

Abstract
Several in vivo techniques have been developed to study and measure the uptake of CNS compounds into the brain. With these techniques, various parameters can be determined after drug administration, including the blood-to-brain influx constant (Kin), the permeability-surface area (PS) product, and the brain uptake index (BUI). These techniques have been mostly used for drugs that are expected to enter the brain via transmembrane diffusion or by carrier-mediated transcytosis. Drugs that have limitations in entering the brain via such pathways have been encapsulated in nanoparticles (based on lipids or synthetic polymers) to enhance brain uptake. Nanoparticles are different from CNS compounds in size, composition and uptake mechanisms. This has led to different methods and approaches to study brain uptake in vivo. Here we discuss the techniques generally used to measure nanoparticle uptake in addition to the techniques used for CNS compounds. Techniques include visualization methods, behavioral tests, and quantitative methods.
KEY WORDS: blood-brain barrier, brain targeting, CNS drugs, nanoparticles
INTRODUCTION
Essentially none of the large-molecule pharmaceutics (e.g. peptides, proteins and nucleic acids) can enter the brain, and over 98% of small-molecule drugs cannot enter the brain either (1). In the past few years, several methods to study brain uptake of drugs have been developed. To enhance brain uptake, nanoparticles have been used to target drugs to the brain. Nanoparticles are different from CNS compounds in size, composition and uptake mechanisms. This has led to different methods and approaches to study brain uptake in vivo. Here we discuss the techniques generally used to measure nanoparticle uptake in addition to the techniques used for CNS compounds.
Drug Transport at the Blood-Brain Barrier
Transport from the blood to the brain is limited by the blood-brain barrier (BBB). The BBB is formed by brain endothelial cells that line the cerebral microvessels. It is supported by other cell types surrounding the endothelium, such as astrocytes and pericytes (2). These surrounding cells contribute to the induction of many barrier characteristics of the endothelium, such as tight junctions, that closely join the endothelial cells together. Next to being a “physical barrier,” the BBB is also a “transport barrier.” This aspect is formed by specific transport proteins and transcytosis mechanisms that mediate the uptake and efflux of molecules. Third, a “metabolic barrier” is formed by the expression of metabolizing enzymes such as peptidases, cytochrome P450 enzymes, and monoamine oxidases (3–5). All of these barrier functions control and regulate both inward and outward transfer of molecules between blood and the brain.
There are several routes for the transport of molecules across the BBB (Fig. 1). Paracellular transport of hydrophilic molecules is highly restricted by the tight junctions present between brain endothelial cells.
Fig. 1.

Lipid-soluble molecules with molecular weights below 400 Da are able to cross by transcellular lipophilic diffusion, provided that they are not bound to plasma proteins to a high extent, or form a substrate for a transport system at the BBB. Based on physicochemical properties, such as molecular weight and hydrogen bonding, predictions can be made whether a compound can cross the BBB via this route (6,7).
For a variety of molecules that are essential for brain function, such as amino acids, glucose, peptides, and proteins, specific endogenous BBB transporters exist. These are expressed at both the luminal and the basolateral membranes of the endothelium (8). These transporters can be either defined as carriers or receptors.
Carriers are membrane-restricted systems. They are generally responsible for the transport of small molecules with a fixed size and mass smaller than 600 Da. Carrier-mediated transcytosis is used for the delivery of nutrients such as glucose, amino acids, and purine bases to the brain. It is substrate selective, and only drugs that closely mimic the endogenous carrier substrates will be taken up (9).
Endocytosis at the BBB is effectuated through adsorption or receptor binding. Adsorptive-mediated endocytosis is initiated by the binding of polycationic substances to negative charges on the plasma membrane (9). Receptor-mediated endocytosis is initiated by the binding of a receptor-specific ligand. Following adsorption or binding, the substance is internalized and transported via the early endosome to the lysosome, or transcytosed to the plasma membrane. The only way for larger molecules and particles such as antibodies, lipoproteins, proteins and nanoparticles to be transported into the brain is via receptor or adsorptive-mediated endocytosis (10), which is different from low-molecular-weight CNS drugs. When compared to the peripheral endothelium, the cerebral endothelium has a much lower endocytotic and transcytotic activity, making BBB passage of larger molecules difficult even when endocytosis is possible. In pathological conditions, the transport mechanism at the BBB might be up- or down-regulated (1).
Next to these influx systems, many efflux mechanisms exist at the BBB as well. These include P-glycoprotein, MDR-related protein, ABC transporters, and several others (1). They restrict entry of molecules into the brain by promoting luminal release of compounds and are important in removing harmful substances from the brain, thereby reducing toxic side effects of CNS drug metabolites. Substrates for efflux transporters include peptides, lipids, cholesterol, hormones, CNS drugs, and metabolites (11).
After in vivo administration, most CNS drugs will enter the brain in their free form via transcellular diffusion. However, many compounds with psychopharmacological activity do not posses the right physicochemical characteristics to be able to cross the BBB. One possible way to mask these characteristics is to package these compounds in nanoparticles. Nanoparticles are, of course, much larger and are only able to enter the brain endothelial cells via adsorptive or receptor-mediated endocytosis. Subsequent transcytosis to the basal side of the brain endothelial cells is required to enter into the brain parenchyma (2). After a drug or nanoparticle formulation has been administered, the concentration that can be measured in the brain depends on several factors, including the plasma concentration-time curve, the extent of plasma protein binding, the permeability across the BBB, the efflux out of the brain by efflux transporters, the metabolic conversion by enzymes, the binding to membranes or intracellular sites in the brain, and the continual secretion and drainage of cerebrospinal fluid (CSF) and brain interstitial fluid (ISF) (5,13).
Many processes are thus involved in uptake and processing of drugs or particles at the BBB. From a pharmaceutical point of view, it is interesting to know at what rate and to what extent uptake occurs. Here we discuss the different techniques that have been used to measure uptake of molecules versus methods to asses uptake of nanoparticles.
IN VIVO TECHNIQUES TO MEASURE COMPOUND PERMEATION INTO THE BRAIN
A number of in vivo techniques have been developed to measure the uptake of CNS drugs into the brain. These techniques are routinely performed in rats or mice. Most of these assays capture the unidirectional uptake phase of a drug, without assumptions about the fate of the drug after it has entered the brain (e.g. cellular binding, degradation, and efflux) (14). Two main parameters for the rate of brain penetration are often determined: Kin and PS product. Kin is the unidirectional influx constant from blood to brain. The PS product (alternatively also referred to as PA product) is the permeability-surface area product and is a measure of unidirectional clearance from blood to brain (15). It represents that volume of plasma which gives up its content of the particular solute to interstitial fluid per unit time (16). Both Kin and PS product are expressed in ml/min/g brain. They are most commonly determined after intravenous injection or after in situ perfusion of the compound. They can also be determined in a specific brain region. In addition to Kin and PS, other pharmacokinetic parameters can be determined, for example by intracerebral microdialysis. Finally, brain-specific parameters like the brain uptake index and the brain/plasma ratio can be determined. The methods to obtain these parameters are described below.
Kin and PS Product Determination by Intravenous Injection
The intravenous injection technique is regarded as the gold standard for brain uptake studies, because it involves fully physiological conditions (14,17). With this technique, a (radiolabeled) compound is injected intravenously. Blood is sampled at various time points. A single brain tissue sample can be obtained at the terminal time point. Kin can be obtained using the following equation:
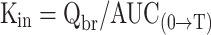 |
1 |
-
where
Kin is the unidirectional influx constant from blood to brain (ml/min/g brain), Qbr is the quantity of compound in the brain, without intravascular content (mass/g brain), and AUC(0→T) is the integral of plasma concentration from t = 0 to t = T.
Note that Qbr should represent the brain concentration without intravascular content (14). The higher the drug concentration at the terminal time point, the more this will contribute to the concentration that is measured for the total brain. One way to remove the intravascular content is by extensively flushing the brain with a (heparinized) buffer before the brain is taken out. Alternatively, the intravascular volume can be determined by co-administration of a vascular marker together with the drug. The marker is a substance that does not penetrate the blood-brain barrier, e.g. radiolabeled sucrose. When such a vascular marker has been included, Qbr can be calculated using the following equation:
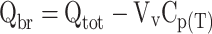 |
2 |
-
where
Qtot is the total quantity of compound in the brain, including vascular content (mass/g brain), Vv is the brain vascular volume (ml/g), and Cp(T) is the concentration of compound in the blood at time point T (mass/ml).
Kin and PS Product Determination by In Situ Perfusion
The in situ perfusion method complements the iv injection method. It was originally developed for rats, but it has been expanded for mice, guinea pigs, and rabbits (18–20). In the original rat method developed by Takasato et al. (21), the animal is anesthetised, and after a series of artery ligations, the perfusion fluid is infused up the common carotid artery (Fig. 2). Perfusion can be stopped at a predetermined time point. Similar to the iv injection method, the perfusion fluid remaining in the brain can be flushed out, or a vascular marker can be included in the fluid. The brain is taken out for analysis of the compound. Kin can be obtained using the following equation:
 |
3 |
-
where
Qbr is the quantity of compound in the brain, without intravascular content (mass/g), Cpf is the concentration of compound in the perfusion fluid (mass/ml), and T is the perfusion time.
Fig. 2.

Schematic representation of in situ brain perfusion. ACA anterior cerebral artery. MCA middle cerebral artery. PCA posterior cerebral artery. Takasato et al. Am J Physiol. 1984 (21). Am Physiol Soc, with permission.
If a vascular marker has been included, Qbr can be calculated similar to the iv injection technique, using the following equation:
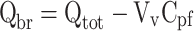 |
4 |
-
where
Qtot is the total quantity of compound in the brain, including vascular content (mass/g brain), Vv is the brain vascular volume (ml/g brain), and Cpf is the concentration of compound in the perfusion fluid (mass/ml).
The main advantages of the iv injection method are the ease of injection, the possibility to simultaneously measure pharmacokinetics, and the fully physiological conditions, enabling all transporters, junction proteins, and enzymes to be present at their physiological concentration. The main advantages of the in situ perfusion method are the ability to tailor the perfusion fluid, the constant infusion concentration, and the absence of compound metabolism in other organs (17,22). The Kin from both the iv injection and the in situ perfusion method can be converted into the cerebrovascular permeability-surface area product. From the PS product, permeability P can be calculated, given that the capillary surface area S is known. However, usually only the PS product is reported. PS calculation can be done using the Renkin-Crone equation:
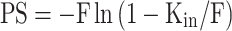 |
5 |
-
where
PS is the permeability-surface area product (ml/min/g brain), and F is the cerebral blood or perfusion flow rate (ml/min/g brain) (23).
Examples of PS product values obtained in vivo range from 0.0003 ml/min/g for sucrose, a compound that is considered BBB impermeable, to 1.2 ml/min/g for caffeine which has a high permeability (24).
Brain Uptake Index (BUI)
The brain uptake index (BUI) represents the relative uptake of a drug compared to a reference substance (25,26). The reference is freely diffusible across the BBB, such as 14C-butanol. The test compound is also radiolabeled, for example with 3H. A small volume of buffer containing both the test compound and the reference is rapidly injected into the common carotid artery of anesthetised animals (e.g. in the rat 0.2 ml in less than 0.5 s). The bolus passes through the brain in less than 2 s after injection. After 5–15 s, the brain is isolated, and the radioactivity in brain tissue and injected buffer is determined. The BUI can be calculated using the following equation:
 |
6 |
The BUI can be expressed as a percentage by multiplying it by 100. The BUI represents the net uptake of the drug normalized by the net uptake of the reference compound. It is therefore a direct function of the single-pass extraction (E) (27). If the extraction of the reference is known (for example 100% for butanol (28)), the extraction of the drug can be calculated:
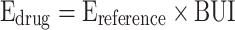 |
7 |
The BUI can be related to the PS product using the Renkin-Crone equation:
 |
8 |
The main advantage of the BUI technique is that it is fast, while its main disadvantage is the low sensitivity. Additionally, drugs that are taken up slowly cannot be studied with this method (14).
Examples of BUI values obtained in vivo are 1.4% for sucrose and 90% for caffeine (24).
Quantitative Autoradiography
Quantitative autoradiography can be used to determine the amount of radioactive test compound in specific regions of the brain, such as stroke-affected areas (29) or brain tumors (30), following oral, intravenous or subcutaneous administration to small animals. Blood is sampled at various time points, and the brain is taken out at the terminal time point. The brain is subsequently sectioned into 20-μm thick sections and exposed to X-ray film along with radioactive standards. Intravascular volume can be determined in a separate experiment using a BBB-impermeable marker, such as radiolabeled sucrose. Kin and PS product can be calculated with equations similar to those used for the intravenous injection method. The strength of quantitative autoradiography lies in the high spatial resolution in the micrometer range (14,31).
Microdialysis
Intracerebral microdialysis involves the implantation of a microdialysis probe in the brain. The probe, which consists of a semipermeable membrane, is continuously perfused with a physiological solution. The test drug is administered to the animal by the desired route (e.g. oral, intravenous or subcutaneous). Drugs that cross the BBB and enter the brain interstitial fluid can traverse the semipermeable membrane by diffusion into the physiological buffer. The buffer is sampled from the probe, and drug concentration is measured. The concentration in the sample reflects the concentration of free drug in the brain. The main advantage of microdialysis is that brain levels, as well as blood levels, of the drug can be determined at many time points in one animal. From these data, pharmacokinetic parameters can be obtained. Drawbacks include the technical difficulties of the implantation and the fact that highly lipophilic compounds are generally difficult to recover (32).
Brain/Plasma Ratio
Commonly used in the pharmaceutical industry is the brain/plasma ratio (33). The test drug is administered to the animal by the desired route. At a predetermined time point, the blood is sampled and the brain is taken out. The brain is homogenized and the drug concentration is determined in both brain and plasma. If multiple animals were used for multiple time points, the AUC of both the brain and plasma can be obtained. The brain concentration is then divided by the plasma concentration. This can be the ratio of one time point or the ratio of the AUCs (34). The ratio provides a measure of the extent of brain penetration, not of the rate of brain penetration. Usually, the presence of drug remaining in the brain vasculature is not taken into account.
External Detection Methods
The techniques described so far involve sampling from the brain. Next to these invasive techniques, several non-invasive external imaging techniques exist, including positron emission tomograpy (PET) and single photon emission computed tomograpy (SPECT). It has been shown that PET can be used to quantitatively measure the PS product (35). However, PET and SPECT are in general used for imaging of transporters, receptors, inflammation, or tumors in the brain, and not for the uptake of compounds (36–38).
More in-depth information on the advantages and disadvantages of each of the above-mentioned techniques can be found in references (14) and (39).
CNS COMPOUNDS VERSUS NANOPARTICLES
As the vast majority of potential CNS compounds have limited brain uptake, they may benefit from the use of advanced delivery systems in order to cross the BBB. Nanoparticles have been widely used as drug carriers to increase uptake of such drugs into the brain. The drug is encapsulated in or associated to the particle, thereby masking its physiochemical characteristics. Particles that have been used include liposomes, solid lipid nanoparticles, nanogels, dendrimers, albumin nanoparticles, and polymeric particles such as poly(lactic-co-glycolic acid) (PLGA) and poly(butyl cyanoacrylate) (PBCA) nanoparticles. In many cases, they are combined with targeting ligands on the particle surface to enhance uptake. Ligands can include peptides, proteins, and antibodies. An overview of recent studies that used nanoparticles to target to the brain in vivo is given in Table I.
Table I.
Methods and Partial Results of Several In Vivo Brain Targeting Studies of the Past Decade
| Species | Administration | vasc corr | Time points | Particle | Ligand | Particle size | Particle charge | Method | Data representation | Times increase | %ID brain | %ID blood | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Balb/c mice | iv jugular and femoral vein | none | 24, 48, 72 h | liposome | 8D3 Mab | – | – | β-gal plasmid staining, microscopy. | images | – | – | – | (44) |
| Rats | iv femoral vein | vasc wash | 0.25, 0.5, 1 h | liposome | RMP-7 | – | – | Evans blue-loaded lipo, abs 623 nm. | ug/100 mg | Non-targ vs targ 4.9 fold | Non-targ 0.002%. Targ 0.012% /braina | – | (45) |
| P18 rats | in situ perfusion and iv tail | vasc wash | perf 2, 5, 15 min. iv 2 h | liposome | OX26 | 150 nm | – | FM. | images | – | – | – | (46) |
| SD rats | iv femoral vein | vasc wash | 0.5 h | liposome | RMP-7 | 70 nm | – | 125I-NGF-loaded lipo. | ng/g PK data | Non-targ vs targ 3.19 fold | Non-targ 0.11% Targ 0.32% /braina | Non-targ 49% Targ 58% totala | (47) |
| Wistar rats | iv jugular vein | volume marker | 1 h | liposome | OX26 | 150 nm | – | 3H-Daunomycin-loaded lipo. | %ID/g PS product | Non-targ vs targ 2 fold | Non-targ 0.005% Targ 0.01% /gram | Both 0.9% /ml | (43) |
| SD rats | iv tail | vasc wash | RA 4, 24 h FM 4 h | liposome | – | 170 nm | –30 mV | Rats 3H lipid and 14C serotonin. Rabbits FM. | %(dpm/g tissue)/dose | Free vs lipo 2-fold | Free drug 0.068%. Lipo drug 0.138%. /gram | Both 1% /ml | (48) |
| Kunming mice | iv tail | none | 0.5, 1, 2 h | liposome | lactoferrin | 130 nm | Non-targ −32 mV Targ −4.3 mV | Coumarin-6-loaded lipo, HPLC. | ug/g PK data | Non-targ vs targ 2.3 fold | npc | PK data | (49) |
| Kunming mice | iv tail | none | 0.25, 0.5, 1, 2, 4, 8, 12, 24, 48, 72 h | SLN | – | 76 nm | – | Prodrug DO-FUdR-encapsulated NP, HPLC | ug/g PK data | Non-targ vs targ0.25 h 1.3fold | Free drug 2.08% SLN drug 2.66% /grama | Free 8.1% SLN 11.3% /mla | (50) |
| Balb/c mice Fischer-344 rats | Rats perfusion, mice iv tail. | vasc wash | Rats 45, 60, 90, 120 s. Mice 2, 6 h | SLN | thiamine | 67 nm | – | Rats 3H-hexadecanol and 3H-thiamine-loaded NP. Mice 111In-thiamine-coated. | mice %ID Rats PS, Kin | no increase | 2 h and 6 h both 0.5% /brain | Non-targ 65% Targ 80% /total | (42) |
| Wistar rats | iv | none | 0.5, 2, 4, 6, 24 h | BSA NP | transferrin | 120 nm | Non-targ −15 mV Targ 8.5 mV | Compound AZT-loaded NP, HPLC. | %ID PK data | Non-targ vs targ 2.3 fold | Non-targ 9.3% Targ 21.1% /brain | Both 25% /totala | (51) |
| ICR mice | iv | n.a. | 15, 30, 45, 60, 90 120 min | HSA NP | ApoE or tween80 | 340 nm | – | Loperamide-loaded NP, tail-flick test. | % MPE | – | – | – | (52) |
| SV129 mice | iv jugular | vasc wash | 15, 30 min | HSA NP | ApoE | 200 to 250 nm | – | EM. | images | – | – | (53) | |
| ICR mice | iv tail | n.a. | 15, 30, 45, 60, 120, 180, 210 min | HSA NP | OX26, R17217 Mab, Tf | 170 nm | −30 mV | Loperamide-loaded NP, tail-flick test. | % MPE | – | – | – | (54) |
| Nude mice | iv tail | none | 0.5, 1, 2, 4, 8, 24, 72, 168 h | PMMA NP | poloxamer 407 or 908 or tween80 | 107 nm | – | 14C-labeled polymer chain. | ng/mg brain PK data | Non-targ vs targ 11 fold | Non-targ 0.088% Targ 0.99% /grama | at 30 min targ 60%. /ml | (55) |
| Balb/c mice | iv tail | none | biodistr 2 h. vivo imag 120 min. pEGFP 48 h. | PAMAMNP | Angiopep-2 | – | – | 125I-labeled PAMAM. pEGFP-loaded NP, FM. In vivo imaging | % ID/g images | Non-targ vs targ 8.4 fold | Non-targ 0.03% Targ 0.25% /gram | – | (56) |
| Nude mice balb/c mice | iv tail | none | TEM 1 h. vivo im 4 h. DNA FM 2d | PAMAM NP | lactoferrin | – | – | GFP-DNA-loaded NP, FM, EM, in vivo imaging. | images | – | – | – | (57) |
| FVB wild type mice | iv tail | none | 1 h | Nanogel | – | 64 to 94 nm | 1.9 to 2.3 mV | 3H-Labeled ODN and 3H labeled nanogel. | % ID/g | Free vs nanogel 15 fold | 2.67% /gram | Nanogel 2.81% /gram | (58) |
| Wistar rats | carotid artery perfusion. Also iv femoral | vasc wash | perfu 3–4 min. iv 1 h | PLGA NP | synthetic opioid peptides | 200 nm | −10 mV | PLGA conjugated rhodamine and fluorescine, FM. | images | – | – | – | (59) |
| Wistar rats | iv tail | none | 15, 30, 45, 60, 90, 120, 240, 360, 420 min | PLGA NP | g7 opoid derived peptide | 150 nm | −17 mV | Loperamide-loaded and rhodamine-labeled NP, hot plate test, FM. | %MPE images | – | – | – | (60) |
| Wild type mice | iv tail | vasc wash | 1, 3, 6 h 1, 7, 14, 28d | PLGA NP | TAT | 320 nm | Non-targ −19 mV Targ 2.4 mV | 3H-ritonavir-loaded NP. FM | ug/g | Non-targ vs targ 6.5 fold | Non-targ 1.08% Targ 7.1% /grama | at 1 h both 1% and remain low /mla | (61) |
| Kunming mice | iv tail | vasc wash | 0.25, 0.5, 1, 2, 4, 8, 12, 24 h | PLGA NP | catBSA | 150 nm | Non-targ −22 mV Targ 11 mV | 6-coumarin-labeled NP, HPLC. FM. | ng/g PK data images | Non-targ vs targ 2.3 fold | npc | PK data | (62) |
| SD rats | iv jugular, carotid, tail, | vasc wash | 1 h | PLGA NP | – | 290 nm | −25 mV | Superoxide dismutase-loaded NP. 6-coumarin-loaded NP, HPLC. | % ROS damage | – | Non-targ NP carot 1.8% jugul 0.13% tail 0.11% /brain | – | (63) |
| Albino rats | iv tail | none | 0.25, 0.5, 1, 1.5, 4, 5, 24 h | PLGA NP | g7 opoid derived peptide | 160 nm | −17 mV | Rhodamine-labeled NP, HPLC. Loperamide-loaded NP, hot plate test. | ug/g (text also %ID/g) | npc | Non-targ low % Targ 15% /gram | 0.25 h: Non-targ undetec Targ ~6% /grama | (64) |
| Wistar rats | iv tail | none | 1, 1.5, 3, 5, 24 h | PLGA NP | g7 opoid derived peptide | 180 nm | −18 mV | Rhodamine-labeled NP, HPLC. Loperamide-loaded NP, hot plate test. FM. | %MPE %ID/g | npc | Non-targ low % Targ 14.3% /gram | – | (65) |
| LiCl-pre-treated Wistar rats | ip | vasc wash | 10, 20, 60 min | PLA NP | – | 250 nm | – | Tacrine-loaded NP. Histological brain damage. | ECoG profiles. Nr of animals with brain damage | – | – | – | (66) |
| ICR mice | iv tail | n.a. | 15, 45, 90, 120, 180 min | PBCA NP | apo AII, B, CII, E, or J | 300 nm | – | Dalargin or loperamide-loaded NP, tail-flick test | % MPE | – | – | – | (67) |
| SD rats | iv tail | vasc wash | 90 min | PBCA NP | – | 145 nm | −5 mV | PMI fluo dye incorporated NP, FM. | images | dependent on conc | – | – | (68) |
| C57Bl/6 mice | iv | none | 15, 45, 90 min, 24 h | PBCA NP | tween80 | 250 nm | – | NGF-loaded NP, NGF ELISA kit and behavior. | pg/mg | Non-targ vs targ 3 fold | Non-targ 8% Targ 24% /braina | – | (69) |
| Balb/c mice | iv tail | none | 2, 5, 15, 30, 60, 120, 240 min | PBCA NP | tween80 | 50 nm | – | 125I-clioquinol-loaded NP, behaviour. In vivo imaging. | %ID/g images | Free vs NP 2.3 fold | Free drug 1% NP drug 2.3% /gram | Free 9.2% NP 12% /gram | (70) |
Underlined Time Points Indicate Time Points of Best Results and of %ID in Table, Unless Indicated Otherwise.
NP nanoparticle. SLN solid lipid nanoparticle. Lipo liposome. FM fluorescence microscopy. EM electron microscopy. Vasc wash vascular washout. n.a. not applicable. npc not possible to calculate by the authors of this review. Non-targ non-targeted particles. Targ targeted particle.
apercentage was calculated by the authors of this review from the data in the referenced article, these values represent estimations
The brain uptake methods described above are excellent methods to determine the brain uptake of compounds that are expected to be brain permeable and are taken up by transcellular diffusion, or for small molecules that will be taken up by carrier-mediated transcytosis (9). However, brain uptake of nanoparticles occurs differently from such small molecules. For most nanoparticles, it has been demonstrated that brain uptake is initiated by adsorptive or receptor-mediated endocytosis into brain endothelial cells. This process is considerably slower than drug permeation and therefore less compatible with some of the common methods described above, like the BUI technique (14).
Of all distribution and kinetic parameters that can be obtained after drug administration, the PS product is in many reviews referred to as the best measure of BBB permeability (5,39). The PS product has been determined for many substances, including potential CNS compounds (19), neurotrophic factors (40), and amino acids (41). However, for nanoparticle formulations, this parameter is not commonly used. Unlike the BUI method that measures permeability after seconds, the iv injection method to measure the PS product allows time points to be selected according the researcher’s own desire, making the PS product compatible with nanoparticles, or any other substance for that matter. The reason why this ‘gold standard’ is not used often in BBB nanoparticle studies may be because the parameter has been developed for drug molecules. When the PS product of a drug has been determined, this is often compared to a brain-impermeable compound (e.g. sucrose), to a completely permeable compound (e.g. butanol), or to another existing drug. In this way, permeabilities of different drugs can be ranked. When studies are performed using nanoparticles, uptake is usually not compared to other drugs. The goal will, rather, be to compare free versus particle-formulated drug or to compare targeted versus non-targeted particles, regardless of any encapsulated drug. Only 2 out of 29 studies shown in Table I included the PS product in their in vivo experiments, either the PS product of the nanoparticle itself (42) or the PS product of a loaded drug (43). The other studies use alternative means of expressing particle uptake into the brain.
IN VIVO TECHNIQUES TO MEASURE NANOPARTICLE UPTAKE INTO THE BRAIN
The design of brain uptake studies of free drugs is generally different from the design of brain uptake studies of nanoparticles, because the size, physicochemical properties, and uptake mechanisms are different. These differences result in different approaches. The methods that have been used for brain targeting studies using nanoparticles in the past decade are described here.
Visualization Methods
Microscopy is the most widely used qualitative method to study BBB uptake of nanoparticles in vivo. Fluorescence microscopy is often used for its sensitivity. It requires loading of the nanoparticles with a fluorescent dye (62) or covalent coupling of the particle building blocks to a dye (59). Commonly used fluorescent dyes include rhodamine-123, fluorescein, and 6-coumarin. Particles are usually injected iv, and after sectioning the brain, they can be localized using fluorescence microscopy. The brain endothelium can be visualized by using an endothelium staining marker such as lectin. If such a marker is included in the staining procedure, a distinction can be made between particles that are associated with the brain endothelium, and particles that have crossed into the brain parenchyma (68).
If gene delivery to the brain is of interest, a plasmid expressing a fluorescent protein can be incorporated into the particle, and gene expression can be assessed by visualizing protein fluorescence in the brain sections (57).
Next to fluorescence microscopy, electron microscopy is commonly used. With this technique, single particles can be visualized in specific regions of the brain. For example, Zensi et al. (53) showed that human serum albumin nanoparticles could be detected by electron microscopy in murine brain sections after iv administration. In the electron microscopic pictures, a distinction can be seen between particles in the capillary lumen, particles within the endothelial cells (Fig. 3), and particles that have been taken up into neuronal cells.
Fig. 3.

Micrographs of the cortex region of mice after injection of Apo E-modified HSA nanoparticles. Fifteen min after intravenous injection, the nanoparticles (dark spheres indicated by arrows) could be seen inside the endothelial cells. Figure 3a shows a cross-section of a blood vessel with two nanoparticles in the endothelial cell and one nanoparticle still in the lumen of the vessel. Figure 3b shows a higher magnification of the same micrograph. Reprinted from Journal of Controlled Release (53). Copyright (2009), with permission from Elsevier.
Besides microscopic section imaging, in vivo fluorescence imaging can be used to visualize uptake of particles into the brain (56,57). In vivo fluorescence imaging involves labeling of the particles with a fluorophore. A sensitive camera detects the fluorescence emission in the whole body of a living small animal. Fluorescent labels that absorb in the near-infrared region are generally preferred, because they limit tissue autofluorescence (71,72).
Fluorescence intensity in the brain area can be compared between different nanoparticle formulations to determine which formulation shows the highest brain accumulation (73). Although in vivo imaging is a rapidly progressing field, detection is at present not sensitive enough to visualize the particles within the microenvironment of the brain, or to provide quantitative information on the usually low concentrations that have accumulated in the brain.
Behavioral Tests
Some drugs that act on the CNS are unique compared to drugs that act on any other organ, because of their ability to interfere with brain signaling and to induce specific behavioral effects. This gives rise to the possibility of not only detecting the level of compound that reaches the brain, but also to determine whether the compound has a pharmacodynamic effect. Drugs that normally do not enter the brain can be encapsulated into nanoparticles to be transported over the BBB. The effect of the drug in the brain can be read out by monitoring the behavior of the animal with specifically designed behavioral tests.
These tests do not register the whole particle inside the brain, but only the free drug that has been released from the particle, as only the free drug will be able to exert an effect. Drug release from nanoparticles at the site of interest is crucial; therefore, behavioral tests are of great value.
Nociceptive Tests
A common model drug that has been used in combination with nanoparticles is the opiate antagonist loperamide. Intracerebral administration of loperamide causes antinociceptive effects. However, after iv injection of loperamide, these effects are not seen. This is due to the efflux membrane transporter P-glycoprotein (74). Loperamide molecules that reach the endothelial cells of the blood-brain barrier are rapidly transported back to the blood circulation. Therefore, loperamide is not able to enter into the brain parenchyma, making it a model drug for nociceptive testing. It can be loaded inside nanoparticles (60) or simply adsorbed onto the surface of the particles (52). Loperamide-containing particles that are successfully taken up by the brain can release their drug content in the brain, causing an analgesic effect of loperamide after iv administration (54). Similar to loperamide, the hexapeptide dalargin has been used for this purpose (67).
To measure nociception, two basic tests have been developed for the use in rats and mice: the tail flick test and the hot plate test. These tests are convenient, because they apply to healthy wild-type animals, requiring no disease induction or transgenic animals. Both tests measure the response to a painful thermal stimulus. In the tail flick test, a light beam is used to apply heat to a small area of the tail. Latency (time) to flick the tail out of the path of the light beam is measured. In the hot plate test, the animal is placed on the surface of a hot plate. Latency to lick the forepaws or to lift one of the hindpaws is recorded. In both tests, latency is usually determined before administration of the drug to determine the background response, and after administration of the drug at one or several time points. To avoid damage if the animal does not respond, a cut-off time is predetermined, typically between 10 and 60 s (54,60,75). This time point is regarded as the maximal possible effect (MPE) (76). The experimental response latencies are usually expressed as percent maximal possible effect (%MPE), which can be calculated using the following formula:
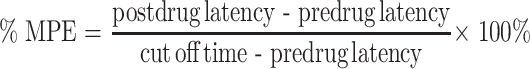 |
9 |
It should be noted that these tests can only be performed with model (analgesic) compounds. The characteristics of other drugs may be different from the tested analgesic. If another drug is used in combination with the nanoparticles, the encapsulation, formulation, and subsequent brain uptake may be different from the model compound.
Motor Function and Learning and Memory Tests
Other models that allow for behavioral testing often include induction of disease states in animals, like MPTP-induced Parkinson syndrome. Drugs that treat these syndromes, but normally do not penetrate the BBB in their free form, are candidates for nanoparticle formulation to enhance brain uptake. An example is nerve growth factor (NGF) (47). NGF has been evaluated for the treatment of neurological diseases such as Alzheimer’s disease and Parkinson’s disease. However, NGF has a low permeability through the BBB following intravenous administration (40). Kurakhmaeva et al. (69) tested MPTP-treated animals for improvement in symptoms of parkinsonism after iv administration of a NGF nanoparticle formulation. The NGF PBCA nanoparticles lead to a significant reduction in symptoms, including rigidity, tremor, locomotor activity, and orientation-research reaction.
Finally, transgenic mice like the APP/PS1 Alzheimer’s disease mice can be used to determine behavioral skills (77). The Cu/Zn-selective metal chelator clioquinol (CQ), which has a poor BBB penetration, has been encapsulated into PBCA nanoparticles by Kulkarni et al. to improve brain uptake (70). The transgenic mice can be tested with the Y-maze test for improvement in cognitive behavioral skills. Furthermore, brains of Alzheimer’s disease mice can be histologically stained for decrease in amyloid plaques.
Quantitative Methods
Quantitative methods to study BBB nanoparticle uptake in vivo involve determination of pharmacokinetic (PK) parameters and biodistribution studies. For both of these studies, either the particle itself or an entrapped compound is labeled. Most commonly, radioisotopes like 3H, 14C or 125I are used. Alternatively, the entrapped compound is quantified by HPLC.
Pharmacokinetic Parameters
In most brain targeting studies, the nanoparticles are administered intravenously. The goal of a pharmacokinetic study is to assess the fraction of the administered dose that is distributed to the brain or is excreted from the body (78). Nanoparticle and/or drug concentrations are measured in the blood (plasma) and in the brain at several time points after administration. From these measurements, the area under the concentration-time curve (AUC) can be obtained for both blood and brain. Pharmacokinetic models quantify the rate and extent of the distribution by mathematically analyzing these data. In this way, a number of PK parameters can be obtained. Commonly, several of these parameters are determined for either blood (51,55), brain or both (49,50). Other organs can also be included in the PK study (62). In current brain targeting studies, the most widely determined PK parameters in both blood and brain are Cmax, peak concentration; Tmax, time to reach peak concentration; AUC, area under the concentration-time curve, from the time of injection (t = 0) to a determined time point t (AUC(0→t)) or extrapolated to infinity  ; Half-life (t1/2), time it takes for 50% to be eliminated; and Mean Residence Time (MRT), the average time a compound remains in the blood or brain.
; Half-life (t1/2), time it takes for 50% to be eliminated; and Mean Residence Time (MRT), the average time a compound remains in the blood or brain.
Biodistribution Studies
For biodistribution studies, the nanoparticles are usually administered intravenously. After administration, the animals are sacrificed at a specific time point, or preferably at several time points. The brain is taken out, along with some of the major organs where the particles are likely to end up, including liver and spleen. If a β-emitting label is used, organs can be dissolved, and radioactivity can be determined by liquid scintillation counting. If a γ-emitting label is used, organs can be directly counted in a γ counter. If the compound itself is to be detected, a tissue extraction can be performed, followed by HPLC determination of the concentration. The amount of drug that was found in the brain and other organs can be represented as mass/organ or mass/gram. In this way, the increase in brain uptake compared to the control sample is obtained. Along with the experimental samples, the injection sample, as it was administered to the animals, can be assayed for drug content using the same analytical method, so that the amount of drug taken up into the brain can be expressed as a percentage of the injected dose (%ID). For example, Ke et al. (56) performed a biodistribution study in mice using dendrimers, which were either unmodified or modified with an angiopep-2 targeting ligand. They found 0.03%ID in the brain for the unmodified dendrimers and 0.25%ID for the angiopep-2 modified dendrimers. This shows the importance of targeting ligands in enhancing brain uptake of nanoparticles. The absolute increase owing to the targeting ligand (over 8 times more) can be exactly assessed by such a biodistribution study.
DISCUSSION
Each method to determine brain uptake of compounds or nanoparticles has advantages and disadvantages. In this section we discuss the possibilities and limitations of the methods.
Percentage of Maximal Possible Effect
As described above, in nociceptive studies the outcomes are usually represented as time to respond, commonly expressed as the percentage of maximal possible effect (%MPE). This parameter tells whether the encapsulated drug has an effect in the brain, but it provides no information on the amount of drug that was taken up. Therefore, it is difficult to compare different formulations or studies. For example, it cannot be concluded that a formulation resulting in 20% MPE, has a two-fold higher brain uptake of the drug than a formulation resulting in 10% MPE. Moreover, formulations that are successfully targeted to the brain will often reach 100% MPE (52,54). This low dynamic range makes it difficult to distinguish between various formulations. In general, the %MPE will provide an indication whether a formulation works to a higher, moderate, or insignificant extent, rather than provide exact drug uptake values.
Percentage of Injected Dose
When a biodistribution study has been performed, presenting the mass of drugs or particles in the brain is frequently preferred over presenting the %ID (see Table I), although it usually provides less information on the extent of uptake into the brain. The reason for this preference is not always clear. It may lie in the fact that the percentages that are found in the brain are generally low, especially compared to other organs. While targeting to the liver can result in 75%ID in the liver (79), and targeting to a tumor can produce values around 10% ID/gram tumor (80,81), percentages that are found in the brain are often below 0.5% and can be as low as 0.01% ID/g brain (43). Although low, uptake may nonetheless be manifold higher compared to an impermeable control. If a low %ID is found, it raises the question what %ID is actually needed to produce an effect. Although this depends on the drug that is eventually going to be used in combination with the nanoparticle for therapeutic purposes, the percentage needed may not be that high. In general, low concentrations of drugs are sufficient in the brain to result in a therapeutic effect. For example, only about 0.02% of a peripherally administered dose of morphine enters the brain, but that is sufficient to produce analgesia. For most CNS therapeutics on the market, less than 0.2% of the peripheral dose is taken up into brain (82). When transferrin is used as a targeting ligand, brain uptake in vivo is limited due to high endogenous concentrations of transferrin in the blood (9). Nevertheless, loperamide-loaded nanoparticles coupled to a transferrin targeting ligand are able to produce significant analgesia compared to non-targeted nanoparticles (54), demonstrating that low levels of uptake in the brain can be sufficient to produce adequate therapeutic effects.
In general, the percentages of the injected dose found in the brain after targeting of nanoparticles are highly variable. As shown in Table I, the %ID generally ranges from 0.01% to 0.5% in case an intravascular content correction (see below) was applied (42,43,45,47,48). In case no intravascular content correction was applied, recorded percentages can be over 15%ID, of which the vascular contribution is unclear (51,64,65,69).
If the %ID is plotted, notion should be taken whether this is %ID/organ or %ID/gram. This also holds for mass/organ or mass/gram. For example, a 20–25 g Balb/c mouse has an average brain weight of about 0.4 g. If a brain dose of 1% ID/g is recorded, the actual percentage of the injected dose in the brain is 0.4%. Therefore, it makes little sense to provide the %ID/gram. In the adult rat, however, the average brain weight is above 1 g, making this expression way more realistic.
Intravascular Content
As mentioned before, particles that are still present in the circulation at the time of sacrifice can contribute to a significant degree to the amount of drug measured in whole brain homogenate. Two methods to correct for intravascular content exist. They are brain perfusion with a buffer and inclusion of a vascular marker.
If administration of the nanoparticles is performed by in situ perfusion, which is usually done under anesthesia, buffer perfusion can be performed directly after infusion of the nanoparticle solution. If the particle administration is performed by iv injection, perfusion can be performed after subsequent anesthesia or directly after sacrifice. In that case, it is recommended to include an anti-coagulant, such as heparin, in the buffer to prevent perfusion obstruction by coagulated blood in the microvasculature.
The vascular washout fluid is usually administered either through the jugular vein or transcardially through the left ventricle of the heart. It should be noted that administration through the heart will result in most of the perfusion fluid distributing to the rest of the body instead of to the brain, if no vessels are clamped. Therefore, in most cases, the aorta is clamped, causing more of the fluid to reach the carotid arteries. Even so, only 10% of the fluid that reaches a common carotid artery reaches the brain if no additional ligations are placed on the external carotid artery and the pterygopalatine artery (25). This should be taken into account when performing a vascular washout, and a sufficient volume should be perfused to ensure that brain vasculature is washed completely. In general, when in situ perfusion is performed with external carotid artery and pterygopalatine artery ligated, perfusion of 1 to 5 ml buffer is considered sufficient (22). When transcardiac perfusion is performed with the descending aorta clamped, 20 ml of buffer is commonly used, as is shown in Table II. Outflow of the perfused fluid can be realized by severing the jugular veins (83,84) or cardiac ventricles (22). Optionally, outflow can be monitored to ensure thorough washout (45,47).
Table II.
Vascular Washout Times and Volumes That Have Been Used in Mice and Rats to Clear the Brain from Intravascular Content
| Species | Nanoparticle or drug administration | Washout administration | Clamps or ligations | Washout volume | Washout time | Ref |
|---|---|---|---|---|---|---|
| Rats | common carotid artery | common carotid artery | common carotid and external carotid arteries | 1–5 ml | 10–15 sec | (22) |
| Rats | femoral vein | jugular arteries | none | 15–20 ml | 30 min | (47) |
| Rats | common carotid artery | common carotid artery | common carotid, external carotid, and occipital arteries | 20–30 ml | 120–180 sec | (59) |
| Rats | femoral vein | carotid artery | carotid artery | 30–35 ml | 60 min | (45) |
| Rats | tail vein | heart | none | 50 ml | 10 min | (48) |
| Mice | common carotid artery | common carotid artery | common carotid artery | 1.15 ml | 60 sec | (85) |
| Mice | tail vein | heart | none | 2 ml | 2 min | (61) |
| Mice | jugular vein | heart | descending aorta | 20 ml | 30 sec | (83) |
| Mice | heart | heart | descending aorta | 20 ml | <1 min | (86) |
| Mice | jugular vein | heart | descending aorta | 20 ml | 1 min | (87) |
| Mice | jugular vein | heart | descending aorta | 20 ml | 1–2 min | (84) |
| Mice | jugular vein | jugular vein | none | 50 ml | – | (53) |
The second intravascular content correction method is co-administration of a vascular marker. The marker is a substance that does not penetrate the blood-brain barrier, e.g. radiolabeled sucrose. This allows for the determination of the total vascular volume in the brain. As described above (Eqs. 2 and 4), brain vascular blood concentration can be subtracted from the total brain concentration, yielding the amount of particles that is associated with the brain endothelial cells and brain parenchyma. The brain vascular volumes of mice and rats have been determined in many studies, as shown in Table III. Typically, this volume ranges between 11 and 14 μl/gram brain and is on average about 12 μl/gram for both mice and rats.
Table III.
Brain Vascular Volumes That Have Been Found in Mice and Rats After Administration of a Vascular Marker
| Species | Method | Vascular volume (μl/gram brain) | Ref |
|---|---|---|---|
| Rats (200–400 g) | Femoral vein injection of [14C]sucrose | 12.5 ± 0.7 | (88) |
| 12.9 ± 1.6 | |||
| 12.2 ± 0.8 | |||
| Rats (220–330 g) | In situ perfusion of [14C]sucrose | 12 ± 1.9 | (23) |
| Rats (220–330 g) | In situ perfusion of [14C]sucrose | 13.3 ± 3.4 | (89) |
| Rats | In situ perfusion of [14C]sucrose | 12 ± 2 | (90) |
| Mice (20–25 g) | In situ perfusion of [3H]inulin | 11.4 ± 0.4 | (19) |
| Mice (20–25 g) | In situ perfusion of [14C]sucrose | 11–14 | (91) |
| Mice (8–22 weeks) | In situ perfusion of [14C]sucrose | 11.9 ± 2.6 | (92) |
| 12.8 ± 1.2 |
Correction for vascular content is in many studies not applied. If the concentration of drugs or particles in the blood is sufficiently low, this may not pose a problem. Otherwise, the quantitative amount or %ID found in the brain is likely to be overestimated. To gain insight in the blood background, it is necessary to measure the blood concentration at the time of sacrifice. Due to clearance, the concentration of circulating particles is generally highest at the first time point measured and will decrease at later time points. If no vascular content correction is applied, the concentration measured in the brain is likely to be highest at the first time point and to decrease as well, due to the blood background. In this case, the brain:blood ratio can be provided in addition to the amount or %ID in the brain (70). If this ratio remains constant over the measured time points, it cannot be concluded that brain uptake was highest at the earliest time point.
Similarly, this holds for experiments in which comparable types of nanoparticles were included, e.g. targeted and non-targeted particles. The concentration in the brain as well as in the blood can be measured for both particle types. The increase in the brain:blood ratio reflects the increase of brain binding/uptake of the targeted particles compared to the non-targeted particles. In this way, contribution of the targeting ligand to the brain uptake of the particle can be seen, although the quantitative amount found in the brain may be overestimated. In most cases the same type of particle is used for both targeted and non-targeted samples. Therefore, circulation times of both particles are often comparable.
As the brain vascular volume for mice and rats is usually about 12 μl/gram brain, this provides an estimate to subtract the brain vascular concentration from the total brain concentration. Of course, blood concentration at the terminal time point needs to be measured in order to accomplish this. Although not common, this approach can be used (87,90).
Capillary Depletion
If the brain is taken out for analysis, the nanoparticle content that is not present within the blood of the brain microvasculature may be bound to the endothelium, be present within the endothelial cells or be transcytosed to the brain parenchyma (39). A distinction can be made between endothelial-associated particles and particles that have transcytosed into the brain parenchyma by performing a capillary depletion (93,94). This technique involves homogenization of the brain, followed by dextran density centrifugation to deplete the homogenate of its vasculature. The nanoparticles can be detected in both the parenchyma and the capillary fraction. It should be noted that particles which are bound to the surface of the capillaries with low affinity may disassociate from the capillaries during centrifugation, and therefore end up in the parenchyma fraction. This may be prevented when the brain vasculature is flushed before the brain is taken out to remove the low affinity bound particles. Particles bound with high affinity to the capillaries should remain associated.
Sampling Time Points
The time points that are chosen to measure brain uptake or drug effect are commonly between 15 min and 24 h. In most cases the earliest time points, between 15 min and 2 h, yield the highest uptake or effect (Table I). After that, the amount of particles measured in the brain generally decreases. This may be due to degradation of the particles, followed by elimination of the label or encapsulated drug by degradation, metabolism, efflux, and permeation through the brain capillaries (95). In many cases, the particle concentration in the brain is highly decreased or undetectable after 24 h (55,62). Measurements at later time points are preferably performed with stable compounds. If a metabolizing drug is used for detection, the concentration in the body will be decreased to undetectable levels at later time points (65).
Intravenous Administration Routes
As mentioned before, nanoparticles are administered intravenously in most studies. The most convenient way to accomplish this is by administration in the tail vein. Other iv administration routes that have been used for brain targeting studies include the femoral vein, the jugular vein, and the carotid artery. Reddy et al. (63) demonstrated that the site of iv administration can have significant influence on the uptake of nanoparticles in the brain. They used a rat model and administered 0.5 ml of a nanoparticle dispersion either through the tail vein, the jugular vein, or the internal carotid artery. After one hour, administration via the internal carotid artery resulted in a 13-fold greater uptake of nanoparticles in the brain compared to either tail or jugular vein administration. The reason may be decreased exposure of the particles to the reticulo-endothelial system, resulting in an increased concentration of circulating particles available for brain uptake. Although administration via the internal carotid artery is technically more difficult, this finding is worthwhile to take into account in BBB targeting studies.
CONCLUSION AND FUTURE PROSPECTS
In recent brain targeting studies involving nanoparticles, a wide range of approaches has been used by researchers to determine the extent, rate, and effect of drug and nanoparticle uptake into the brain. Choosing the right method depends on whether you want to obtain quantitative uptake values, visualize the uptake mechanism, or test a drug effect. Each technique has its own advantages and disadvantages which should be considered before experiments are started. Many parameters, like administration route, sampling time points, and the way in which the data are presented, should be taken into account. Because contribution of cerebral vascular content can be high, it is recommended to determine the amount of drugs or particles present in the blood at the terminal time point and to properly correct for this.
Many studies have shown that nanoparticles can reach the brain. Within the brain microenvironment, targeting of nanoparticles starts with binding and uptake of the particles into the endothelial cells. By using techniques such as brain capillary depletion or electron microscopy, it can be shown that particles have crossed the endothelial barrier and reached the brain parenchyma. However, the intracellular fate of nanoparticles in vivo often remains unclear. This is due to limitations of in vivo detection methods and techniques. Therefore, intracellular processes such as endocytotic mechanisms currently have to be studied in vitro (96,97) in an artificial environment. Progress in the field of in vivo detection methods is ongoing (98,99). In the future, faster and more sensitive detection methods are required to study uptake and trafficking of nanoparticles within the brain.
ACKNOWLEDGEMENTS
We thank Mike Langeveld for graphic design of Fig. 1. This work was supported by Top Institute Pharma, project T5-105-1: nanoscience as a tool for improving bioavailability and blood-brain barrier penetration.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
REFERENCES
- 1.Pardridge WM. Blood-brain barrier delivery. Drug Discov Today. 2007;12(1–2):54–61. doi: 10.1016/j.drudis.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 2.de Boer AG, van der Sandt IC, Gaillard PJ. The role of drug transporters at the blood-brain barrier. Annu Rev Pharmacol Toxicol. 2003;43:629–656. doi: 10.1146/annurev.pharmtox.43.100901.140204. [DOI] [PubMed] [Google Scholar]
- 3.Ghersi-Egea JF, Leninger-Muller B, Suleman G, Siest G, Minn A. Localization of drug-metabolizing enzyme activities to blood-brain interfaces and circumventricular organs. J Neurochem. 1994;62(3):1089–1096. doi: 10.1046/j.1471-4159.1994.62031089.x. [DOI] [PubMed] [Google Scholar]
- 4.Ghersi-Egea JF, Leininger-Muller B, Cecchelli R, Fenstermacher JD. Blood-brain interfaces: relevance to cerebral drug metabolism. Toxicol Lett. 1995;82–83:645–653. doi: 10.1016/0378-4274(95)03510-9. [DOI] [PubMed] [Google Scholar]
- 5.Abbott NJ. Prediction of blood-brain barrier permeation in drug discovery from in vivo, in vitro and in silico models. Drug Discov Today: Technologies. 2004;1(4):337–463. doi: 10.1016/j.ddtec.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 6.Cecchelli R, Berezowski V, Lundquist S, Culot M, Renftel M, Dehouck MP, et al. Modelling of the blood-brain barrier in drug discovery and development. Nat Rev Drug Discov. 2007;6(1474–17768; 8):650–661. doi: 10.1038/nrd2368. [DOI] [PubMed] [Google Scholar]
- 7.Pardridge WM. Drug targeting to the brain. Pharm Res. 2007;24(0724–8741; 9):1733–1744. doi: 10.1007/s11095-007-9324-2. [DOI] [PubMed] [Google Scholar]
- 8.de Boer AB, de Lange EL, van der Sandt ICJ, Breimer DD. Transporters and the blood-brain barrier (BBB) Int J Clin Pharmacol Ther. 1998;36(0946–19651; 1):14–15. [PubMed] [Google Scholar]
- 9.de Boer AG, Gaillard PJ. Drug targeting to the brain. Annu Rev Pharmacol Toxicol. 2007;47:323–355. doi: 10.1146/annurev.pharmtox.47.120505.105237. [DOI] [PubMed] [Google Scholar]
- 10.Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2(1545–53431; 1):3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37(1):13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 12.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7(1471–003; 1):41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 13.Abbott NJ, Dolman DE, Patabendige AK. Assays to predict drug permeation across the blood-brain barrier, and distribution to brain. Curr Drug Metab. 2008;9(1389–2002; 9):901–910. doi: 10.2174/138920008786485182. [DOI] [PubMed] [Google Scholar]
- 14.Bickel U. How to measure drug transport across the blood-brain barrier. NeuroRx. 2005;2(1545–5343; 1):15–26. doi: 10.1602/neurorx.2.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pardridge WM. Log(BB), PS products and in silico models of drug brain penetration. Drug Discov Today. 2004;9(1359–6446; 9):392–393. doi: 10.1016/S1359-6446(04)03065-X. [DOI] [PubMed] [Google Scholar]
- 16.Pinter GG, Atkins JL, Bell DR. Albumin permeability times surface area (PS) product of peritubular capillaries in kidney. Experientia. 1974;30(9):1045. doi: 10.1007/BF01939000. [DOI] [PubMed] [Google Scholar]
- 17.Smith QR. A review of blood-brain barrier transport techniques. Methods Mol Med. 2003;89:193–208. doi: 10.1385/1-59259-419-0:193. [DOI] [PubMed] [Google Scholar]
- 18.Hervonen H, Steinwall O. Endothelial surface sulfhydryl-groups in blood-brain barrier transport of nutrients. Acta Physiol Scand. 1984;121(4):343–351. doi: 10.1111/j.1748-1716.1984.tb07465.x. [DOI] [PubMed] [Google Scholar]
- 19.Murakami H, Takanaga H, Matsuo H, Ohtani H, Sawada Y. Comparison of blood-brain barrier permeability in mice and rats using in situ brain perfusion technique. Am J Physiol Heart Circ Physiol. 2000;279(3):H1022–H1028. doi: 10.1152/ajpheart.2000.279.3.H1022. [DOI] [PubMed] [Google Scholar]
- 20.Zlokovic BV, Begley DJ, Djuricic BM, Mitrovic DM. Measurement of solute transport across the blood-brain barrier in the perfused guinea pig brain: method and application to N-methyl-alpha-aminoisobutyric acid. J Neurochem. 1986;46(5):1444–1451. doi: 10.1111/j.1471-4159.1986.tb01760.x. [DOI] [PubMed] [Google Scholar]
- 21.Takasato Y, Rapoport SI, Smith QR. An in situ brain perfusion technique to study cerebrovascular transport in the rat. Am J Physiol. 1984;247(3 Pt 2):H484–H493. doi: 10.1152/ajpheart.1984.247.3.H484. [DOI] [PubMed] [Google Scholar]
- 22.Smith QR, Allen DD. In situ brain perfusion technique. Methods Mol Med. 2003;89:209–218. doi: 10.1385/1-59259-419-0:209. [DOI] [PubMed] [Google Scholar]
- 23.Lockman PR, Koziara J, Roder KE, Paulson J, Abbruscato TJ, Mumper RJ, et al. In vivo and in vitro assessment of baseline blood-brain barrier parameters in the presence of novel nanoparticles. Pharm Res. 2003;20(0724–8741; 5):705–713. doi: 10.1023/a:1023492015851. [DOI] [PubMed] [Google Scholar]
- 24.Lundquist S, Renftel M, Brillault J, Fenart L, Cecchelli R, Dehouck MP. Prediction of drug transport through the blood-brain barrier in vivo: a comparison between two in vitro cell models. Pharm Res. 2002;19(0724–8741; 7):976–981. doi: 10.1023/a:1016462205267. [DOI] [PubMed] [Google Scholar]
- 25.Bonate PL. Animal models for studying transport across the blood-brain barrier. J Neurosci Methods. 1995;56(1):1–15. doi: 10.1016/0165-0270(94)00081-q. [DOI] [PubMed] [Google Scholar]
- 26.Oldendorf WH. Measurement of brain uptake of radiolabeled substances using a tritiated water internal standard. Brain Res. 1970;24(2):372–376. doi: 10.1016/0006-8993(70)90123-x. [DOI] [PubMed] [Google Scholar]
- 27.Riant P, Urien S, Albengres E, Renouard A, Tillement JP. Effects of the binding of imipramine to erythrocytes and plasma proteins on its transport through the rat blood-brain barrier. J Neurochem. 1988;51(2):421–425. doi: 10.1111/j.1471-4159.1988.tb01055.x. [DOI] [PubMed] [Google Scholar]
- 28.Pardridge WM, Sakiyama R, Fierer G. Transport of propranolol and lidocaine through the rat blood-brain barrier. Primary role of globulin-bound drug. J Clin Invest. 1983;71(4):900–908. doi: 10.1172/JCI110844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knight RA, Karki K, Ewing JR, Divine GW, Fenstermacher JD, Patlak CS, et al. Estimating blood and brain concentrations and blood-to-brain influx by magnetic resonance imaging with step-down infusion of Gd-DTPA in focal transient cerebral ischemia and confirmation by quantitative autoradiography with Gd-[(14)C]DTPA. J Cereb Blood Flow Metab. 2009;29(5):1048–1058. doi: 10.1038/jcbfm.2009.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferrier MC, Sarin H, Fung SH, Schatlo B, Pluta RM, Gupta SN, et al. Validation of dynamic contrast-enhanced magnetic resonance imaging-derived vascular permeability measurements using quantitative autoradiography in the RG2 rat brain tumor model. Neoplasia. 2007;9(7):546–555. doi: 10.1593/neo.07289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lemaire M, Desrayaud S. The priorities/needs of the pharmaceutical industry in drug delivery to the brain. Int Congr Ser. 2005;1277:32–46. [Google Scholar]
- 32.de Lange EC, de Boer BA, Breimer DD. Microdialysis for pharmacokinetic analysis of drug transport to the brain. Adv Drug Deliv Rev. 1999;36(2–3):211–227. doi: 10.1016/s0169-409x(98)00089-1. [DOI] [PubMed] [Google Scholar]
- 33.Reichel A. Addressing central nervous system (CNS) penetration in drug discovery: basics and implications of the evolving new concept. Chem Biodivers. 2009;6(11):2030–2049. doi: 10.1002/cbdv.200900103. [DOI] [PubMed] [Google Scholar]
- 34.Doran A, Obach RS, Smith BJ, Hosea NA, Becker S, Callegari E, et al. The impact of P-glycoprotein on the disposition of drugs targeted for indications of the central nervous system: evaluation using the MDR1A/1B knockout mouse model. Drug Metab Dispos. 2005;33(1):165–174. doi: 10.1124/dmd.104.001230. [DOI] [PubMed] [Google Scholar]
- 35.Schlageter NL, Carson RE, Rapoport SI. Examination of blood-brain barrier permeability in dementia of the Alzheimer type with [68 Ga]EDTA and positron emission tomography. J Cereb Blood Flow Metab. 1987;7(1):1–8. doi: 10.1038/jcbfm.1987.1. [DOI] [PubMed] [Google Scholar]
- 36.Urakami T, Sakai K, Asai T, Fukumoto D, Tsukada H, Oku N. Evaluation of O-[(18)F]fluoromethyl-D-tyrosine as a radiotracer for tumor imaging with positron emission tomography. Nucl Med Biol. 2009;36(3):295–303. doi: 10.1016/j.nucmedbio.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 37.Emond P, Guilloteau D, Chalon S. PE2I: a radiopharmaceutical for in vivo exploration of the dopamine transporter. CNS Neurosci Ther. 2008;14(1):47–64. doi: 10.1111/j.1527-3458.2007.00033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doorduin J, Klein HC, de Jong JR, Dierckx RA, de Vries EF. Evaluation of [11C]-DAA1106 for imaging and quantification of neuroinflammation in a rat model of herpes encephalitis. Nucl Med Biol. 2010;37(1):9–15. doi: 10.1016/j.nucmedbio.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 39.Nicolazzo JA, Charman SA, Charman WN. Methods to assess drug permeability across the blood-brain barrier. J Pharm Pharmacol. 2006;58(3):281–293. doi: 10.1211/jpp.58.3.0001. [DOI] [PubMed] [Google Scholar]
- 40.Poduslo JF, Curran GL. Permeability at the blood-brain and blood-nerve barriers of the neurotrophic factors: NGF, CNTF, NT-3, BDNF. Brain Res Mol Brain Res. 1996;36(2):280–286. doi: 10.1016/0169-328x(95)00250-v. [DOI] [PubMed] [Google Scholar]
- 41.Oldendorf WH. Brain uptake of radiolabeled amino acids, amines, and hexoses after arterial injection. Am J Physiol. 1971;221(6):1629–1639. doi: 10.1152/ajplegacy.1971.221.6.1629. [DOI] [PubMed] [Google Scholar]
- 42.Lockman PR, Oyewumi MO, Koziara JM, Roder KE, Mumper RJ, Allen DD. Brain uptake of thiamine-coated nanoparticles. J Control Release. 2003;93(3):271–282. doi: 10.1016/j.jconrel.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 43.Schnyder A, Krahenbuhl S, Drewe J, Huwyler J. Targeting of daunomycin using biotinylated immunoliposomes: pharmacokinetics, tissue distribution and in vitro pharmacological effects. J Drug Target. 2005;13(5):325–335. doi: 10.1080/10611860500206674. [DOI] [PubMed] [Google Scholar]
- 44.Shi N, Zhang Y, Zhu C, Boado RJ, Pardridge WM. Brain-specific expression of an exogenous gene after i.v. administration. Proc Natl Acad Sci USA. 2001;98(22):12754–12759. doi: 10.1073/pnas.221450098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang X, Xie Y, Jin Y, Hou X, Ye L, Lou J. The effect of RMP-7 and its derivative on transporting Evans blue liposomes into the brain. Drug Deliv. 2004;11(5):301–309. doi: 10.1080/10717540490494078. [DOI] [PubMed] [Google Scholar]
- 46.Gosk S, Vermehren C, Storm G, Moos T. Targeting anti-transferrin receptor antibody (OX26) and OX26-conjugated liposomes to brain capillary endothelial cells using in situ perfusion. J Cereb Blood Flow Metab. 2004;24(11):1193–1204. doi: 10.1097/01.WCB.0000135592.28823.47. [DOI] [PubMed] [Google Scholar]
- 47.Xie Y, Ye L, Zhang X, Cui W, Lou J, Nagai T, et al. Transport of nerve growth factor encapsulated into liposomes across the blood-brain barrier: in vitro and in vivo studies. J Control Release. 2005;105(1–2):106–119. doi: 10.1016/j.jconrel.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 48.Afergan E, Epstein H, Dahan R, Koroukhov N, Rohekar K, Danenberg HD, et al. Delivery of serotonin to the brain by monocytes following phagocytosis of liposomes. J Control Release. 2008;132(2):84–90. doi: 10.1016/j.jconrel.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 49.Chen H, Tang L, Qin Y, Yin Y, Tang J, Tang W, et al. Lactoferrin-modified procationic liposomes as a novel drug carrier for brain delivery. Eur J Pharm Sci. 2010;40(2):94–102. doi: 10.1016/j.ejps.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 50.Wang JX, Sun X, Zhang ZR. Enhanced brain targeting by synthesis of 3′, 5′-dioctanoyl-5-fluoro-2′-deoxyuridine and incorporation into solid lipid nanoparticles. Eur J Pharm Biopharm. 2002;54(3):285–290. doi: 10.1016/s0939-6411(02)00083-8. [DOI] [PubMed] [Google Scholar]
- 51.Mishra V, Mahor S, Rawat A, Gupta PN, Dubey P, Khatri K, et al. Targeted brain delivery of AZT via transferrin anchored pegylated albumin nanoparticles. J Drug Target. 2006;14(1):45–53. doi: 10.1080/10611860600612953. [DOI] [PubMed] [Google Scholar]
- 52.Michaelis K, Hoffmann MM, Dreis S, Herbert E, Alyautdin RN, Michaelis M, et al. Covalent linkage of apolipoprotein e to albumin nanoparticles strongly enhances drug transport into the brain. J Pharmacol Exp Ther. 2006;317(3):1246–1253. doi: 10.1124/jpet.105.097139. [DOI] [PubMed] [Google Scholar]
- 53.Zensi A, Begley D, Pontikis C, Legros C, Mihoreanu L, Wagner S, et al. Albumin nanoparticles targeted with Apo E enter the CNS by transcytosis and are delivered to neurones. J Control Release. 2009;137(1):78–86. doi: 10.1016/j.jconrel.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 54.Ulbrich K, Hekmatara T, Herbert E, Kreuter J. Transferrin- and transferrin-receptor-antibody-modified nanoparticles enable drug delivery across the blood-brain barrier (BBB) Eur J Pharm Biopharm. 2009;71(2):251–256. doi: 10.1016/j.ejpb.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 55.Lode J, Fichtner I, Kreuter J, Berndt A, Diederichs JE, Reszka R. Influence of surface-modifying surfactants on the pharmacokinetic behavior of 14C-poly (methylmethacrylate) nanoparticles in experimental tumor models. Pharm Res. 2001;18(11):1613–1619. doi: 10.1023/a:1013094801351. [DOI] [PubMed] [Google Scholar]
- 56.Ke W, Shao K, Huang R, Han L, Liu Y, Li J, et al. Gene delivery targeted to the brain using an Angiopep-conjugated polyethyleneglycol-modified polyamidoamine dendrimer. Biomaterials. 2009;30(36):6976–6985. doi: 10.1016/j.biomaterials.2009.08.049. [DOI] [PubMed] [Google Scholar]
- 57.Huang R, Ke W, Han L, Liu Y, Shao K, Jiang C, et al. Lactoferrin-modified nanoparticles could mediate efficient gene delivery to the brain in vivo. Brain Res Bull. 2010;81(6):600–604. doi: 10.1016/j.brainresbull.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 58.Vinogradov SV, Batrakova EV, Kabanov AV. Nanogels for oligonucleotide delivery to the brain. Bioconjug Chem. 2004;15(1):50–60. doi: 10.1021/bc034164r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Costantino L, Gandolfi F, Tosi G, Rivasi F, Vandelli MA, Forni F. Peptide-derivatized biodegradable nanoparticles able to cross the blood-brain barrier. J Control Release. 2005;108(1):84–96. doi: 10.1016/j.jconrel.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 60.Tosi G, Costantino L, Rivasi F, Ruozi B, Leo E, Vergoni AV, et al. Targeting the central nervous system: In vivo experiments with peptide-derivatized nanoparticles loaded with Loperamide and Rhodamine-123. J Control Release. 2007;122(1):1–9. doi: 10.1016/j.jconrel.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 61.Rao KS, Reddy MK, Horning JL, Labhasetwar V. TAT-conjugated nanoparticles for the CNS delivery of anti-HIV drugs. Biomaterials. 2008;29(33):4429–4438. doi: 10.1016/j.biomaterials.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu F, Lu W, Wu H, Fan L, Gao X, Jiang X. Brain delivery and systemic effect of cationic albumin conjugated PLGA nanoparticles. J Drug Target. 2009;17(6):423–434. doi: 10.1080/10611860902963013. [DOI] [PubMed] [Google Scholar]
- 63.Reddy MK, Labhasetwar V. Nanoparticle-mediated delivery of superoxide dismutase to the brain: an effective strategy to reduce ischemia-reperfusion injury. FASEB J. 2009;23(5):1384–1395. doi: 10.1096/fj.08-116947. [DOI] [PubMed] [Google Scholar]
- 64.Vergoni AV, Tosi G, Tacchi R, Vandelli MA, Bertolini A, Costantino L. Nanoparticles as drug delivery agents specific for CNS: in vivo biodistribution. Nanomedicine. 2009;5(4):369–377. doi: 10.1016/j.nano.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 65.Tosi G, Vergoni AV, Ruozi B, Bondioli L, Badiali L, Rivasi F et al. Sialic acid and glycopeptides conjugated PLGA nanoparticles for central nervous system targeting: In vivo pharmacological evidence and biodistribution. J Control Release. 2010. [DOI] [PubMed]
- 66.Iannone M, Cosco D, Cilurzo F, Celia C, Paolino D, Mollace V, et al. A novel animal model to evaluate the ability of a drug delivery system to promote the passage through the BBB. Neurosci Lett. 2010;469(1):93–96. doi: 10.1016/j.neulet.2009.11.051. [DOI] [PubMed] [Google Scholar]
- 67.Kreuter J, Shamenkov D, Petrov V, Ramge P, Cychutek K, Koch-Brandt C, et al. Apolipoprotein-mediated transport of nanoparticle-bound drugs across the blood-brain barrier. J Drug Target. 2002;10(4):317. doi: 10.1080/10611860290031877. [DOI] [PubMed] [Google Scholar]
- 68.Weiss CK, Kohnle MV, Landfester K, Hauk T, Fischer D, Schmitz-Wienke J, et al. The first step into the brain: uptake of NIO-PBCA nanoparticles by endothelial cells in vitro and in vivo, and direct evidence for their blood-brain barrier permeation. ChemMedChem. 2008;3(9):1395–1403. doi: 10.1002/cmdc.200800130. [DOI] [PubMed] [Google Scholar]
- 69.Kurakhmaeva KB, Djindjikhashvili IA, Petrov VE, Balabanyan VU, Voronina TA, Trofimov SS, et al. Brain targeting of nerve growth factor using poly(butyl cyanoacrylate) nanoparticles. J Drug Target. 2009;17(8):564–574. doi: 10.1080/10611860903112842. [DOI] [PubMed] [Google Scholar]
- 70.Kulkarni PV, Roney CA, Antich PP, Bonte FJ, Raghu AV, Aminabhavi TM. Quinoline-n-butylcyanoacrylate-based nanoparticles for brain targeting for the diagnosis of Alzheimer’s disease. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2010;2(1):35–47. doi: 10.1002/wnan.59. [DOI] [PubMed] [Google Scholar]
- 71.Rao J, Dragulescu-Andrasi A, Yao H. Fluorescence imaging in vivo: recent advances. Curr Opin Biotechnol. 2007;18(1):17–25. doi: 10.1016/j.copbio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 72.Frangioni JV. In vivo near-infrared fluorescence imaging. Curr Opin Chem Biol. 2003;7(5):626–634. doi: 10.1016/j.cbpa.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 73.Gaydess A, Duysen E, Li Y, Gilman V, Kabanov A, Lockridge O, et al. Visualization of exogenous delivery of nanoformulated butyrylcholinesterase to the central nervous system. Chem Biol Interact. 2010;187(1–3):295–298. doi: 10.1016/j.cbi.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sadeque AJ, Wandel C, He H, Shah S, Wood AJ. Increased drug delivery to the brain by P-glycoprotein inhibition. Clin Pharmacol Ther. 2000;68(3):231–237. doi: 10.1067/mcp.2000.109156. [DOI] [PubMed] [Google Scholar]
- 75.Karl T, Pabst R, von Horsten S. Behavioral phenotyping of mice in pharmacological and toxicological research. Exp Toxicol Pathol. 2003;55(1):69–83. doi: 10.1078/0940-2993-00301. [DOI] [PubMed] [Google Scholar]
- 76.Brady LS, Holtzman SG. Analgesic effects of intraventricular morphine and enkephalins in nondependent and morphine-dependent rats. J Pharmacol Exp Ther. 1982;222(1):190–197. [PubMed] [Google Scholar]
- 77.Scholtzova H, Wadghiri YZ, Douadi M, Sigurdsson EM, Li YS, Quartermain D, et al. Memantine leads to behavioral improvement and amyloid reduction in Alzheimer’s-disease-model transgenic mice shown as by micromagnetic resonance imaging. J Neurosci Res. 2008;86(12):2784–2791. doi: 10.1002/jnr.21713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Riviere JE. Pharmacokinetics of nanomaterials: an overview of carbon nanotubes, fullerenes and quantum dots. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1(1):26–34. doi: 10.1002/wnan.24. [DOI] [PubMed] [Google Scholar]
- 79.Opanasopit P, Sakai M, Nishikawa M, Kawakami S, Yamashita F, Hashida M. Inhibition of liver metastasis by targeting of immunomodulators using mannosylated liposome carriers. J Control Release. 2002;80(1–3):283–294. doi: 10.1016/s0168-3659(02)00006-8. [DOI] [PubMed] [Google Scholar]
- 80.Gabizon A, Price DC, Huberty J, Bresalier RS, Papahadjopoulos D. Effect of liposome composition and other factors on the targeting of liposomes to experimental tumors: biodistribution and imaging studies. Cancer Res. 1990;50(19):6371–6378. [PubMed] [Google Scholar]
- 81.Zheng JG, Tan TZ. Antisense imaging of colon cancer-bearing nude mice with liposome-entrapped 99m-technetium-labeled antisense oligonucleotides of c-myc mRNA. World J Gastroenterol. 2004;10(17):2563–2566. doi: 10.3748/wjg.v10.i17.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Banks W. Developing drugs that can cross the blood-brain barrier: applications to Alzheimer’s disease. BMC Neurosci. 2008;9(Suppl 3):S2. doi: 10.1186/1471-2202-9-S3-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kastin AJ, Akerstrom V. Mahogany (1377–1428) enters brain by a saturable transport system. J Pharmacol Exp Ther. 2000;294(2):633–636. [PubMed] [Google Scholar]
- 84.Jaeger LB, Banks WA, Varga JL, Schally AV. Antagonists of growth hormone-releasing hormone cross the blood-brain barrier: a potential applicability to treatment of brain tumors. Proc Natl Acad Sci USA. 2005;102(35):12495–12500. doi: 10.1073/pnas.0504163102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Regina A, Demeule M, Che C, Lavallee I, Poirier J, Gabathuler R, et al. Antitumour activity of ANG1005, a conjugate between paclitaxel and the new brain delivery vector Angiopep-2. Br J Pharmacol. 2008;155(2):185–197. doi: 10.1038/bjp.2008.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Banks WA, Coon AB, Robinson SM, Moinuddin A, Shultz JM, Nakaoke R, et al. Triglycerides induce leptin resistance at the blood-brain barrier. Diabetes. 2004;53(5):1253–1260. doi: 10.2337/diabetes.53.5.1253. [DOI] [PubMed] [Google Scholar]
- 87.Price TO, Farr SA, Yi X, Vinogradov S, Batrakova E, Banks WA, et al. Transport across the blood-brain barrier of pluronic leptin. J Pharmacol Exp Ther. 2010;333(1):253–263. doi: 10.1124/jpet.109.158147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ennis SR, Betz AL. Sucrose permeability of the blood-retinal and blood-brain barriers. Effects of diabetes, hypertonicity, and iodate. Invest Ophthalmol Vis Sci. 1986;27(7):1095–1102. [PubMed] [Google Scholar]
- 89.Lockman PR, McAfee G, Geldenhuys WJ, Van der Schyf CJ, Abbruscato TJ, Allen DD. Brain uptake kinetics of nicotine and cotinine after chronic nicotine exposure. J Pharmacol Exp Ther. 2005;314(2):636–642. doi: 10.1124/jpet.105.085381. [DOI] [PubMed] [Google Scholar]
- 90.Lockman PR, Manda VK, Geldenhuys WJ, Mittapalli RK, Thomas F, Albayati ZF, et al. Carrier-mediated transport of the quaternary ammonium neuronal nicotinic receptor antagonist n, n’-dodecylbispicolinium dibromide at the blood-brain barrier. J Pharmacol Exp Ther. 2008;324(1):244–250. doi: 10.1124/jpet.107.130906. [DOI] [PubMed] [Google Scholar]
- 91.Murakami H, Sawada N, Koyabu N, Ohtani H, Sawada Y. Characteristics of choline transport across the blood-brain barrier in mice: correlation with in vitro data. Pharm Res. 2000;17(12):1526–1530. doi: 10.1023/a:1007613326759. [DOI] [PubMed] [Google Scholar]
- 92.Lee YJ, Kusuhara H, Jonker JW, Schinkel AH, Sugiyama Y. Investigation of efflux transport of dehydroepiandrosterone sulfate and mitoxantrone at the mouse blood-brain barrier: a minor role of breast cancer resistance protein. J Pharmacol Exp Ther. 2005;312(1):44–52. doi: 10.1124/jpet.104.073320. [DOI] [PubMed] [Google Scholar]
- 93.Triguero D, Buciak J, Pardridge WM. Capillary depletion method for quantification of blood-brain barrier transport of circulating peptides and plasma proteins. J Neurochem. 1990;54(6):1882–1888. doi: 10.1111/j.1471-4159.1990.tb04886.x. [DOI] [PubMed] [Google Scholar]
- 94.Gutierrez EG, Banks WA, Kastin AJ. Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. J Neuroimmunol. 1993;47(2):169–176. doi: 10.1016/0165-5728(93)90027-v. [DOI] [PubMed] [Google Scholar]
- 95.Misra A, Ganesh S, Shahiwala A, Shah SP. Drug delivery to the central nervous system: a review. J Pharm Pharm Sci. 2003;6(2):252–273. [PubMed] [Google Scholar]
- 96.Chang J, Jallouli Y, Kroubi M, Yuan X, Feng W, Kang C, et al. Characterization of endocytosis of transferrin-coated PLGA nanoparticles by the blood–brain barrier. Int J Pharm. 2009;379(2):285–292. doi: 10.1016/j.ijpharm.2009.04.035. [DOI] [PubMed] [Google Scholar]
- 97.Jallouli Y, Paillard A, Chang J, Sevin E, Betbeder D. Influence of surface charge and inner composition of porous nanoparticles to cross blood-brain barrier in vitro. Int J Pharm. 2007;344(1–2):103–109. doi: 10.1016/j.ijpharm.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 98.de Kemp RA, Epstein FH, Catana C, Tsui BM, Ritman EL. Small-animal molecular imaging methods. J Nucl Med. 2010;51(Suppl 1):18S–32S. doi: 10.2967/jnumed.109.068148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 2010;90(3):1103–1163. doi: 10.1152/physrev.00038.2009. [DOI] [PubMed] [Google Scholar]