

Abstract
Acetaminophen is the leading cause of acute hepatic failure in many developed nations. Acetaminophen hepatotoxicity is mediated by the reactive metabolite N-acetyl-p-benzoquinonimine (NAPQI). We performed a “discovery” genome-wide association study using a cell line–based model system to study the possible contribution of genomics to NAPQI-induced cytotoxicity. A total of 176 lymphoblastoid cell lines from healthy subjects were treated with increasing concentrations of NAPQI. Inhibiting concentration 50 values were determined and were associated with “glutathione pathway” gene single nucleotide polymorphisms (SNPs) and genome-wide basal messenger RNA expression, as well as with 1.3 million genome-wide SNPs. A group of SNPs in linkage disequilibrium on chromosome 3 was highly associated with NAPQI toxicity. The p value for rs2880961, the SNP with the lowest p value, was 1.88 × 10−7. This group of SNPs mapped to a “gene desert,” but chromatin immunoprecipitation assays demonstrated binding of several transcription factor proteins including heat shock factor 1 (HSF1) and HSF2, at or near rs2880961. These chromosome 3 SNPs were not significantly associated with variation in basal expression for any of the genome-wide genes represented on the Affymetrix U133 Plus 2.0 GeneChip. We have used a cell line–based model system to identify a SNP signal associated with NAPQI cytotoxicity. If these observations are validated in future clinical studies, this SNP signal might represent a potential biomarker for risk of acetaminophen hepatotoxicity. The mechanisms responsible for this association remain unclear.
Keywords: acetaminophen, N-acetyl-p-benzoquinonimine, NAPQI, cytotoxicity, single nucleotide polymorphisms, SNPs, expression array, mRNA, Human Variation Panel, GWAS
Acetaminophen is widely used as an over-the-counter analgesic and antipyretic agent. Although it is considered a “safe” drug, acetaminophen is the leading cause of acute liver failure in the United States (Larson et al., 2005; Lee, 2004). As a result, the U.S. Food and Drug Administration has recently recommended a stronger warning on the acetaminophen label (2009). Acetaminophen use can also result in elevations in serum aminotransferase levels in healthy adults when administered at the upper limit of currently recommended dosages (Harrill et al., 2009; Watkins et al., 2006). Mortality rates for patients with acetaminophen-induced hepatotoxicity who present with hepatic failure range from 20 to 40% (Makin et al., 1995; Schiodt et al., 1997).
When taken in therapeutic doses, acetaminophen is metabolized primarily by sulfation and glucuronidation (Vermeulen et al., 1992). However, cytochrome P450 (CYP) enzymes, including CYP2E1, CYP1A2, and CYP3A4, convert 5–9% of acetaminophen to a highly reactive metabolite, N-acetyl-p-benzoquinonimine (NAPQI) (Corcoran et al., 1980; Dahlin et al., 1984) (Fig. 1). NAPQI detoxification occurs primarily by glutathione (GSH) conjugation. After GSH depletion, it is thought that NAPQI causes hepatotoxicity by binding to cellular macromolecules, although the exact mechanism of cellular toxicity remains a subject of controversy (Coles et al., 1988; Kaplowitz, 2004; Mitchell et al., 1973; Rogers et al., 1997). N-acetylcysteine treatment can prevent or limit liver injury by restoring hepatic GSH concentrations. Therefore, N-acetylcysteine is used to treat acetaminophen overdose (Mitchell et al., 1973; Prescott et al., 1974). Unfortunately, after hepatic failure has developed, N-acetylcysteine administration is associated with only a 21–28% reduction in mortality (Harrison et al., 1990; Keays et al., 1991). Recent studies have even suggested that delayed administration of N-acetylcysteine may impair hepatic regeneration (Athuraliya and Jones, 2009; Yang et al., 2009). In the clinic, the major predictor for hepatotoxicity is plasma acetaminophen concentration. The Rumack-Matthew nomogram is used clinically to determine whether a patient who presents within 24 h after a single acute acetaminophen drug ingestion should be treated with N-acetylcysteine by predicting the likelihood of hepatotoxicity based on plasma acetaminophen concentration (Rumack and Matthew, 1975). However, this nomogram is not useful if the time of ingestion is unknown or if toxicity is the result of repeated supratherapeutic doses (Heard, 2008).
FIG. 1.

Acetaminophen metabolism. Acetaminophen is metabolized primarily by sulfation and glucuronidation. However, it can also be oxidized by CYPs to form NAPQI. NAPQI can then interact with cellular macromolecules, resulting in toxicity, or it can be detoxified by undergoing GSH conjugation.
Inheritance may contribute to individual variation in susceptibility to acetaminophen hepatotoxicity. However, “pharmacogenetic” studies of acetaminophen hepatotoxicity have generally focused on drug metabolism, e.g., genetic variation in sulfation, glucuronidation, or CYP-mediated biotransformation (Adjei et al., 2008; Court et al., 2001; Herd et al., 1991; Miners et al., 1986, 1990; Patel et al., 1992). In the present study, we set out to test the hypothesis that genetic variation “downstream” of the formation of NAPQI might contribute to variation in cytotoxicity risk and, as a result, provide insight into mechanisms responsible for acetaminophen-induced hepatotoxicity. To test that hypothesis, we used a “Human Variation Panel” of lymphoblastoid cell lines (LCLs), a pharmacogenomic model system with a demonstrated potential to generate valid pharmacogenomic hypotheses as well as novel mechanistic insights (Li et al., 2008a; Niu et al., 2010; Pei et al., 2009). Studying associations between NAPQI-induced cytotoxicity and single nucleotide polymorphisms (SNPs) in this cell line–based model system could also identify biomarkers for predicting the severity of acetaminophen cytotoxicity and to help individualize the therapy of overdose. Our use of NAPQI rather than acetaminophen itself allowed us to directly study variation in the toxicity of the active metabolite, rather than the parent drug. Specifically, we obtained NAPQI IC50 values, genome-wide SNPs, and basal messenger RNA (mRNA) expression microarray data for 176 LCLs. After adjusting for race and gender, genotype-phenotype association analyses and mRNA expression-IC50 association analyses were performed. We were able to use this model system to study both variation in the “GSH pathway,” the major metabolic pathway known to be responsible for NAPQI detoxification, as well as the possible association of genome-wide SNPs with NAPQI IC50 values. These studies resulted in the identification of a “SNP signal” composed of a group of SNPs in linkage disequilibrium (LD) on chromosome 3 that was highly associated with cytotoxicity in this cell line–based pharmacogenomic model system.
MATERIALS AND METHODS
Cell lines.
LCLs derived from 60 Caucasian-American (CA), 56 African-American (AA), and 60 Han Chinese-American (HCA) healthy subjects were obtained from the Coriell Cell Repository (Camden, NJ) (sample sets HD100CAU, HD100AA, and HD100CHI). The National Institute of General Medical Sciences had anonymized these cell lines before deposit, and all subjects provided written informed consent for the use of their samples for research purposes. These cell lines have been used previously to perform multiple pharmacogenetic studies, with the data deposited in public databases. Our studies were reviewed and approved by the Mayo Clinic Institutional Review Board.
NAPQI cytotoxicity experiments.
NAPQI was purchased from Dalton Pharma Services (Toronto, ON, Canada) and was dissolved in dimethyl sulfoxide (DMSO) immediately prior to use. Upon receipt, the NAPQI was aliquoted into single-use airtight vials under nitrogen. The aliquots were stored in the dark at −80°C. Cytotoxicity experiments were performed as previously described (Li et al., 2008a). Specifically, after plating each cell line at a concentration of 5 × 104 cells per well, the cells were incubated with NAPQI for 24 h at seven concentrations ranging from 0 to 100μM dissolved in 1% DMSO. The growth inhibitory effect of NAPQI was evaluated by determining the concentration required to inhibit cell growth by 50% (IC50) with the CellTiter Blue assay (Promega, Madison, WI). All experiments were performed in triplicate, and the IC50 values reported are averages of those three determinations.
SNP genotyping.
DNA from each cell line studied was obtained from the Coriell Cell Repository. Over 1.6 million genome-wide SNPs were genotyped in this DNA for each LCL using Illumina Infinium HumanHap 550K and 510S Bead Chips and Affymetrix 6.0 GeneChips. SNPs within five GSH pathway genes, GSTT1, GSTM1, GSTP1, GSTO1, and GSTO2, had been determined in these same cell lines previously by in-depth gene resequencing, and those SNPs were also included in the analysis (Moyer et al., 2007, 2008; Mukherjee et al., 2006). Specifically, analysis of the GSH pathway included 763 SNPs obtained during the in-depth gene resequencing studies and during genome-wide SNP genotyping. The GSH pathway was defined, as in our previous studies (Moyer et al., 2010), to include genes involved in GSH synthesis (GSS, GCLC, and GCLM), GSH redox status (GSR, GPX1, GPX2, GPX3, GPX4, GPX5, GPX6, and GPX7), GSH S-transferases (GSTA1, GSTA2, GSTA3, GSTA4, GSTA5, GSTM1, GSTM2, GSTM3, GSTM4, GSTM5, GSTO1, GSTO2, GSTP1, GSTT1, GSTT2, and GSTZ1), and GSH conjugate transporters (ABCC1, ABCC2, ABCC3, and ABCC4).
mRNA microarray analysis.
mRNA microarray analysis was performed as described previously (Li et al., 2008a). Specifically, total RNA was extracted from LCLs at baseline using the RNeasy kit (Qiagen, Valencia, CA). Prior to microarray analysis, RNA quality assessment was performed using the Agilent 2100 bioanalyzer. All RNA samples had Agilent RNA Integrity Number values greater than 9.0. The RNA was then reverse transcribed and biotin labeled for hybridization with Affymetrix U133 Plus 2.0 GeneChips (Affymetrix, Santa Clara, CA). The microarray images were analyzed using quality control techniques established in the Mayo Clinic Microarray Core Facility. Fifty-two probe sets for GSH pathway genes, as described above, were utilized for the pathway analysis. There were no probe sets available for GPX6, GSTA2, or GSTA5.
Chromatin immunoprecipitation assays.
Chromatin immunoprecipitation (ChIP) assays were performed using the ChampionChIP one-day kit (SABiosciences, Frederick, MD) for the genomic region containing the rs2880961 SNP. Both real-time and standard PCR were used to monitor ChIP assay results. The following DNA sequences were used to design PCR primers that amplified 275 bp of sequence flanking rs2880961 (Forward primer: 5′-tgtggtttttgatctggattatg-3′ and Reverse primer: 5′-ttgcacagcagaactcttgg-3′). ChIP-grade antibodies for transcription factors HSF1 (SC-17756X) and HSF2 (SC-8062X) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), whereas nuclear factor (NF)-kappaB antibody (ab7970) was purchased from Abcam (Cambridge, MA). Two microliters DNA from each ChIP assay was used as template for the PCR reactions, and 20 μl of the PCR products were loaded on 1.2% agarose gels for electrophoresis.
Data analysis.
The cytotoxicity data were fitted to dose-response curves using the R package (R Development Core Team, 2010), and IC50 values were estimated using the Cedergreen-Ritz-Streibig five-parameter model (Cedergreen et al., 2005). This model, which is a modification of the four-parameter logistic curve, was utilized to take into account the hormesis observed with NAPQI. SNPs were excluded from the analysis if the call rate was less than 95%, if the minor allele frequency (MAF) was less than 5%, or if the SNP was out of Hardy-Weinberg equilibrium (HWE) (p < 0.001). One sample for an AA subject was removed from the analysis because the SNP genotype call rate across the sample was less than 95%. After quality control, a total of 1,348,798 SNPs were included in the analysis. To control for population stratification, eigen analysis of the SNP data was performed within each race (Price et al., 2006). The top five eigenvectors for each race were used together with race to adjust the genotype data. The estimated IC50 values were log transformed and adjusted for gender, race, and the top five eigenvectors. Association between adjusted IC50 values and adjusted genotypes was computed using the test statistic F = (r2/1 − r2) × df, where r represents the correlation coefficient and df represents the degrees of freedom, a function of the sample size. Finally, p values were calculated based on the F distribution. Pairwise LD was estimated using LD r2 statistics and was displayed graphically using the Haploview software (Barrett et al., 2005). To refine the chromosome 3 region associated with NAPQI IC50, genotype imputation was performed using MACH 1.0 (Li et al., 2008b, 2009), with HapMap data (http://hapmap.ncbi.nlm.nih.gov/) as the reference panel. Specifically, SNPs for AAs were imputed using both CEU and YRI data, SNPs for CAs were imputed based on CEU data, and SNP markers for HCA subjects were imputed based on the Chinese Han Beijing and JPT data.
mRNA expression data were normalized on a log2 scale using GCRMA (Bolstad et al., 2003; Wu et al., 2004). The normalized expression data were regressed on gender and race. Pearson correlation coefficients were calculated for the adjusted IC50, expression levels, and the test statistic, given by 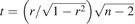 , where r represents the Pearson correlation coefficient used to test for a nonzero correlation. This test statistic follows a t distribution, with degrees of freedom of n − 2 under the null hypothesis. Q values, a type of false discovery rate control, were computed for each SNP (Storey, 2002). The percentage of variation in IC50 explained by GSH pathway variation was calculated based on the coefficient of determination, r2, using a multiple regression model between IC50 values and expression of individual probe sets.
, where r represents the Pearson correlation coefficient used to test for a nonzero correlation. This test statistic follows a t distribution, with degrees of freedom of n − 2 under the null hypothesis. Q values, a type of false discovery rate control, were computed for each SNP (Storey, 2002). The percentage of variation in IC50 explained by GSH pathway variation was calculated based on the coefficient of determination, r2, using a multiple regression model between IC50 values and expression of individual probe sets.
RESULTS
NAPQI Cytotoxicity
NAPQI cytotoxicity studies were performed for each cell line to determine the extent of interindividual variation in IC50 values as a measure of NAPQI toxicity and as a phenotype for association studies designed to identify potential biomarkers for the prediction of toxicity risk. Figure 2 shows representative NAPQI cytotoxicity curves for two LCLs (Fig. 2A and 2B) as well as a graphical representation of the IC50 values calculated for all 176 cell lines (Fig. 2C). Large individual variations in NAPQI IC50 values were observed (Fig. 2C). The average NAPQI IC50 for these cell lines was 6.5 ± 4.5μM (mean ± SD). No differences were observed in NAPQI IC50 values between males and females, p = 0.63, or among ethnic groups, p = 0.24. At low concentrations of NAPQI, a slight increase in proliferation, suggestive of hormesis, was observed in many of the cell lines (see Fig. 2A). Hormesis refers to a dose-response relationship that is characterized by increased proliferation in response to low doses of toxic substances that normally cause growth inhibition at higher doses (Calabrese, 2010).
FIG. 2.

Cytotoxicity curves and IC50 distribution by ethnic group. Two representative NAPQI cytotoxicity dose-response curves are shown on the left in (A) and (B). Each point in (A) and (B) represents the mean ± SEM for three separate determinations. The percentage of viable cells after a 24-h incubation with NAPQI is plotted on the y-axis, with the concentration of NAPQI on the x-axis. IC50 values represent the concentration of NAPQI at which 50% of the cells, relative to control, were viable. The top curve in (A) shows an example of the apparent “hormesis” observed with some cell lines. The panel on the right shows the distribution of IC50 values for all 176 cell lines studied, with the bars for each sample color coded by ethnic group (AA, red; CA, blue; HCA, yellow). The height of each bar represents the IC50 value for that cell line.
GSH Pathway Analyses
Although GSH conjugation is the only known mechanism for the biotransformation of NAPQI, variation in the basal expression of GSH pathway genes could explain only 37.3% of the variation in NAPQI IC50 in this Human Variation Panel model system. Specifically, 716 SNPs in 31 genes in the GSH pathway were examined. Of these SNPs, 41 had p values < 0.05 (Supplementary table 1). Particularly striking was the heavy representation of SNPs in the ATP-binding cassette transporter gene, ABCC4. When the 52 expression probe sets for 28 of these genes (there were no probe sets available for GPX6, GSTA2, or GSTA5) were examined, five had p values for association with NAPQI cytotoxicity that were less than 0.05 (Fig. 3A, red dots; Supplementary table 2). However, none of these associations remained significant after correction for multiple comparisons. Therefore, we moved beyond the known GSH pathway to an “unbiased” genome-wide approach in an attempt to identify SNPs and/or mRNA expression that might be associated with NAPQI toxicity.
FIG. 3.

Manhattan plots for expression and SNPs. The y-axis of each graph represents the −log10(p value) for the association, whereas the x-axis gives the chromosomal location of the expression array probe set or SNP. (A) Expression array data versus IC50. Red dots represent “GSH pathway” probe sets. (B) SNP versus IC50 associations.
Genome-Wide SNP Analyses
Correlations between SNP genotypes and NAPQI IC50 values were determined in an attempt to identify biomarkers that might help to predict risk for NAPQI-induced cytotoxicity. In total, 1,348,798 SNPs passed quality control checks and were included in the analysis. Specifically, 48,754 SNPs were excluded because of call rates < 95%, 277,598 were excluded for MAFs less than 0.05, and 14,244 were excluded because of deviation from HWE, with p values < 0.001. Table 1 lists the top 15 SNPs for NAPQI IC50 associations (all SNPs with p values < 10−4 are listed in Supplementary table 3), and Figure 3B shows a Manhattan plot for all the SNPs studied. A Q-Q plot for these data is included as Supplementary figure 1. The SNP with the lowest p value was rs2880961 on chromosome 3, with p = 1.88 × 10−7. Ten of the top 15 SNPs with the lowest p values among the 1.3 million SNPs analyzed were located in the same region of chromosome 3 (see Table 1). This group of SNPs was in high LD, and the top SNPs were located in a region of chromosome 3 without known genes, 275 kb downstream from C3orf38 and 624 kb upstream of EPHA3 (Fig. 4). The most highly associated SNP, rs2880961, had a permutation p value of 0.1542, with a q value of 0.107. The MAF for this SNP varied by population, with AA = 0.19, CA = 0.35, and HCA = 0.44. In addition to SNPs represented on the genotyping platforms used to study the DNA from these cell lines, we also imputed the SNPs in this region (Fig. 4). Although this region contained no known genes, examination of the region with the Vista Genome Browser (Frazer et al., 2004) revealed many evolutionarily conserved segments. The region encompassing rs2880961, corresponding to chromosome 3 positions 88,606,732 to 88,606,874, is very highly conserved with the rhesus macaque. The area in which the chromosome 3 SNP signal is located also contains multiple clusters of predicted transcription factor binding sites that are conserved with the dog, mouse, and chicken. One of those conserved regions, predicted to be a portion of an unidentified gene with ATP-binding cassette domains, was located only about 7 kb upstream from rs2880961, the SNP with the strongest signal in our genome-wide association study (GWAS).
TABLE 1.
Top 15 SNPs Associated with NAPQI IC50
| SNP ID | Chromosome | Gene | Position | MAF | p Value | q Value | Correlation |
| rs2880961 | 3 | C3orf38 | 88606865 | 0.34 | 1.88 × 10−7 | 0.107 | 0.40 |
| rs17559005 | 3 | C3orf38 | 88595229 | 0.33 | 3.18 × 10−7 | 0.107 | 0.39 |
| rs33966381 | 3 | C3orf38 | 88595312 | 0.33 | 3.18 × 10−7 | 0.107 | 0.39 |
| rs10511137 | 3 | C3orf38 | 88594892 | 0.33 | 3.18 × 10−7 | 0.107 | 0.39 |
| rs1532815 | 1 | LMX1A | 163446713 | 0.34 | 6.04 × 10−7 | 0.163 | 0.39 |
| rs12107308 | 3 | C3orf38 | 88565043 | 0.23 | 4.09 × 10−6 | 0.742 | 0.36 |
| rs5936441 | 23 (X) | LOC100129661 | 147131948 | 0.36 | 5.38 × 10−6 | 0.742 | 0.35 |
| rs6809413 | 3 | C3orf38 | 88595139 | 0.49 | 5.47 × 10−6 | 0.742 | 0.36 |
| rs2344953 | 3 | C3orf38 | 88614905 | 0.26 | 5.59 × 10−6 | 0.742 | 0.35 |
| rs6852435 | 4 | KIAA1712 | 175537354 | 0.27 | 5.71 × 10−6 | 0.742 | −0.35 |
| rs6795028 | 3 | C3orf38 | 88563780 | 0.51 | 6.05 × 10−6 | 0.742 | 0.35 |
| rs3795578 | 1 | ETNK2 | 202379607 | 0.40 | 7.18 × 10−6 | 0.807 | 0.35 |
| rs13101122 | 3 | C3orf38 | 88590539 | 0.48 | 8.35 × 10−6 | 0.867 | 0.35 |
| rs1876381 | 8 | SNX16 | 83120878 | 0.19 | 9.60 × 10−6 | 0.925 | −0.34 |
| rs1982562 | 3 | C3orf38 | 88591057 | 0.48 | 1.28 × 10−5 | 0.991 | 0.34 |
FIG. 4.

The region of chromosome 3 with the SNPs most highly associated with NAPQI IC50 values. The x-axis shows the position of the SNPs on chromosome 3, whereas the y-axis indicates the −log10(p value) of the SNP-IC50 association. The relative positions of transcripts in the expressed sequence tag database (small red bars), novel genes predicted to be encoded in this region (green bars), and an evolutionarily conserved region with multiple predicted transcription factor binding sites (black bar) are also shown.
Genome-Wide Basal mRNA Expression Analysis
In an attempt to better understand baseline characteristics of the cells that might predispose to NAPQI-mediated cytotoxicity, basal mRNA expression was also correlated with IC50 values. Nineteen probe sets were associated with NAPQI IC50 with p < 0.0001 (Table 2). None of the genes close to the chromosome 3 SNPs identified during the genome-wide SNP analysis had transcripts that were associated with IC50. However, that does not rule out the possibility that the SNPs may be related to expression through trans effects.
TABLE 2.
Expression Array Probe Sets Associated with NAPQI IC50 with p < 10−4
| Probe set | Gene | Chromosome | p Value | q Value | Correlation |
| 226989_at | RGMB | 5 | 8.00 × 10−6 | 0.25039 | −0.32948 |
| 229016_s_at | TRERF1 | 6 | 1.77 × 10−5 | 0.25039 | −0.31736 |
| 206037_at | CCBL1 | 9 | 2.46 × 10−5 | 0.25039 | −0.31225 |
| 229017_s_at | RIPK5 | 1 | 2.97 × 10−5 | 0.25039 | −0.30924 |
| 212698_s_at | SEPT10 | 2 | 3.27 × 10−5 | 0.25039 | −0.30765 |
| 202741_at | PRKACB | 1 | 3.43 × 10−5 | 0.25039 | 0.306891 |
| 227339_at | RGMB | 5 | 3.64 × 10−5 | 0.25039 | −0.30595 |
| 205270_s_at | LCP2 | 5 | 3.73 × 10−5 | 0.25039 | −0.30554 |
| 225792_at | HOOK1 | 1 | 4.17 × 10−5 | 0.25039 | 0.303737 |
| 205204_at | NMB | 15 | 4.64 × 10−5 | 0.25101 | −0.30197 |
| 244063_at | BTN2A1 | 6 | 6.09 × 10−5 | 0.27044 | −0.29745 |
| 205965_at | BATF | 14 | 6.22 × 10−5 | 0.27044 | −0.2971 |
| 223835_x_at | OTP | 5 | 7.18 × 10−5 | 0.27044 | −0.2947 |
| 228583_at | C14orf46 | 14 | 7.70 × 10−5 | 0.27044 | 0.293519 |
| 212270_x_at | RPL17 | 18 | 7.87 × 10−5 | 0.27044 | −0.29313 |
| 227370_at | FAM171B | 2 | 8.00 × 10−5 | 0.27044 | −0.29286 |
| 214472_at | HIST1H3D | 6 | 8.91 × 10−5 | 0.27941 | 0.291016 |
| 219976_at | HOOK1 | 1 | 9.46 × 10−5 | 0.27941 | 0.29 |
| 241813_at | MBD1 | 18 | 9.82 × 10−5 | 0.27941 | −0.28936 |
Characterization of rs2880961
The conserved segments near the chromosome 3 SNPs associated with NAPQI IC50 values could potentially represent binding sites for proteins, such as transcription factors. Therefore, a transcription factor binding site search was performed with TFSEARCH (Heinemeyer et al., 1998) to identify possible alterations by rs2880961 of transcription factor binding motifs. When the nucleotide present at that locus was the wild type (WT), CCAAT enhancer-binding protein (C/EBP) was predicted to bind (score = 86.2 out of 100). However, when the variant nucleotide was present, HSF2 (score = 90.4), NF-kappaB (89.6), and HSF1 (87.7) were predicted to bind, but the C/EBP binding site was predicted to remain intact (score = 86.2). Therefore, as the next step in our analysis, ChIP assays for possible binding of HSF1, HSF2, and NF-kappaB were performed. We observed, as predicted, binding of all three of these transcription factors (Fig. 5). However, there was not a striking difference in binding between WT and variant sequences for any of the three transcription factors tested.
FIG. 5.

rs2880961 ChIP assays. The figure shows ChIP assays performed using lymphoblastoid cells with known genotypes at rs2880961. V, variant genotype; WT, wild-type genotype.
Finally, the binding of transcription factors at this locus could potentially affect the expression of other genes either close to this location or elsewhere in the genome. Because no candidate genes were located near this position, we also performed a GWAS of genotype at rs2880961 with genome-wide mRNA expression probes. However, none of the 54,000 Affymetrix probe sets displayed significant variation in expression that was associated with rs2880961 genotype.
DISCUSSION
Acetaminophen is metabolized to form NAPQI in vivo, and NAPQI is primarily responsible for the cytotoxic effects of this widely used drug. It is known that GSH conjugation plays an important role in the biotransformation of NAPQI (Mitchell et al., 1973). However, very little is known with regard to genes that might influence susceptibility or resistance to this highly reactive metabolite. Therefore, we utilized a Human Variation Panel cell line–based model system that included both genome-wide expression and SNP data to perform a study of the association of NAPQI IC50 values with genomic biomarkers. By studying NAPQI rather than acetaminophen, we were able to focus exclusively on genes and pathways related to the metabolism and toxicity of this highly reactive acetaminophen metabolite rather than its metabolic generation, which is subject to variation as a result of interindividual differences in sulfation, glucuronidation, and the activity of CYP enzymes.
It had been reported previously that proliferation is one way in which hepatocytes may respond to toxic stimuli (Bell et al., 1988; Calabrese and Mehendale, 1996; Mehendale, 1991) and that low concentrations of acetaminophen stimulate cellular proliferation (Gadd et al., 2002; Schonberg and Skorpen, 1997). Of interest is the fact that we observed proliferation at low doses of NAPQI (Fig. 2A). Variation in ability to stimulate proliferation among the cell lines in our model system may be one factor contributing to the variation in NAPQI IC50 values that we observed.
As mentioned previously, the GSH pathway has been implicated as an important drug-metabolizing system for the detoxification of NAPQI (Coles et al., 1988; Mitchell et al., 1973; Rogers et al., 1997). Therefore, we began by studying the contribution of this important pathway to variation in NAPQI toxicity. Although GSH pathway genes play an important role in the detoxification of NAPQI, individually they did not appear to be useful biomarkers for variation in NAPQI-dependent cytotoxicity. Although variation in the expression of the GSH pathway as a whole, i.e., the combined effect of variation in expression of all these genes, could explain 37.3% of the variation in NAPQI IC50 values, no individual probe set or SNP had extremely low p values or would have been selected as a biomarker for NAPQI cytotoxicity in this cell line–based model system. These observations may be due, in part, to the fact that the GSH pathway includes many genes and gene families with overlapping substrate specificities. As a result, even after analysis of the GSH pathway, the majority of the variation in IC50 values remained unexplained.
In an attempt to identify novel candidates that might potentially be useful biomarkers for risk of NAPQI-induced cytotoxicity, we next performed genome-wide analyses of SNP-IC50 associations. The striking SNP signal that we observed on chromosome 3 (Figs. 3 and 4) did represent a potential biomarker for NAPQI-dependent cytotoxicity. The association of NAPQI IC50 with genotype at rs2880961 and the group of SNPs in LD with rs2880961 shown in Figure 4 had very low p values and warrant further evaluation. However, the identification of mechanisms by which this group of SNPs might be associated with IC50 values remains a challenge. These SNPs are located approximately 275 kb away from the nearest gene, C3orf38. Understanding of mechanisms responsible for this association could result in the identification of novel drug targets and/or the development of new treatment modalities and/or protocols for treating acetaminophen overdose. However, as has been demonstrated by many other GWAS, when a “signal” is detected in a region devoid of annotated genes, it is difficult to determine underlying mechanisms responsible for that signal (Ghoussaini et al., 2008; McPherson et al., 2007). We were able to detect transcription factor binding to the region immediately surrounding rs2880961, but we did not observe striking differences in binding to WT and variant SNP sequences based on ChIP assays performed with three transcription factors (Fig. 5). We also attempted to identify variation in the basal expression of genes that might be associated with rs2880961. However, no transcripts were identified that were significantly associated with genotype at rs2880961. Because GWAS utilize tagging SNPs, it is unclear whether rs2880961, the observed SNP with the lowest p value, is the causal SNP or if a linked SNP—one not on the genotyping platform—might be responsible for the association with NAPQI IC50.
A recent study by Harrill et al. (2009) identified CD44 as a potential biomarker for acetaminophen-induced liver injury in humans. We were unable to replicate this finding in our study, which included 12 expression array probe sets for CD44 (lowest p value 1.3 × 10−3). However, our study differed significantly from the study of Harrill et al. (2009) both in the model system that we utilized and the compound studied—acetaminophen for the study of Harrill et al. (2009) versus NAPQI in our experiments.
Acetaminophen overdose and resultant hepatotoxicity is a common, life-threatening medical problem, but the algorithm utilized in the clinical setting to direct treatment is not optimal in many situations. Therefore, the identification of valid biomarkers that might predict risk for toxicity could help to improve patient care. Those biomarkers might also lead to the identification of novel drug targets, better treatment strategies, and a more complete understanding of sensitivity to drug-induced hepatotoxicity. In the present “discovery” study, we have performed GSH pathway–based and genome-wide analyses of SNP genotypes versus IC50 values, utilizing 176 Human Variation Panel LCLs. The strongest association detected involved a group of SNPs in tight LD on chromosome 3. Those SNPs, polymorphisms that are not located near any known genes, map to a region that displays significant evolutionary conservation and that may contain transcription factor binding sites or encode transcripts that are involved in the trans-regulation of the transcription of distant genes or the regulation of not yet identified genes. Future studies will be required to confirm the association of rs2880961 and the surrounding SNPs with NAPQI toxicity, to understand the mechanisms underlying that association, and to elucidate the possible relevance of this region of chromosome 3 for drug-induced hepatic injury.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institutes of Health (R01 GM28157, U19 GM61388) (The Pharmacogenomics Research Network); PhRMA Foundation “Center of Excellence in Clinical Pharmacology” Award.
Acknowledgments
We thank Drs Alison Harrill and Paul Watkins, Center for Drug Safety Sciences, Hamner Institutes for Health Sciences, Research Triangle Park, NC, for their helpful advice and Luanne Wussow for her assistance with the manuscript.
References
- Adjei AA, Gaedigk A, Simon SD, Weinshilboum RM, Leeder JS. Interindividual variability in acetaminophen sulfation by human fetal liver: implications for pharmacogenetic investigations of drug-induced birth defects. Birth Defects Res. A Clin. Mol. Teratol. 2008;82:155–165. doi: 10.1002/bdra.20535. [DOI] [PubMed] [Google Scholar]
- Athuraliya TN, Jones AL. Prolonged N-acetylcysteine therapy in late acetaminophen poisoning associated with acute liver failure—a need to be more cautious? Crit. Care. 2009;13:144. doi: 10.1186/cc7800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Bell AN, Young RA, Lockard VG, Mehendale HM. Protection of chlordecone-potentiated carbon tetrachloride hepatotoxicity and lethality by partial hepatectomy. Arch. Toxicol. 1988;61:392–405. doi: 10.1007/BF00334621. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Calabrese EJ. Hormesis is central to toxicology, pharmacology and risk assessment. Hum. Exp. Toxicol. 2010;29:249–261. doi: 10.1177/0960327109363973. [DOI] [PubMed] [Google Scholar]
- Calabrese EJ, Mehendale HM. A review of the role of tissue repair as an adaptive strategy: why low doses are often non-toxic and why high doses can be fatal. Food Chem. Toxicol. 1996;34:301–311. doi: 10.1016/0278-6915(95)00101-8. [DOI] [PubMed] [Google Scholar]
- Cedergreen N, Ritz C, Streibig JC. Improved empirical models describing hormesis. Environ. Toxicol. Chem. 2005;24:3166–3172. doi: 10.1897/05-014r.1. [DOI] [PubMed] [Google Scholar]
- Coles B, Wilson I, Wardman P, Hinson JA, Nelson SD, Ketterer B. The spontaneous and enzymatic reaction of N-acetyl-p-benzoquinonimine with glutathione: a stopped-flow kinetic study. Arch. Biochem. Biophys. 1988;264:253–260. doi: 10.1016/0003-9861(88)90592-9. [DOI] [PubMed] [Google Scholar]
- Corcoran GB, Mitchell JR, Vaishnav YN, Horning EC. Evidence that acetaminophen and N-hydroxyacetaminophen form a common arylating intermediate, N-acetyl-p-benzoquinoneimine. Mol. Pharmacol. 1980;18:536–542. [PubMed] [Google Scholar]
- Court MH, Duan SX, von Moltke LL, Greenblatt DJ, Patten CJ, Miners JO, Mackenzie PI. Interindividual variability in acetaminophen glucuronidation by human liver microsomes: identification of relevant acetaminophen UDP-glucuronosyltransferase isoforms. J. Pharmacol. Exp. Ther. 2001;299:998–1006. [PubMed] [Google Scholar]
- Dahlin DC, Miwa GT, Lu AY, Nelson SD. N-acetyl-p-benzoquinone imine: a cytochrome P-450-mediated oxidation product of acetaminophen. Proc. Natl. Acad. Sci. U.S.A. 1984;81:1327–1331. doi: 10.1073/pnas.81.5.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32:W273–W279. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd SL, Hobbs G, Miller MR. Acetaminophen-induced proliferation of estrogen-responsive breast cancer cells is associated with increases in c-myc RNA expression and NF-kappaB activity. Toxicol. Sci. 2002;66:233–243. doi: 10.1093/toxsci/66.2.233. [DOI] [PubMed] [Google Scholar]
- Ghoussaini M, Song H, Koessler T, Al Olama AA, Kote-Jarai Z, Driver KE, Pooley KA, Ramus SJ, Kjaer SK, Hogdall E, et al. Multiple loci with different cancer specificities within the 8q24 gene desert. J. Natl. Cancer Inst. 2008;100:962–966. doi: 10.1093/jnci/djn190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrill AH, Watkins PB, Su S, Ross PK, Harbourt DE, Stylianou IM, Boorman GA, Russo MW, Sackler RS, Harris SC, et al. Mouse population-guided resequencing reveals that variants in CD44 contribute to acetaminophen-induced liver injury in humans. Genome Res. 2009;19:1507–1515. doi: 10.1101/gr.090241.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PM, Keays R, Bray GP, Alexander GJ, Williams R. Improved outcome of paracetamol-induced fulminant hepatic failure by late administration of acetylcysteine. Lancet. 1990;335:1572–1573. doi: 10.1016/0140-6736(90)91388-q. [DOI] [PubMed] [Google Scholar]
- Heard KJ. Acetylcysteine for acetaminophen poisoning. N. Engl. J. Med. 2008;359:285–292. doi: 10.1056/NEJMct0708278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, Ignatieva EV, Ananko EA, Podkolodnaya OA, Kolpakov FA, et al. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998;26:362–367. doi: 10.1093/nar/26.1.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herd B, Wynne H, Wright P, James O, Woodhouse K. The effect of age on glucuronidation and sulphation of paracetamol by human liver fractions. Br. J. Clin. Pharmacol. 1991;32:768–770. [PMC free article] [PubMed] [Google Scholar]
- Kaplowitz N. Acetaminophen hepatoxicity: what do we know, what don't we know, and what do we do next? Hepatology. 2004;40:23–26. doi: 10.1002/hep.20312. [DOI] [PubMed] [Google Scholar]
- Keays R, Harrison PM, Wendon JA, Forbes A, Gove C, Alexander GJ, Williams R. Intravenous acetylcysteine in paracetamol induced fulminant hepatic failure: a prospective controlled trial. BMJ. 1991;303:1026–1029. doi: 10.1136/bmj.303.6809.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS, Reisch JS, Schiodt FV, Ostapowicz G, Shakil AO, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42:1364–1372. doi: 10.1002/hep.20948. [DOI] [PubMed] [Google Scholar]
- Lee WM. Acetaminophen and the U.S. Acute Liver Failure Study Group: lowering the risks of hepatic failure. Hepatology. 2004;40:6–9. doi: 10.1002/hep.20293. [DOI] [PubMed] [Google Scholar]
- Li L, Fridley B, Kalari K, Jenkins G, Batzler A, Safgren S, Hildebrandt M, Ames M, Schaid D, Wang L. Gemcitabine and cytosine arabinoside cytotoxicity: association with lymphoblastoid cell expression. Cancer Res. 2008a;68:7050–7058. doi: 10.1158/0008-5472.CAN-08-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Willer C, Sanna S, Abecasis G. Genotype imputation. Annu. Rev. Genomics Hum. Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Willer CJ, Ding J, Scheet P, Abecasis G. Markov Model for Rapid Haplotyping and Genotype Imputation in Genome Wide Studies. Ann Arbor, MI: University of Michigan School of Public Health; 2008b. [Google Scholar]
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993) Gastroenterology. 1995;109:1907–1916. doi: 10.1016/0016-5085(95)90758-0. [DOI] [PubMed] [Google Scholar]
- McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehendale HM. Role of hepatocellular regeneration and hepatolobular healing in the final outcome of liver injury. A two-stage model of toxicity. Biochem. Pharmacol. 1991;42:1155–1162. doi: 10.1016/0006-2952(91)90249-5. [DOI] [PubMed] [Google Scholar]
- Miners JO, Lillywhite KJ, Yoovathaworn K, Pongmarutai M, Birkett DJ. Characterization of paracetamol UDP-glucuronosyltransferase activity in human liver microsomes. Biochem. Pharmacol. 1990;40:595–600. doi: 10.1016/0006-2952(90)90561-x. [DOI] [PubMed] [Google Scholar]
- Miners JO, Robson RA, Birkett DJ. Paracetamol metabolism in pregnancy. Br. J. Clin. Pharmacol. 1986;22:359–362. doi: 10.1111/j.1365-2125.1986.tb02901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J. Pharmacol. Exp. Ther. 1973;187:211–217. [PubMed] [Google Scholar]
- Moyer AM, Salavaggione OE, Hebbring SJ, Moon I, Hildebrandt MA, Eckloff BW, Schaid DJ, Wieben ED, Weinshilboum RM. Glutathione S-transferase T1 and M1: gene sequence variation and functional genomics. Clin. Cancer Res. 2007;13:7207–7216. doi: 10.1158/1078-0432.CCR-07-0635. [DOI] [PubMed] [Google Scholar]
- Moyer AM, Salavaggione OE, Wu TY, Moon I, Eckloff BW, Hildebrandt MA, Schaid DJ, Wieben ED, Weinshilboum RM. Glutathione S-transferase P1: gene sequence variation and functional genomic studies. Cancer Res. 2008;68:4791–4801. doi: 10.1158/0008-5472.CAN-07-6724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer AM, Sun Z, Batzler AJ, Li L, Schaid DJ, Yang P, Weinshilboum RM. Glutathione pathway genetic polymorphisms and lung cancer survival after platinum-based chemotherapy. Cancer Epidemiol. Biomarkers Prev. 2010;19:811–821. doi: 10.1158/1055-9965.EPI-09-0871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee B, Salavaggione OE, Pelleymounter LL, Moon I, Eckloff BW, Schaid DJ, Wieben ED, Weinshilboum RM. Glutathione S-transferase omega 1 and omega 2 pharmacogenomics. Drug Metab. Dispos. 2006;34:1237–1246. doi: 10.1124/dmd.106.009613. [DOI] [PubMed] [Google Scholar]
- Niu N, Qin Y, Fridley BL, Hou J, Kalari KR, Zhu M, Wu T-Y, Jenkins GD, Batzler A, Wang L. Radiation pharmacogenomics: a genome-wide association approach to identify radiation response biomarkers using human lymphoblastoid cell lines. Genome Res. 2010;20:1482–1492. doi: 10.1101/gr.107672.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M, Tang BK, Kalow W. Variability of acetaminophen metabolism in Caucasians and Orientals. Pharmacogenetics. 1992;2:38–45. doi: 10.1097/00008571-199202000-00007. [DOI] [PubMed] [Google Scholar]
- Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, Petersen G, Lou Z, Wang L. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell. 2009;16:259–266. doi: 10.1016/j.ccr.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott LF, Newton RW, Swainson CP, Wright N, Forrest AR, Matthew H. Successful treatment of severe paracetamol overdosage with cysteamine. Lancet. 1974;1:588–592. doi: 10.1016/s0140-6736(74)92649-x. [DOI] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. In R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2010. [Google Scholar]
- Rogers LK, Moorthy B, Smith CV. Acetaminophen binds to mouse hepatic and renal DNA at human therapeutic doses. Chem. Res. Toxicol. 1997;10:470–476. doi: 10.1021/tx960159i. [DOI] [PubMed] [Google Scholar]
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics. 1975;55:871–876. [PubMed] [Google Scholar]
- Schiodt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N. Engl. J. Med. 1997;337:1112–1117. doi: 10.1056/NEJM199710163371602. [DOI] [PubMed] [Google Scholar]
- Schonberg SA, Skorpen F. Paracetamol counteracts docosahexaenoic acid-induced growth inhibition of A-427 lung carcinoma cells and enhances tumor cell proliferation in vitro. Anticancer Res. 1997;17:2443–2448. [PubMed] [Google Scholar]
- Storey JD. A direct approach to false discovery rates. J. R. Stat. Soc. Series B Methodol. 2002;64:479–498. [Google Scholar]
- U.S. Department of Health and Human Services, FDA Organ-specific warnings; internal analgesic, antipyretic, and antirheumatic drug products for over-the-counter human use; final monograph. Final rule. Fed. Regist. 2009;74:19385–19409. [PubMed] [Google Scholar]
- Vermeulen NP, Bessems JG, Van de Straat R. Molecular aspects of paracetamol-induced hepatotoxicity and its mechanism-based prevention. Drug Metab. Rev. 1992;24:367–407. doi: 10.3109/03602539208996298. [DOI] [PubMed] [Google Scholar]
- Watkins PB, Kaplowitz N, Slattery JT, Colonese CR, Colucci SV, Stewart PW, Harris SC. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA. 2006;296:87–93. doi: 10.1001/jama.296.1.87. [DOI] [PubMed] [Google Scholar]
- Wu Z, Irizarry RA, Gentleman R, Martinez-Murillo F, Spencer F. A model-based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc. 2004;99:909–917. [Google Scholar]
- Yang R, Miki K, He X, Killeen ME, Fink MP. Prolonged treatment with N-acetylcystine delays liver recovery from acetaminophen hepatotoxicity. Crit. Care. 2009;13:R55. doi: 10.1186/cc7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.