified for 4–6 h using rapid decalcification using RapidCal Immuno, BBC Biochemical, paraffin embedded, and sectioned. Samples were incubated with different primary antibodies (1:100 dilutions) for 1 h at 37°C followed by incubation with Dylight488 or TRITC-labeled secondary antibody (1:200 dilution). For double-immunolabeling, samples were incubated with both the fluorescent-tagged antibodies at same time. Slides were then imaged using an Olympus confocal microscope (Olympus America Inc.). Images captured were analyzed using ImageJ analysis software from NIH (http://rsbweb.nih.gov/ij/index.html).
siRNA sequences
The rodent set of siRNAs was based on the homologous sequences in the rat and mouse cDNA sequences. Custom siRNA was purchased from Qiagen. Scrambled siRNA was also procured from Human/Mouse starter kit (Qiagen).
The target sequence of rodent NOX3 siRNA was 5′-AAGGTGGTGAGTCACCCATCT-3′; that of rodent STAT1 siRNA was 5′-AAGGAAAAGCAAGCGTAATCT-3′; that of rodent TRPV1 siRNA was 5′-GCGCATCTTCTACTTCAACTT-3′ (5).
RNA isolation
Cochleae were pared down to the bone, cracked open, and the soft tissue scooped out in 500 μl of TRI reagent. About 0.1 ml of chloroform was added, and the tube was shaken vigorously for 15 s and centrifuged at 12,000 g for 15 min. RNA was extracted by washing the pellet with 0.5 ml ice-cold isopropanol followed by cold 75% diethylpyrocarbonate-treated ethanol. The ethanol was removed and the tube was air dried briefly. The RNA pellet was resuspended in nuclease-free water and RNA levels were determined using optical density readings corresponding to wavelengths of 260, 280, and 320 nm using a spectrophotometer (Eppendorf BioPhotometer).
Real-time reverse transcriptase polymerase chain reaction
One microgram of total RNA was converted to cDNA using iScript cDNA Synthesis Kit (Bio-Rad). The reaction mixture was set up as follows: 1 μg of total RNA, 4 μl of iScript reaction mix, 1 μl of iScript reverse transcriptase, and nuclease-free water to bring the total volume to 20 μl. The reaction mix was incubated at 25°C for 5 min, 42°C for 30 min, and 85°C for 5 min. This cDNA reaction mix was used for real-time polymerase chain reaction (PCR), as described previously (22). Gene-specific primer pairs were used for the various reactions and mRNA expression levels were normalized to the levels of GAPDH. The primer sets were purchased from Sigma Genosys, and were as follows:
Rodent-Bax: 5′-ATGGCTGGGGAGACACCTGA-3′ (sense); 5′-GCAAAGTAGAAGAGGGCAACC-3′ (antisense)
Rodent-Bcl2: 5′-CCTTCTTTGAGTTCGGTG-3′ (sense); 5′-GAGACAGCCAGGAGAAAT-3′ (antisense)
Rodent-COX-2: 5′-TGATCGAAGACTACGTGCAAC-3′ (sense); 5′-GTACTCCTGGTCTTCAATGTT-3′ (antisense)
Rodent-GAPDH: 5′-ATGGTGAAGGTCGGTGTGAAC-3′ (sense); 5′-TGTAGTTGAGGTCAATGAAGG-3′ (antisense)
Rodent NOX3: 5′-GTGAACAAGGGAAGGCTCAT-3′ (sense); 5′-GACCCACAGAAGAACACGC-3′ (antisense)
Rodent p53: 5′-GGACGACAGGCAGACTTTTC-3′ (sense); 5′-GGCACAAACACGAACCTCAAA-3′ (antisense)
Rodent-STAT1: 5′-CATGGAAATCAGACAGTACCT-3′ (sense); 5′-TCTGTACGGGATCTTCTTGGA-3′ (antisense)
Rodent-TNF-α: 5′-CAGACCCTCACACTCAGATCA-3′ (sense); 5′-TGAAGAGAACCTGGGAGTAGA-3′ (antisense)
Rodent TRPV1: 5′-CAAGGCTGTCTTCATCATCC-3′ (sense); 5′-AGTCCAGTTTACCTCGTCCA-3′ (antisense)
Cell culture
Immortalized organ of Corti cells derived from the mouse, UB/OC-1 cells, were obtained from Dr. Matthew Holley (Institute of MolecularPhysiology, Addison Building, Western Bank, Sheffield, United Kingdom) and cultured in RPMI 1640 supplemented with 10% Fetalclone II serum (Hyclone) and penicillin–streptomycin. Cultures were grown at 33°C in an incubator with 10% CO2.
H2DCFDA assay
ROS generation was measured with green-fluorescent dye H2DCFDA as described by Jajoo et al. (11). Briefly, UB/OC-1 cells were pretreated with different inhibitors (for 30 min) or transfected with siRNAs (for 48 h). Cells were then treated with capsaicin for 15 min followed by incubation with 5 μM H2DCFDA dye for 15 min. ROS generation was detected as green fluorescence by confocal microscopy.
Western blot analysis
UB/OC-1 cells were homogenized in ice-cold 50 mM Tris HCl, 10 mM MgCl2, and 1 mM ethylenediaminetetraacetic acid in the presence of protease inhibitors cocktail (Sigma). Nuclear lysates were prepared according to the protocol described previously (10), and used for Western blotting experiments for the detection of activated STAT1. After transfer to nitrocellulose membranes, blots were probed with a p-STAT1 primary antibody, followed by a horse radish peroxidase-tagged secondary antibody. The proteins were then observed by LAS 4000 chemiluminescence imaging system (GE Healthcare Biosciences) using ECL reagent (Pierce Biotechnology). The bands were normalized to total STAT1 levels, and quantitative analysis was performed with Multi Gauge software (version 3.0) from Fujifilm.
Statistical analyses
Statistical significance differences among groups were evaluated using Student's t-tests and analysis of variance, followed by Tukey's post hoc test.
Results
Transtympanic administration of capsaicin produces transient hearing loss
ABRs were first determined in male Wistar rats that were then administered vehicle or capsaicin (0.1 μM) by the trans-tympanic route. Animals were again tested at 24 and 72 h and their post-treatment ABRs compared to their pretreatment ABR values to determine the degree of hearing loss. This is reflected as changes in ABR thresholds (Fig. 1A). The trans-tympanic route of administration produced a consistent but statistically insignificant change in ABR threshold at 24 h, as indicated in the vehicle-treated group assessed at 24 h. However, this ABR threshold shift was not seen at 72 h after trans-tympanic injection in the control group. This suggests that trans-tympanic injections produced a small degree of trauma/damage to the tympanic membrane, that healed by 72 h (a needle puncture was observed 24 h postinjection on the tympanic membrane, which completely healed by 72 h). Animals tested 24 h after administration of capsaicin exhibited increased ABR thresholds ranging from 25 to 30 dB at the frequencies of 8, 16, and 32 kHz. The same animals when tested for ABR thresholds 72 h after capsaicin administration exhibited statistically indistinguishable threshold shifts from the vehicle (∼5 dB shifts at all frequencies tested)-treated rats (Fig. 1A). These observations suggest that capsaicin produces transient hearing loss and therefore differs from cisplatin that produces permanent hearing loss (33). SEM images of the organ of Corti were used to assess potential damage to the cochlea produced by capsaicin to account for the ABR shifts, and show normal OHC morphology at 24 h after capsaicin administration. This is in contrast to images obtained for cisplatin-treated rats, where damage to the stereocilia and loss of hair cells were observed at 72 h using a 11 mg/kg intraperitoneal dose of cisplatin (Fig. 1B). Thus, the temporary shift in ABR produced by capsaicin was independent of gross morphological changes in or loss of OHCs.
FIG. 1.

Transtympanic administration of capsaicin produces transient hearing loss. Capsaicin was administered by trans-tympanic injections to anesthetized male Wistar rats with the aid of a Zeiss operating microscope. The drug or vehicle was administered with 28G–30G needles in a volume of 50 μl. (A) ABR measurements performed 24 h later showed increased threshold shifts at all frequencies tested, but significant reductions from the 24 h levels at 72 h. *Statistically significant difference from vehicle-treated rats (p < 0.05, analysis of variance, n = 5–9 rats per group); **statistically significant difference from capsaicin (24 h)-treated group. (B) Scanning electron microscopy images of OHCs show no significant changes in morphology or loss 24 h after capsaicin administration. This is in contrast to cisplatin that produced significant degree of OHC damage or loss 72 h after intraperitoneal administration. Arrows indicate evidence of damage or loss of OHCs. Scanning electron microscopy images are a representative from one cochlea each from three different rats, with each showing similar changes. ABR, auditory brainstem response; OHC, outer hair cell.
Capsaicin increases ROS generation by inducing NOX3 activity and intracellular Ca2+ release
To determine the downstream events triggered by TRPV1 activation, we used UB/OC-1 cell line, an in vitro model of OHC's in culture (23). Exposure of UB/OC-1 cells to capsaicin (1 μM) resulted in increased ROS levels as measured by H2DCFDA fluorescence. The increase in ROS was dose dependent, as evident by a greater fluorescent intensity obtained with 2.5 versus 1 μM capsaicin (Fig. 2A). This increase was attenuated by TRPV1 antagonist capsazepine (Supplementary Fig. S1; Supplementary Data are available online at www.liebertonline.com/ars), or after knockdown of TRPV1 in these cells by siRNA against TRPV1 (Fig. 2B). Knockdown of TRPV1 was shown by real-time reverse transcriptase (RT)-PCR (data not shown).
FIG. 2.

Capsaicin increases ROS generation in UB/OC-1 cells. UB/OC-1 cells exposed to capsaicin demonstrated increased ROS generation as measured by H2DCFDA dye. (A) Cells were pretreated with different concentrations of capsaicin (0–2.5 μM) for 15 min, followed by incubation with H2DCFDA dye. The fluorescence intensity increased with increasing capsaicin concentrations. (B) Capsaicin-mediated increase in ROS generation was abrogated after knockdown of TRPV1 channels using siRNA. UB/OC-1 cells were transfected with TRPV1-siRNA for 48 h, and ROS generation was assessed in these cells after 15 min capsaicin treatment. TRPV1-siRNA alone did not alter the level of ROS generation. Data presented in (A) and (B) were replicated at least three times with similar results. Scale bar (lower right panels) represent 10 μm. H2DCFDA, 2′,7′-dichlorodihydrofluorescein diacetate; ROS, reactive oxygen species; siRNA, short interfering RNA; TRPV1, transient receptor potential 1. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
In our previous study (23), we showed cochlear-specific NADPH oxidase (NOX3) as the primary source of ROS generation in UB/OC-1 cells. Therefore, we next determined if NADPH oxidase (and the cochlear-specific NOX3 isoform) was involved in capsaicin-induced ROS generation. As observed previously, capsaicin increased ROS generation in UB/OC-1 cells. However, this increase was significantly abrogated in the cells by pretreatment with diphenyliodonium, an inhibitor of NADPH oxidase (Supplementary Fig. S2) and by knockdown of NOX3 by siRNA (Fig. 3A). NOX3 siRNA produced a ∼70% knockdown of NOX3 protein, as determined by real-time RT-PCR (data not shown). Further, the increase in NOX3 activity was dependent on intracellular Ca2+ release, as this increase in NOX3 was inhibited by pretreatment of cells with the intracellular Ca2+ chelator BAPTA-AM (Fig. 3B). Taken together, these data support the conclusion that TRPV1 can stimulate ROS generation in the UB/OC-1 cells via NOX3 by increasing intracellular Ca2+ release.
FIG. 3.

Capsaicin-induced ROS generation involves NOX3 activation. (A) UB/OC-1 cells exposed to capsaicin demonstrated increased ROS generation, as measured by H2DCFDA fluorescence. UB/OC-1 cells were pretreated with scramble or NOX3 siRNA for 48 h, the culture media were replaced with fresh media, and cultures were treated with vehicle or capsaicin (2.5 μM). Capsaicin increased ROS was abrogated by NOX3 siRNA. The transfection of cells with NOX3 siRNA alone did not alter H2DCFDA fluorescence. (B) Cells pretreated with vehicle or BAPTA-AM (10 μM) for 30 min before capsaicin administration showed reduced capsaicin-mediated ROS generation. The addition of BAPTA-AM alone was not associated with any appreciable change in ROS generation. Scale bar (lower right panel) represents 10 μm. Data presented in (A) and (B) were replicated at least three times with similar results. BAPTA-AM, 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis (acetoxymethyl ester). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
Capsaicin increases STAT1 activity
Previous studies have indicated that trauma to the cochlea leads to inflammation that can result in hearing loss (38). In addition, a recent study implicated STAT1 in cisplatin-induced killing of utricular hair cell in vitro (36). Therefore, we examined whether STAT1 mediates some of the downstream events after TRPV1 activation. As shown in Figure 4A, capsaicin (2.5 μM) produced a time-dependent increase in STAT1 Ser727 phosphorylation in UB/OC-1 cells, which peaked at 60 min and remained at this level up to 120 min after drug administration.
FIG. 4.

Capsaicin increases STAT1 activity in UB/OC-1 cells and in the rat cochlea. (A) UB/OC-1 cells were incubated with vehicle (0) or capsaicin (2.5 μM) for up to 2 h and nuclear lysates were prepared from each time point and used for determination of Ser727 p-STAT1 and total STAT1 by Western blotting. Capsaicin increased Ser727 p-STAT1 levels over total STAT1, indicative of STAT1 activation. *Statistically significant difference from vehicle-treated cells (p < 0.05; n = 4). (B) Capsaicin increased Ser727 p-STAT1 in the rat cochlea. Rats were anesthetized and then administered capsaicin by trans-tympanic injections. Cochleae were isolated 24 or 72 h later, perfused with 4% glutaraldehyde, decalcified, and paraffin imbedded, and the resulting sections used for Ser727 p-STAT1 immunocytochemistry. Capsaicin increased Ser727 p-STAT1 levels throughout the cochlea, but especially in the SVA, OHCs, and SG cells (see insets). (C) Similar increases in total STAT1 were observed in adjacent cochlear sections. Data presented in (B) and (C) are representative sections from three different rats (one cochlea each) per group. Insets show enlarged sections of OHCs and SG. Scale bars (right lower panel) represent 50 and 10 μm (for insets). SG, spiral ganglion; STAT1, signal transducer and activator of transcription 1; SVA, stria vascularis. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
Trans-tympanic administration of capsaicin also increased STAT1 Ser727 phosphorylation in the rat cochlea by 24 h and this increase in p-STAT1 Ser727 was abolished by 72 h postcapsaicin treatment. Increases in STAT1 phosphorylation (by approximately three- to fourfold) were evident in the OHCs, stria vascularis (SVA) and spiral ganglion (SG) cells (Fig. 4B). Interestingly, p-STAT Tyr701 was also increased at 24 h after capsaicin treatment, and was abolished by 72 h (see Supplementary Fig. S3). However, the changes in STAT1 phosphorylation were likely due to an increase in the level of total STAT1 observed in cochlea assessed at 24 h after trans-tympanic administration of capsaicin (Fig. 4C). These data suggest that induction of STAT1 contributes to the increased STAT1 phosphorylation observed with capsaicin in the cochlea.
Capsaicin increased STAT1 activity via NOX3
To determine the mechanism underlying the activation of STAT1, we pretreated rats with trans-tympanic injections of scrambled, STAT1 or NOX3 siRNAs 2 days before trans-tympanic administration of vehicle or capsaicin. STAT1 and NOX3 siRNAs decreased expression of these genes by 80%–90%, as determined by real-time RT-PCR. As shown above, capsaicin increased STAT1 activity in the OHC, SG, and SVA within 24 h, whereas this increase was abolished by 72 h (Fig. 5A). As expected, the increased in STAT1 protein was abolished by STAT1 and NOX3 siRNAs, implicating NOX3 in mediating the increase in p-STAT1 (Fig. 5A, C). Interestingly, STAT1 siRNA also reduced the increase in TRPV1 expression observed with capsaicin, implicating this transcription factor in the induction of TRPV1 (Fig. 5A, B). As shown previously (23), NOX3 siRNA also reduced the levels of TRPV1 in the cochlea (Fig. 5A). Quantification of TRPV1 and STAT1 mRNA revealed changes consistent with the immunocytochemical data. The induction of both of these genes was blocked by STAT1 and NOX3 siRNAs (Fig. 5D, E). Taken together, these data support a role of NOX3 in the activation of STAT1 and induction of TRPV1.
FIG. 5.

Capsaicin-mediated STAT1 activation in vivo is transient and involves ROS generation. (A) Rats were anesthetized and administered scrambled siRNA (scramble), STAT1, or NOX3 siRNAs by trans-tympanic injections. Two days later capsaicin was administered by intratympanic injections for 24 or 72 h. Cochleae were isolated, sectioned, and used for p-STAT1 Ser727 and TRPV1 immunolabeling studies. Capsaicin increased both TRPV1 (green) and p-STAT1 Ser727 (red) immunoreactivity at 24 h in SVA, OHC, and SG. However, these increases recovered to baseline levels by 72 h. Animals pretreated with STAT1 or NOX3 siRNA did not show any induction of p-STAT1 Ser727 and TRPV1 immunolabeling. Merged images (yellow) show colocalization of TRPV1 and p-STAT1 Ser727, suggesting possible functional interactions between these two proteins. Images shown are representatives of cochleae from four different animals showing similar results. Insets show enlarged sections of OHCs and SGs. Scale bars (lower right panel) represent 50 and 10 μm for insets. (B, C) Quantification of TRPV1 and p-STAT1 Ser727 immunolabeling indicate significant increases in the levels at the 24 h period after capsaicin administration but substantially reduced levels of these proteins were evident at 72 h or in STAT1 or NOX3 siRNAs pretreated groups. (D, E) Quantification of mRNA from rat cochleae by real-time polymerase chain reaction indicated significant increases in STAT1 and TRPV1 expression at 24 h but recovery to essentially baseline levels at 72 h. Knockdown of STAT1 or NOX3 by siRNAs abolished the increases in these transcripts. *Statistically significant difference from scramble siRNA-treated rats; **statistically significant difference from scramble + capsaicin-24 h treated rats (p < 0.05; n = 4). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
Capsaicin increased inflammatory mediators by activating STAT1
Since STAT1 regulates genes encoding proteins mediating the inflammatory response (28), we determined selected downstream targets of STAT1 that could be regulated by capsaicin. These include TNF-α, iNOS, and COX-2. For these experiments, rats were administered scrambled, STAT1 or NOX3 siRNA by trans-tympanic injections, followed by vehicle or capsaicin 2 days later. Cochleae were assessed for TNF-α and NOX3 immunoreactivity 24 and 72 h later. We observed low levels of TNF-α immunoreactivity throughout the cochleae of vehicle-treated mice. Trans-tympanic administration of capsaicin (scrambled siRNA + capsaicin-24 h) led to increased TNF-α immunolabeling in the OHCs, SG, and SVA at 24 h (Fig. 6A). However, TNF-α immunolabeling was substantially reduced from these levels in sections at 72 h after capsaicin administration (scrambled siRNA + capsaicin-72 h). In rats pretreated with STAT1 or NOX3 siRNA before capsaicin, TNF-α immunolabeling measured at 24 h was not different from sections obtained from rats treated with scrambled siRNA alone (Fig. 6A). Quantification of the TNF-α immunoreactivity is presented in Figure 6B. A similar profile of COX-2, NOX3 (Supplementary Figs. S4 and S5) and iNOS (Supplementary Fig. S6) immunolabeling was observed after trans-tympanic administration of capsaicin in rats pretreated with scrambled, STAT1 or NOX3 siRNA. The increases in TNF-α, COX-2, and iNOS immunolabeling observed with capsaicin and the suppression observed with STAT1 siRNA pretreatment were consistent with similar changes in their mRNAs (Fig. 6C and Supplementary Figs. S6C and S7). Overall, these changes support a role of capsaicin in the induction of STAT1-regulated genes involved in inflammation.
FIG. 6.

Capsaicin increases expression of STAT1-targeted genes in the cochlea. Rats were administered scrambled (scramble), STAT1, or NOX3 siRNAs via the trans-tympanic route, followed by vehicle or capsaicin. Cochleae were retrieved 24 or 72 h later, processed for coimmunolabeling for CD14 (green) and TNF-α (red) and RNA preparation. (A) In control animals (scramble), CD14 and TNF-α immunoreactivity was localized to the SVA and less to the OHC and SG cells. These proteins showed colocalization in cochlear merged images (yellow). Capsaicin produced substantial increases in labeling of these two proteins in these regions at 24 h but not at 72 h. However, pretreatment with STAT1 and NOX3 siRNAs abolished the regional increases in these proteins. Both STAT1 and NOX3 siRNAs reduced capsaicin-stimulated CD14 and TNF-α immunolabeling. Insets are enlarged sections of OHCs and SGs. Scale bars (right lower panels) represent 50 and 10 μm for insets. Images shown are representatives from cochleae from four rats showing similar results. (B, D) Quantification of immunofluorescence depicted in (A). (C) TNF-α mRNA obtained from the groups of rats treated similarly as in (A). *Statistically significant difference from scramble + vehicle-treated rats; **statistically significant difference from scramble + capsaicin-24 h treated rats (p < 0.05; n = 4). TNF-α, tumor necrosis factor-α. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
To determine whether capsaicin increased the migration of inflammatory cells into the cochlea, we utilized CD14 to assess infiltration of immune cells (43) into the cochlea. Cochlear sections were colabeled with a monoclonal antibody against TNF-α (as described above). We observed low basal CD14 in the cochleae obtained from rats administered scrambled siRNA or STAT1 siRNA added alone (Fig. 6A). However, capsaicin increased CD14 immunoreactivity in the SVA, SG, SL, and OHC. These increases were reduced after knockdown of STAT1 or NOX3 by their respective siRNAs administered by the trans-tympanic route (Fig. 6A, D). The increases in CD14 immunoreactivity returned to baseline levels 72 h after capsaicin administration (Fig. 6A, D). These findings are consistent with a role of TRPV1 in stimulating migration of inflammatory cells to the cochlea that contribute to increased TNF-α levels.
Transtympanic delivery of capsaicin did not alter the expression of apoptotic markers
To determine whether in vivo capsaicin administration was linked to cell apoptosis, we examined expression of p53 and Bax. Quantification of p53 and Bax expression by real-time PCR indicated that capsaicin did not increase expression of p53 or Bax (data not shown). Thus, the transient activation of TRPV1 does not seem to activate an apoptotic pathway, which is in contrast to cisplatin-induced TRPV1 activation, associated with increased p53 and Bax, and decreased Bcl2 levels (22). These data are consistent with our observations that capsaicin treatment does not produce any hair cell damage or permanent hearing loss as shown in Figure 1.
Trans-tympanic delivery of STAT1 siRNA protects against capsaicin ototoxicity
To test the efficacy of STAT1 siRNA against capsaicin ototoxicity, rats were administered siRNA against STAT1 (0.6 μg) by trans-tympanic injections, before administering capsaicin 2 days later. Auditory thresholds and OHC morphology were determined 24 h after trans-tympanic capsaicin administration. The effectiveness of STAT1 siRNA in reducing STAT1 expression in the cochlea was previously demonstrated in Figure 4. This was associated with a significant attenuation of ABR threshold shifts, compared to rats pretreated with scrambled siRNA before capsaicin administration (Fig. 7). Capsaicin consistently produced an ∼30 dB ABR threshold shift in rats pretreated with scrambled siRNA, but <5 dB threshold shifts after pretreatment with STAT1 siRNA at all frequencies tested. SEM images of the base of the cochlea indicated no significant damage or loss of OHCs after trans-tympanic administration of capsaicin in all treatment groups (see Fig. 1B). These results suggest that STAT1 mediates capsaicin-induced hearing loss in rats that was independent of obvious changes in OHC morphology.
FIG. 7.

Trans-tympanic delivery of STAT1 siRNA protects against hearing loss. Rats were anesthetized and then administered scrambled (scramble) STAT1 or NOX3 siRNAs. Two days later, these animals were administered vehicle or capsaicin and tested 24 h later. Capsaicin produced a ∼30 dB threshold shift at all frequencies examined, which was attenuated by trans-tympanic administration of STAT1 or NOX3 siRNAs. *Statistically significant difference from animals administered a scramble siRNA sequence; **statistically significant reduction in the ABR threshold shifts from capsaicin-24 h treated group (p < 0.05; n = 6).
Discussion
The data presented in this study implicate TRPV1 in activation of NOX3 and inflammatory processes leading to hearing loss. The data demonstrate that activation of TRPV1 promotes inflammation via STAT1, a regulator of inflammatory gene expression. As such, knockdown of STAT1 attenuated the deleterious effect of TRPV1 activation by reducing inflammation and protecting against hearing loss. These data support the contention that activation of TRPV1 initiates an ROS signal, which stimulates an inflammatory response in the cochlea, leading to temporary hearing loss.
The present results show that activation of TRPV1 promotes ROS generation via the NOX3 isoform of NADPH oxidase. NOX3 represents the primary source of ROS generation in the cochlea, which contributes to cisplatin-induced ROS production in the cochlea (2, 23). Its activation appears dependent on intracellular Ca2+ release that is provided via TRPV1 activation. It is likely that Ca2+ contributes to the activation of protein kinase C that phosphorylates p47phox, a critical step in its translocation to the membrane and activation of NADPH oxidase (7). One prominent role of ROS is the activation (29) and induction of TRPV1 (23), thereby contributing to a positive feedback loop to increase more ROS generation. An increase in ROS in the cochlea is associated with damage to lipid membranes and enzymes by lipid peroxidation (33). This information has served as the basis for using antioxidants to treat cisplatin ototoxicity (33). In this study, we highlighted a novel role of ROS, which involves phosphorylation and activation of STAT1. Such a pathway provides a link between TRPV1, STAT1 activation, and initiation of an inflammatory response. Accordingly, the inflammatory process initiated via TRPV1 involves the sequential induction of ROS, STAT1 activation, the increases in inflammatory cytokines, and the recruitment of inflammatory cells into the cochlea.
We show that capsaicin stimulates rapid Ser727 phosphorylation of STAT1 that followed the course of ROS generation, implicating ROS in this process. Immunocytochemical studies in the cochlea showing colocalization of TRPV1 and Ser727-phosphorylated STAT1 provide support for a role of TRPV1 in the activation of STAT1 in vivo. STAT1 promotes the transcription of genes regulating inflammation, such as COX-2 (45), iNOS (27), and TNF-α (40). STAT1 also promotes expression of genes linked to apoptosis, such as Fas, and TNF-related apoptosis inducing ligand and caspases (16). This latter observation could account for STAT1-dependent cell killing by cisplatin, which also involves p53 activation (42). In fact, cisplatin activation of STAT1 is dependent on p53 since it is not observed in p53-null cells (48). The mechanisms underlying STAT1 Ser727 phosphorylation by various stressors are only recently being defined. Bacterial liposaccharide-mediated phosphorylation of STAT1 Ser727 in macrophages involves p38 activation in macrophages (16). Calcium-calmodulin-dependent protein kinase II (18) and phosphatidyl inositol 3-kinase/Akt (24) are also linked to stress-induced phosphorylation of STAT1 Ser727. Besides STAT1 Ser727 phosphorylation, we observed increased STAT1701 phosphorylation, suggesting that capsaicin can mediate full activation of STAT1.
In addition to increased STAT1 phosphorylation, capsaicin also increased the levels of the STAT1 protein at 24 h after trans-tympanic capsaicin administration. Since the capsaicin-mediated induction of STAT1 was attenuated by STAT1 siRNA, the data suggest that STAT1 can positively regulate its own expression.
Our data show that STAT1 also mediates expression of inflammatory genes induced by capsaicin in the cochlea. These genes include COX-2, iNOS, and TNF-α. The induction of these genes was transient, as it was observed within 24 h after initiation of capsaicin treatment, but returned to basal levels within 72 h. We also show increased migration of CD14-positive immune cells into the cochlea after capsaicin administration. The accumulation of CD14 immunoreactivity was concentrated in the SVA and spiral ligament, the latter representing an area associated with inflammatory cytokine production (47). The induction of these genes was also dependent on NOX3, since knockdown of this enzyme by siRNA reduced their expression. These findings support the contention that activation of TRPV1 mediates inflammation through generation of ROS and activation of STAT1 and are consistent with an ROS-dependent immune activation into the cochlea (31).
Inflammation is now being viewed as an important component of hearing loss. The role of inflammation in the development of hearing loss was suggested by an early study showing that the administration of steroid (anti-inflammatory agent) protected against sensorineural hearing loss (13). Later studies have shown that the cochlea can mount an inflammatory response as a result of immune challenges or toxic insults. For example, proinflammatory cytokines, such as interleukin-1β and TNF-α, increase the expression of inflammatory mediators in cultured spiral ligament fibrocytes (47). Induction of an inflammatory response in the cochlea of mice was observed after antigen challenge to the inner ear (35). Recent studies have implicated inflammatory pathways in cisplatin-induced cell death. For example, cisplatin increased expression of inflammatory cytokines in UB/UE1 utricular epithelial cells and in the cristae ampullae and utricles isolated from mice (15). Cisplatin also mediates inflammation in the cochlea that is attenuated by flunarizine, an activator of NF-E2-related factor 2/heme oxygenase-1 (38). However, a detailed mechanism that links cisplatin-induced inflammation and death of OHCs in the inner ear has not been defined. A recent study has implicated STAT1 in cisplatin-mediated apoptosis of utricular cells in vitro (36). The current data show that activation of TRPV1 significantly increased expression of iNOS, COX-2, and TNF-α, genes mediating the inflammatory response (21), by activating STAT1. iNOS (44), COX-2 (9), and TNF-α (8) have been linked to various experimental models of hearing loss. Our previous data showing that cisplatin produces hearing loss through activation of TRPV1 and induction of ROS and the current findings that TRPV1, via ROS, activates STAT1 support a role of STAT1 in mediating cisplatin ototoxicity. Therefore, we predict that cisplatin-induced hearing loss is produced, at least in part, through activation of a downstream inflammatory process. As such, targeting STAT1 or its downstream inflammatory mediators for inhibition could represent a novel mechanism of treating hearing loss.
The data presented in this study show that, unlike cisplatin, capsaicin promotes temporary hearing loss. We show that capsaicin induces significant hearing loss at 24 h but that this resolved within 72 h. This was associated with transient STAT1 phosphorylation and expression of inflammatory genes. Significant elevations of these parameters were also observed 24 h after trans-tympanic injections of capsaicin, but these returned to baseline levels within 72 h. A significant finding is that the hearing loss induced by capsaicin was not associated with any obvious effects on OHC morphology. Unlike capsaicin, cisplatin produces permanent hearing loss, damage to OHCs, and prolonged activation of STAT1 (unpublished data). The mechanism underlying the anticancer therapeutic action of cisplatin involves alkylation of DNA, induction in the cell cycle regulator p53, and apoptosis (32). In addition, we have attributed cisplatin ototoxicity to the induction of ROS, activation of TRPV1, and induction of apoptosis (23). These latter findings imply that direct activation of TRPV1 by capsaicin should mimic cisplatin in producing ototoxicity. The differences in ototoxicity profiles produced by cisplatin and capsaicin may indicate that damage or loss of OHCs require a more sustained activation of STAT1 and induction of inflammation. We observed that capsaicin did not produce any change in the induction of p53, whereas cisplatin produced a more persistent induction of p53 (23). Further, unlike cisplatin, capsaicin did not induce expression of Bax. Overall, these results suggest that differences in STAT1 activation and the induction of p53 and Bax could explain the different ototoxicity profiles of capsaicin and cisplatin. The transient nature of capsaicin-induced hearing loss is akin to that produced by noise preconditioning and suggests that capsaicin could serve as a preconditioning stimulus against noise or drug-induced ototoxicity.
In summary, the data presented in this study indicate that STAT1 couples TRPV1 activation and ROS production to induction of inflammation in the cochlea. A model describing our concept of TRPV1-mediated inflammation is presented in Figure 8. STAT1-induced inflammatory processes were transient and are likely implicated in temporary hearing loss, as it is relieved after its knockdown by siRNA. Further, TRPV1-mediated temporary hearing loss was not associated with damage to OHCs, attesting to its transient nature. These data suggest that although activation of TRPV1 alone contributes to temporary hearing loss, it does not fully account for cisplatin ototoxicity. Either a more persistent activation of STAT1 and/or recruitment of the apoptotic pathway might be required for the manifestation of cisplatin ototoxicity. Overall, this study provides novel insight into the roles of TRPV1 in the cochlea, serving as a mediator of inflammation and temporary hearing loss.
FIG. 8.
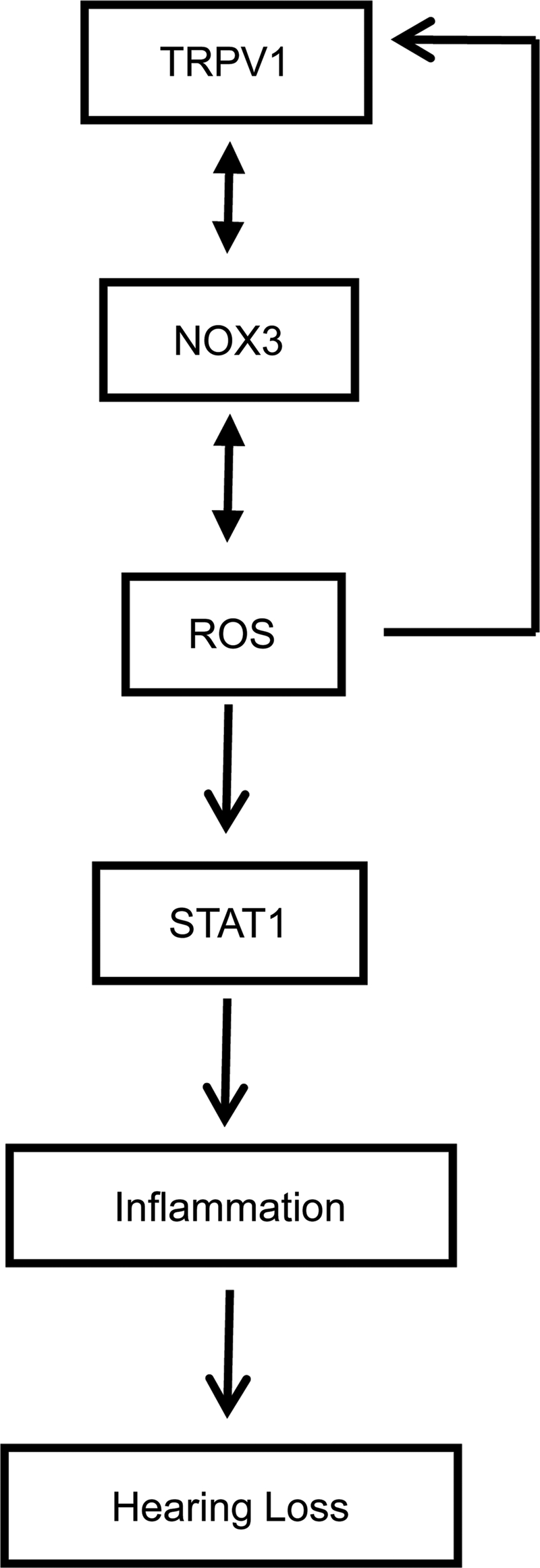
Model of capsaicin-induced inflammation in the cochlea. Capsaicin activates TRPV1, which allows entry of Ca2+ into the cell and increased ROS production via NOX3 activation of STAT1. ROS also produce positive feedback regulation of NOX3 and TRPV1 expression. STAT1 promotes induction of a number of proinflammatory genes that contribute to cochlear inflammation and hearing loss.
Supplementary Material
Abbreviations Used
- ABR
auditory brainstem response
- BAPTA-AM
1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis (acetoxymethyl ester)
- COX-2
cyclooxygenase 2
- DPI
diphenyleneiodonium
- H2DCFDA
2′,7′-dichlorodihydrofluorescein diacetate
- iNOS
inducible nitric oxide synthase
- OHC
outer hair cell
- ROS
reactive oxygen species
- RT-PCR
reverse transcriptase-polymerase chain reaction
- SEM
scanning electron microscopy
- SG
spiral ganglion
- siRNA
short interfering RNA
- STAT1
signal transducer and activator of transcription 1
- SVA
stria vascularis
- TNF-α
tumor necrosis factor-α
- TRPV1
transient receptor potential 1
Acknowledgments
This work was supported in part by a grant from the National Institutes of Health (DC02396) to L.P.R.; a grant from the National Organization for Hearing Research, SIU School of Medicine Excellence in Academic Medicine Award, and NIH grant (CA135494) to V.R.; and an NRSA F32 award (DC009950) to D.M.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Alawi K. Keeble J. The paradoxical role of the transient receptor potential vanilloid 1 receptor in inflammation. Pharmacol Ther. 2010;125:181–195. doi: 10.1016/j.pharmthera.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 2.Bánfi B. Malgrange B. Knisz J. Steger K. Dubois-Dauphin M. Krause K-H. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J Biol Chem. 2004;279:46065–46072. doi: 10.1074/jbc.M403046200. [DOI] [PubMed] [Google Scholar]
- 3.Bielefeld EC. Hu BH. Harris KC. Henderson D. Damage and threshold shift resulting from cochlear exposure to paraquat-generated superoxide. Hear Res. 2005;207:35–42. doi: 10.1016/j.heares.2005.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birder LA. Nakamura Y. Kiss S. Nealen ML. Barrick S. Kanai AJ. Wang E. Ruiz G. De Groat WC. Apodaca G. Watkins S. Caterina MJ. Altered urinary bladder function in mice lacking the vanilloid receptor TRPV1. Nat Neurosci. 2002;5:856–860. doi: 10.1038/nn902. [DOI] [PubMed] [Google Scholar]
- 5.Christoph T. Grünweller A. Mika J. Schäfer MK. Wade EJ. Weihe E. Erdmann VA. Frank R. Gillen C. Kurreck J. Silencing of vanilloid receptor TRPV1 by RNAi reduces neuropathic and visceral pain in vivo. Biochem Biophys Res Commun. 2006;350:238–243. doi: 10.1016/j.bbrc.2006.09.037. [DOI] [PubMed] [Google Scholar]
- 6.Davis JB. Gray J. Gunthorpe MJ. Hatcher JP. Davey PT. Overend P. Harries MH. Latcham J. Clapham C. Atkinson K. Hughes SA. Rance K. Grau E. Harper AJ. Pugh PL. Rogers DC. Bingham S. Randall A. Sheardown SA. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- 7.el Benna J. Faust LP. Babior BM. The phosphorylation of the respiratory burst oxidase component p47phox during neutrophil activation. Phosphorylation of sites recognized by protein kinase C and by proline-directed kinases. J Biol Chem. 1994;269:23431–23436. [PubMed] [Google Scholar]
- 8.Haake SM. Dinh CT. Chen S. Eshraghi AA. Van De Water TR. Dexamethasone protects auditory hair cells against TNFalpha-initiated apoptosis via activation of PI3K/Akt and NFκB signaling. Hear Res. 2009;255:22–32. doi: 10.1016/j.heares.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 9.Hoshino T. Tabuchi K. Hirose Y. Uemaetomari I. Murashita H. Tobita T. Hara A. The non-steroidal anti-inflammatory drugs protect mouse cochlea against acoustic injury. Tohoku J Exp Med. 2008;216:53–59. doi: 10.1620/tjem.216.53. [DOI] [PubMed] [Google Scholar]
- 10.Jajoo S. Mukherjea D. Pingle S. Sekino Y. Ramkumar V. Induction of adenosine A1 receptor expression by pertussis toxin via an ADP ribosylation independent pathway. J Pharmacol Exp Ther. 2006;317:1–10. doi: 10.1124/jpet.105.096255. [DOI] [PubMed] [Google Scholar]
- 11.Jajoo S. Mukherjea D. Watabe K. Ramkumar V. Adenosine A(3) receptor suppresses prostate cancer metastasis by inhibiting NADPH oxidase activity. Neoplasia. 2009;11:1132–1145. doi: 10.1593/neo.09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamimura T. Whitworth CA. Rybak LP. Effect of 4-methylthiobenzoic acid on cisplatin-induced ototoxicity in the rat. Hear Res. 1999;131:117–127. doi: 10.1016/s0378-5955(99)00017-9. [DOI] [PubMed] [Google Scholar]
- 13.Kanzaki J. Ouchi T. Steroid-responsive bilateral sensorineural hearing loss and immune complexes. Arch Otorhinolaryngol. 1981;230:5–9. doi: 10.1007/BF00665374. [DOI] [PubMed] [Google Scholar]
- 14.Kaygusuz I. Oztürk A. Ustündağ B. Yalçin S. Role of free oxygen radicals in noise-related hearing impairment. Hear Res. 2001;162:43–47. doi: 10.1016/s0378-5955(01)00365-3. [DOI] [PubMed] [Google Scholar]
- 15.Kim HJ. So HS. Lee JH. Park C. Lee JB. Youn MJ. Kim SJ. Yang SH. Lee KM. Kwon KB. Park BH. Park R. Role of proinflammatory cytokines in cisplatin-induced vestibular hair cell damage. Head Neck. 2008;30:1445–1456. doi: 10.1002/hed.20892. [DOI] [PubMed] [Google Scholar]
- 16.Kim HS. Lee MS. Essential role of STAT1 in caspase-independent cell death of activated macrophages through the p38 mitogen-activated protein kinase/STAT1/reactive oxygen species pathway. Mol Cell Biol. 2005;25:6821–6833. doi: 10.1128/MCB.25.15.6821-6833.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korver KD. Rybak LP. Whitworth C. Campbell KM. Round window application of D-methionine provides complete cisplatin otoprotection. Otolaryngol Head Neck Surg. 2002;126:683–689. doi: 10.1067/mhn.2002.125299. [DOI] [PubMed] [Google Scholar]
- 18.Lim WS. Timmins JM. Seimon TA. Sadler A. Kolodgie FD. Virmani R. Tabas I. Signal transducer and activator of transcription-1 is critical for apoptosis in macrophages subjected to endoplasmic reticulum stress in vitro and in advanced atherosclerotic lesions in vivo. Circulation. 2008;117:940–951. doi: 10.1161/CIRCULATIONAHA.107.711275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma W. Quirion R. Inflammatory mediators modulating the transient receptor potential vanilloid 1 receptor: therapeutic targets to treat inflammatory and neuropathic pain. Expert Opin Ther Targets. 2007;11:307–320. doi: 10.1517/14728222.11.3.307. [DOI] [PubMed] [Google Scholar]
- 20.McCabe BF. Autoimmune sensorineural hearing loss. Ann Otol Rhinol Laryngol. 2007;116:875–879. [PubMed] [Google Scholar]
- 21.Medzhitov R. Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. 2009;9:692–703. doi: 10.1038/nri2634. [DOI] [PubMed] [Google Scholar]
- 22.Mukherjea D. Jajoo S. Kaur T. Sheehan KE. Ramkumar V. Rybak LP. Transtympanic administration of short interfering (si)RNA for the NOX3 isoform of NADPH oxidase protects against cisplatin-induced hearing loss in the rat. Antioxid Redox Signal. 2010;13:589–598. doi: 10.1089/ars.2010.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mukherjea D. Jajoo S. Whitworth CA. Bunch JR. Turner JG. Rybak LP. Ramkumar V. Inhibition of cisplatin ototoxicity by short interfering (si)RNA against transient receptor potential vanilloid-1 (TRPV1) in the rat cochlea. J Neurosci. 2008;28:13056–13065. doi: 10.1523/JNEUROSCI.1307-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen H. Ramana CV. Bayes J. Stark GR. Role of phosphatidylinositol 3-kinase in interferon-γ dependent phosphorylation of STAT1 on serine 727 and activation of gene expression. J Biol Chem. 2001;276:33361–33368. doi: 10.1074/jbc.M105070200. [DOI] [PubMed] [Google Scholar]
- 25.Ohinata Y. Miller JM. Altschuler RA. Schacht J. Intense noise induces formation of vasoactive lipid peroxidation products in the cochlea. Brain Res. 2000;878:163–173. doi: 10.1016/s0006-8993(00)02733-5. [DOI] [PubMed] [Google Scholar]
- 26.Ohlemiller YY. Wright JS. Dugan LL. Early elevation of cochlear reactive oxygen species following noise exposure. Audiol Neurootol. 1999;4:229–236. doi: 10.1159/000013846. [DOI] [PubMed] [Google Scholar]
- 27.Ohmori Y. Hamilton TA. Requirement for STAT1 in LPS-induced gene expression in macrophages. J Leukoc Biol. 2001;69:598–604. [PubMed] [Google Scholar]
- 28.O'Shea JJ. Targeting the Jak/STAT pathway for immunosuppression. Ann Rheum Dis. 2005;63(Suppl 2):ii67–ii71. doi: 10.1136/ard.2004.028290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pabbidi RM. Cao DS. Parihar A. Pauza ME. Premkumar LS. Direct role of streptozotocin in inducing thermal hyperalgesia by enhanced expression of transient receptor potential vanilloid 1 in sensory neurons. Mol Pharmacol. 2008;73:995–1004. doi: 10.1124/mol.107.041707. [DOI] [PubMed] [Google Scholar]
- 30.Ramkumar V. Whitworth CA. Pingle SC. Hughes LF. Rybak LP. Noise induces A1 adenosine receptor expression in the chinchilla cochlea. Hear Res. 2004;188:47–56. doi: 10.1016/S0378-5955(03)00344-7. [DOI] [PubMed] [Google Scholar]
- 31.Riva C. Donadieu E. Magnan J. Lavieille JP. Age-related hearing loss in CD/1 mice is associated to ROS formation and HIF target proteins up-regulation in the cochlea. Exp Gerontol. 2007;42:327–336. doi: 10.1016/j.exger.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 32.Rybak LP. Mukherjea D. Jajoo S. Ramkumar V. Cisplatin ototoxicity and protection: clinical and experimental studies. Tohoku J Exp Med. 2009;219:177–186. doi: 10.1620/tjem.219.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rybak LP. Ramkumar V. Ototoxicity. Kidney Int. 2007;72:931–935. doi: 10.1038/sj.ki.5002434. [DOI] [PubMed] [Google Scholar]
- 34.Rybak LP. Whitworth CA. Mukherjea D. Ramkumar V. Mechanisms of cisplatin-induced ototoxicity and prevention. Hear Res. 2007;226:157–167. doi: 10.1016/j.heares.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 35.Satoh H. Firestein GS. Billings PB. Harris JP. Keithley EM. Proinflammatory cytokine expression in the endolymphatic sac during inner ear inflammation. J Assoc Res Otolaryngol. 2003;4:139–147. doi: 10.1007/s10162-002-3025-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmitt NC. Rubel EW. Nathanson NM. Cisplatin-induced hair cell death requires STAT1 and is attenuated by epigallocatechin gallate. J Neurosci. 2009;29:3843–3851. doi: 10.1523/JNEUROSCI.5842-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharif Naeini R. Witty MF. Séguéla P. Bourque CW. An N-terminal variant of Trpv1 channel is required for osmosensory transduction. Nat Neurosci. 2006;9:93–98. doi: 10.1038/nn1614. [DOI] [PubMed] [Google Scholar]
- 38.So H. Kim H. Kim Y. Kim E. Pae HO. Chung HT. Kim HJ. Kwon KB. Lee KM. Lee HY. Moon SK. Park R. Evidence that cisplatin-induced auditory damage is attenuated by downregulation of pro-inflammatory cytokines via Nrf2/HO-1. J Assoc Res Otolaryngol. 2008;9:290–306. doi: 10.1007/s10162-008-0126-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steiner AA. Turek VF. Almeida MC. Burmeister JJ. Oliveira DL. Roberts JL. Bannon AW. Norman MH. Louis JC. Treanor JJ. Gavva NR. Romanovsky AA. Nonthermal activation of transient receptor potential vanilloid-1 channels in abdominal viscera tonically inhibits autonomic cold-defense effectors. J Neurosci. 2007;27:7459–7468. doi: 10.1523/JNEUROSCI.1483-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sugawara I. Yamada H. Mizuno S. STAT1 knockout mice are highly susceptible to pulmonary mycobacterial infection. Tohoku J Exp Med. 2004;202:41–50. doi: 10.1620/tjem.202.41. [DOI] [PubMed] [Google Scholar]
- 41.Tanaka F. Whitworth CA. Rybak LP. Influence of pH on the ototoxicity of cisplatin: a round window application study. Hear Res. 2003;177:21–31. doi: 10.1016/s0378-5955(02)00771-2. [DOI] [PubMed] [Google Scholar]
- 42.Townsend PA. Scarabelli TM. Davidson SM. Knight RA. Latchman DS. Stephanou A. STAT-1 interacts with p53 to enhance DNA damage-induced apoptosis. J Biol Chem. 2004;279:5811–5820. doi: 10.1074/jbc.M302637200. [DOI] [PubMed] [Google Scholar]
- 43.Ulevitch RJ. Tobias PS. Receptor-dependent mechanism of cell stimulation by bacterial endotoxin. Annu Rev Immunol. 1995;13:437–457. doi: 10.1146/annurev.iy.13.040195.002253. [DOI] [PubMed] [Google Scholar]
- 44.Watanabe KI. Hess A. Bloch W. Michel O. Nitric oxide synthase inhibitor suppresses the ototoxic side effect of cisplatin in guinea pigs. Anticancer Drugs. 2000;11:401–406. doi: 10.1097/00001813-200006000-00011. [DOI] [PubMed] [Google Scholar]
- 45.Xuan YT. Guo Y. Zhu Y. Han H. Langenbach R. Dawn B. Bolli R. Mechanism of cyclooxygenase-2 upregulation in late preconditioning. J Mol Cell Cardiol. 2003;35:525–537. doi: 10.1016/s0022-2828(03)00076-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamashita D. Jiang HY. Schacht J. Miller JM. Delayed production of free radicals following noise exposure. Brain Res. 1995;1019:201–209. doi: 10.1016/j.brainres.2004.05.104. [DOI] [PubMed] [Google Scholar]
- 47.Yoshida K. Ichimiya I. Suzuki M. Mogi G. Effect of proinflammatory cytokines on cultured spiral ligament fibrocytes. Hear Res. 1999;137:155–159. doi: 10.1016/s0378-5955(99)00134-3. [DOI] [PubMed] [Google Scholar]
- 48.Youlyouz-Marfak I. Gachard N. Le Clorennec C. Najjar I. Baran-Marszak F. Reminieras L. May E. Bornkamm GW. Fagard R. Feuillard J. Identification of a novel p53-dependent activation pathway of STAT1 by antitumour genotoxic agents. Cell Death Differ. 2008;15:376–385. doi: 10.1038/sj.cdd.4402270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.